")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 17
Similarities and Differences of Vascular Calcification in Diabetes and Chronic Kidney Disease
Received 4 September 2023
Accepted for publication 21 December 2023
Published 10 January 2024 Volume 2024:17 Pages 165—192
DOI https://doi.org/10.2147/DMSO.S438618
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Xiabo Wang,1 Zhongqun Wang,1 Jianqiang He2
1Department of Cardiology, Affiliated Hospital of Jiangsu University, Zhenjiang, 212001, People’s Republic of China; 2Department of Nephrology, Affiliated Hospital of Jiangsu University, Zhenjiang, 212001, People’s Republic of China
Correspondence: Jianqiang He, Department of Nephrology, Affiliated Hospital of Jiangsu University, Zhenjiang, 212001, People’s Republic of China, Email [email protected] Zhongqun Wang, Department of Cardiology, Affiliated Hospital of Jiangsu University, Zhenjiang, 212001, People’s Republic of China, Email [email protected]; [email protected]
Abstract: Presently, the mechanism of occurrence and development of vascular calcification (VC) is not fully understood; a range of evidence suggests a positive association between diabetes mellitus (DM) and VC. Furthermore, the increasing burden of central vascular disease in patients with chronic kidney disease (CKD) may be due, at least in part, to VC. In this review, we will review recent advances in the mechanisms of VC in the context of CKD and diabetes. The study further unveiled that VC is induced through the stimulation of pro-inflammatory factors, which in turn impairs endothelial function and triggers similar mechanisms in both disease contexts. Notably, hyperglycemia was identified as the distinctive mechanism driving calcification in DM. Conversely, in CKD, calcification is facilitated by mechanisms including mineral metabolism imbalance and the presence of uremic toxins. Additionally, we underscore the significance of investigating vascular alterations and newly identified molecular pathways as potential avenues for therapeutic intervention.
Keywords: chronic kidney disease, vascular calcification, diabetes mellitus
Graphical Abstract:
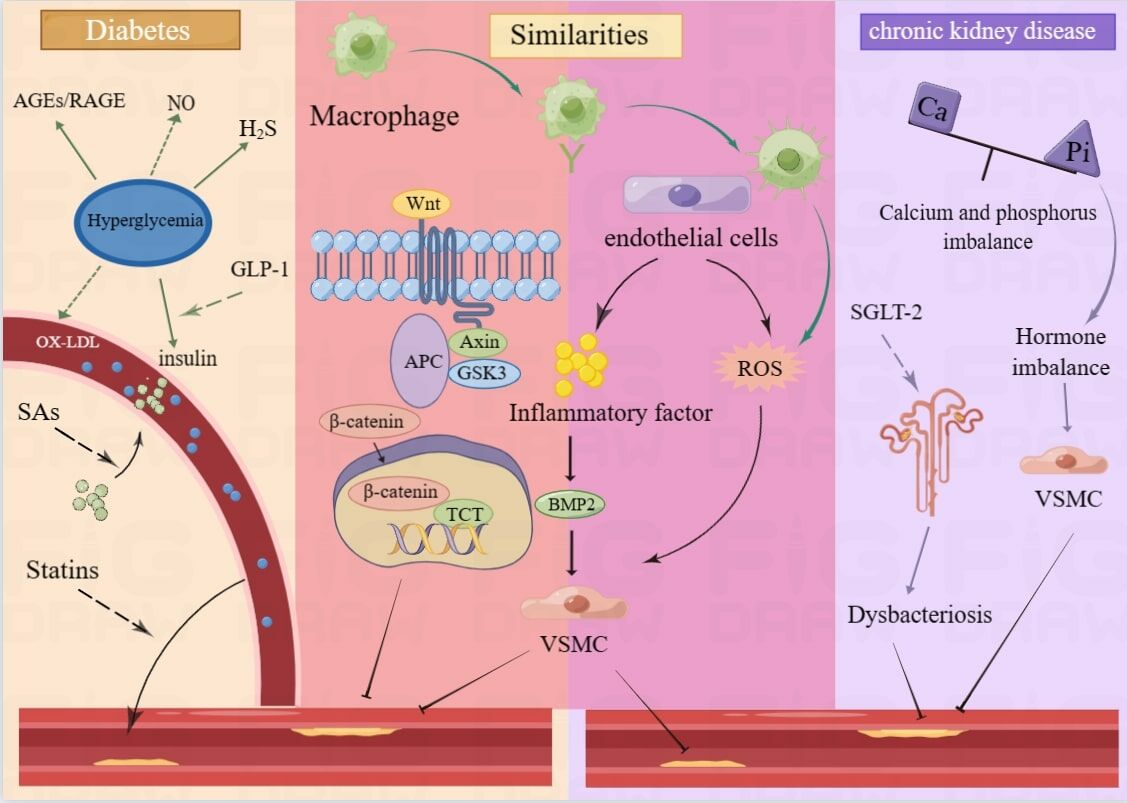
Introduction
The VC procedure parallels the bone morphogenesis process and could frequently be delineated as progressive atherosclerosis, culminating in diminished vascular elasticity.1 Ultimately increasing the risk of future cardiovascular events. There are two primary forms of calcification; intimal calcification, which is present in atherosclerotic lesions of the intima, and medial calcification, also known as Mönckeberg sclerosis, which involves calcification of the smooth muscle layer of the vascular mesentery.2 Cardiovascular disease is an essential terminal phase alteration in type 2 diabetes mellitus (T2DM) and CKD, and it is the most prevalent cause of mortality in the populace, wherein the high frequency of VC is a significant contributing risk factor.3
At present, an array of investigations have demonstrated that both DM and CKD are intimately interconnected with VC. Recent evidence has revealed a multitude of shared mechanisms that accelerate VC in the milieu of both diseases. These include mitochondrial oxidative stress, pro-inflammatory cytokine release, endothelial impairment, lipid metabolic aberrations, hypoxia, common signalling cascades (such as Advanced Glycation End products (AGEs)/Receptor for AGEs (RAGE)), extracellular vesicles, miRNAs, etc. Despite these shared mechanisms, the differences between the two in VC conditions are equally noticeable. VC is primarily caused by metabolic disorders such as hyperglycemia and hyperlipidemia, whereas in CKD chronology, VC is exacerbated by calcium and phosphorus imbalances, hormonal disorders, and intestinal leakage of large amounts of uremic toxins. This review will summarize recent advances in VC mechanisms in the context of CKD and diabetes. We will also highlight vascular alterations and newly detected molecular pathways as promising novel therapeutic targets. Insights into molecular mechanisms and the identification of novel targeted agents for calcification will foster the discovery of novel biomarkers and therapeutic strategies for cardiovascular disease.
Relationship Between Two Different Environments and Vascular Calcification
Although many aspects of VC pathogenesis remain elusive, it is currently recognized as a multi-factorial process that requires careful regulation and continuous suppression. Numerous pathognomonic features have been extensively documented as being correlated with VC. Prolonged hyperglycemia serves as a hallmark feature of diabetes mellitus, with its clinical association with VC firmly established. There is mounting evidence that numerous elements of metabolic syndrome (Mets) linked to type 2 diabetes may promote VC, including oxidative stress, endothelial abnormalities, dyslipidemia, disordered mineral metabolism, and elevated synthesis of pro-inflammatory cytokines. In CKD patients, VC is amplified by multiple interconnected mechanisms, including aberrant mineral metabolism, inflammatory pathways, hormonal dysregulation, and uremic toxins, all of which could potentially augment VC directly in such patients. Therefore, we will delineate recent advances in VC pathogenesis in the context of diabetes and nephropathy. The similarities and correlations of these pathways will be discussed in more detail. VC plays a critical role in the progression of both diseases.
Similarities in the Mechanisms of Vascular Calcification in Both Environments
VC is a common risk factor for both diabetes and chronic kidney disease. The mechanisms of VC in these two environments have many similarities (Table 1).
 |
Table 1 Similarity of Vascular Calcification Mechanisms in the Two Environments |
Pro-Inflammatory Factors
Chronic inflammation is a well-documented phenomenon in the development of VC and DM, in addition to Renal Disease. The present scientific consensus suggests that VC is essentially a chronic inflammatory state. There is substantial evidence to demonstrate that pro-inflammatory cytokines, such as interleukin-1 (IL-1), interleukin-6 (IL-6),46 interleukin-19 (IL-19), angiopoietin-2 (Ang-2), transforming growth factor beta (TGF-β),47 and tumor necrosis factor-alpha (TNF-α)12 play crucial roles in the etiopathogenesis of VC.48 Furthermore, evidence has also linked the augmented levels of TNF-α, IL-6, and interleukin-1β (IL-1β) with the initiation of VC by catalyzing the osteochondral differentiation of vascular smooth muscle cells (VSMCs).4,5 Additionally, it is well-established that the activation of t tissue nonspecific alkaline phosphatase (TNAP) is instrumental in the formation of hydroxyapatite crystals,49 and TNF-α promotes the expression of matrix metalloproteinase 2 (MSX2) via the nuclear factor-κB (NF-κB) signalling pathway, thereby enhancing TNAP activity and thereby promoting VC.6,7 Furthermore, TNF-α stimulates the production of IL-6, which, in turn, induces osteochondral maturation of VSMCs through bone morphogenetic protein-2 (BMP2) mediated amplification of IL-6,7,50 Notably, reactive ROS released by IL-6, IL-1β, and TNF-α are shown to regulate VC through activation of AKT signalling and regulation of the associated transcription factor 2 (Runx2) / core-binding factor alpha 1 (CBFA1).8 Reactive ROS also stimulate NF-κB and NF-κB dependent bone-induced signalling pathways9 and augment pro-inflammatory reactions to VSMCs.51 In addition, protein kinase A (PKA) is demonstrated to suppress the calcification of human aortic smooth muscle cells that alleviate inflammatory reactions.52 Research indicates that finerone (an Angiotensin-converting enzyme II receiver antagonist (MRA) mitigates the mRNA expression of M1 macrophage markers IL-1β and the pro-inflammatory factor TNF-α.52 Therefore, the suppression of inflammatory cytokines may serve as a promising strategy to mitigate cardiovascular risk among individuals with diabetic nephropathy.
Decreased Expression of Silent Mating Type Information Regulation 2 Homolog- 1(SIRT1)
SIRT1, a NAD-dependent deacetylase, is strongly associated with cellular metabolism, energy stress, and longevity. Acetylation of Runx2 promoter significantly controls Runx2 expression, leading to osteogenic differentiation. In an environment of heightened glucose levels, SIRT1 expression in the serum of diabetic subjects is notably decreased due to increased acetylation of Runx2 promoter in comparison with healthy individuals and under osteogenic conditions.53 This implies that an increased acetylation of Runx2 promoter in cells is associated with reduced SIRT1 expression (Figure 1). Additionally, Liu et al54 conducted an experiment demonstrating that treatment of human VSMCs with spermidine (Spd) resulted in a decrease in BMP2 and Runx2 activity on the seventh day. This, in turn, led to an up-regulation of SIRT1 in VSMCs, which ultimately reduced the expression of endoplasmic reticulum (ER) stress signal transduction molecules, contributing to CKD arterial calcification. These findings highlight the potential of SIRT1 activators in managing VC.
 |
Figure 1 Mechanism of oxidative stress promoting vascular calcification in diabetes. In the environment of diabetes, AGEs bind to advanced glycosylation receptor (RAGE), which activates NF-kB and promotes vascular calcification by enhancing the expression of VEGF, VCAM-1 and other genes. PKC pathway increases ROS level of endothelial cells by reducing NO production.Oxidative stress promotes VC by activating p21, JNK, p38 and NF-kB. ROS excess inhibits insulin gene transcription by opening ATP-sensitive K+channels, thus inhibiting insulin production and secretion. Oxidative stress promotes VC by activating p21, JNK, p38 and NF-kB. Upregulation of SIRT1 in VSMC leads to downregulation of endoplasmic reticulum (ER) stress signal transduction components, regulating SIRT1 and ER stress signals to inhibit arterial calcification in CKD. Graphics by Figdraw. |
Similarities in Oxidative Stress
Oxidative stress has been demonstrated to be strongly associated with vascular endothelial dysfunction. The primary mechanistic pathways involved in vascular endothelial cell dysfunction include PKC, AGE/RAGE, and additional pathways. Specifically, PKC reduces the production of nitric oxide (NO),14 which is the key regulatory factor for the normalization of vascular function, which is a critical regulatory factor for the proper function of the vascular system. Furthermore, the AGEs/RAGE pathway promotes vascular endothelial cell inflammation and thrombosis by increasing the content of reactive oxygen species (ROS) within endothelial cells (ECs), stimulating the activation of NF-κB, and enhancing the expression of vascular endothelial growth factor (VEGF), vascular cell adhesion molecule-1 (VCAM-1), and other genes.13,14 (Figures 1) Antioxidant stress is also implicated in VC by stimulating the expression of p21, c-Jun N-terminal kinase (JNK), p38, and NF-κB. Furthermore, other animal experiments have confirmed the role of NADPH oxidase family protein 1 (Nox1) in oxidative stress associated with diabetes-induced atherosclerosis,15 (Figures 1) while a similar role is observed in CKD. NADPH oxidase inhibitors, such as Dextromethorphan (DXM), effectively decrease ROS production and impede calcifying mediators-induced VSMCs calcification.55,56
Oxidative Stress - Downstream Signaling Pathway
Adequate scientific research suggests that an increase in oxidative stress is often associated with an increase in the expression of RUNX2 and alkaline phosphatase (ALP), and the calcification of VSMCs.57 Furthermore, the generation of hydrogen peroxide from a variety of sources in blood vessel cells is considered a significant contributor to oxidative stress in atherosclerotic lesions.19 As such, inhibiting the activation of the H2O2-activated AKT signalling pathway has the potential to suppress the increased expression of RUNX2 and the calcification of VSMCs.8 Concurrently, it has been established that the accumulation of advanced oxidation protein products (AOPPs) can promote the osteoblast differentiation of human VSMCs through the activation of the ERK signalling pathway, thereby stimulating VC.16 Other studies have revealed that the oxidative stress triggered by the ERK1/2 MAP kinase17,58 and p38 MAPK pathway can promote bone induction17 of VSMCs. Furthermore, oxidative stress can trigger the apoptosis of VSMCs and the progression of VC.59,60 It is increasingly being recognized that the upregulation of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) can lead to a decrease in antioxidant enzyme expression (including superoxide dismutase (SOD)) and an increase in mitochondrial reactive oxygen species (mtROS) production, which are associated with VC.61, In addition, recent studies have identified an increase in posttranslational protein modification via oxidative stress and O-linked β-n-acetylglucosamine modification (o-glcnac acylation), which is linked to calcium flux62,63 and the regulation of calcium-dependent signalling.61,64 The calcium-sensing protein STIM1 is known to regulate intracellular calcium flux and intracellular calcium influx through store-operated calcium portals.65 Additional research has demonstrated that an increase in calcium flux, calcium signalling activation and ER stress in STIM1δ / ΔVSMCs can lead to STIM1-deficient O-GlcNAcylation and VSMC calcification.18 Sirtuin 3 (SIRT3) also plays a crucial role in mediating the protective effects of PGC-1α on mitochondrial oxidative stress and VC.66 It is worth noting that a recent study has highlighted the essential role of antioxidants in defending against ROS attacks.67 Therefore, antioxidants should be included as an integral component of modern treatment strategies. Antioxidant therapy has shown promising results in preventing and slowing the development of VC in patients with CKD and DM.
Similarities in Endothelial Dysfunction
The role of endothelial dysfunction in the development of cardiovascular disease has been extensively investigated. Endothelial cells serve as vital regulators of vascular tone through the release of vasodilators such as NO and vasoconstrictors like endothelin-1 (ET-1). In response to physical strain and autonomic molecules, ECs control the behaviour of VSMCs and contribute to the maintenance of vascular health.10,68 The potential regulatory roles of NO and ET-1 in VC have been the focus of recent studies. Evidence suggests that NO can inhibit TGF-induced Smad2/3 phosphorylation, thereby preventing SMC calcification and osteochondrocyte transformation.10 Conversely, ET-1 has been shown to contribute to VC by suppressing calcification inhibitors like matrix Gla protein (MGP) and osteoprotegerin (OPG). Moreover, ET-1 also promotes the production of pro-calcification mediators like BMP2 and osteopontin (OPN).11 In an in vitro study conducted by Sanchez-Duffhues et al.12 IL-1β and TNF-α were found to suppress the expression of bone morphogenetic protein receptor type II (BMPR2) in endothelial cells. This suppression was associated with activation of the BMPR-c-Jun N-terminal kinase (JNK) signaling axis, a pathway that plays a crucial role in inflammation-induced endochondral metaplasia and promotes endothelial osteogenic transformation. Individuals with CKD and hyperglycemia are at a relatively high risk of developing VC. High levels of glucose can induce mineral metabolic imbalance in ECs by increasing calcium and potassium uptake, leading to an irregular phosphorylation of endothelial nitric oxide synthase (eNOS) and overexpression of osteopontin through the p38 pathway.69 These alterations ultimately undermine endothelial function and lead to the development of VC.
The beneficial effects of metformin, an anti-diabetic medication, on VC have been demonstrated in animal models. Metformin has been shown to mitigate VC through activation of the adenosine monophosphate kinase (AMPK)/eNOS/TGF-β1 signaling pathway.70 In summary, the regulation of vascular tone by ECs in response to autonomic molecules and physical strain is critical for maintaining vascular health. The identification of key pathways such as NO and ET-1 as potential targets for the prevention and treatment of VC provides important insights into the development of novel therapeutic strategies for this prevalent disease.
Oxygen Deficit and Vascular Calcification
Hypoxia significantly enhanced the protein expression of HIF-1α (Hypoxia-Inducible Factor-1α), GLUT1 (Glucose Transporter 1), and VEGFA (Vascular Endothelial Growth Factor A) mRNA expression. The activity of RUNX2, SOX9 (Sex-determining region Y (SRY)-box 9), OCN (Osteocalcin), and ALP (Alkaline phosphatase) proteins was also increased. A time-dependent accumulation of calcium was detected within the extracellular matrix (ECM) of VSMCs in response to hypoxia.36 Hypoxia-induced osteochondrogenesis and differentiation of VSMCs is triggered by HIF-1-dependent and mitochondria-derived reactive oxygen species, subsequently promoting VC. The expression of OCN and ALP genes, regulated by osteochondrogenic transcription factors, also increased in VSMCs under hypoxia.20 Several studies have indicated that hypoxia-induced ROS generation is mediated by HIF-1 activation in VSMCs.71 In addition, hypoxia enhances osteochondral differentiation and extracellular matrix calcification through ROS. It is believed that excess ROS generated during hypoxic conditions elevates the expression of HIF-1α.19 Furthermore, Zhao et al9 reported that mitigation of mitochondrial ROS generation can hinder phosphate-induced VSMCs osteogenic differentiation.
Lipid Metabolism Disorders
VC is often associated with disruptions in lipid metabolism. A well-researched correlation between diabetes and atherosclerosis is the concentration of small, dense, low-density lipoprotein (sdLDL-C).72 Notably, the prevalence of type B (with small-sized LDL particles) is higher in patients with diabetes,73 which contributes to the development of VC.74 Furthermore, the formation of sdLDL is strongly associated with insulin resistance and elevated triglycerides.73 Although the concentration of LDLC in patients with moderate to advanced CKD does not appreciably increase, oxidized low-density lipoprotein cholesterol (oxLDLC) and sdLDL-C tend to rise.22,23 In experimental conditions of high phosphate exposure, fat cells have been found to induce vascular smooth muscle cell calcification by secreting adipokines such as VEGF.75 PGC-1α, as a metabolic master regulator, controls downstream target genes in the nucleus and mitochondria through the recruitment of coactivators. It plays a pivotal role in mitochondrial biogenesis, fatty acid oxidation, and adipogenesis.76,77 In a pioneering study, Feng et al21 demonstrated that the activation of the PGC-1α/SIRT3 pathway can inhibit VC. This suggests that the PGC-1α/SIRT3 pathway may be a promising therapeutic target for managing VC. Additionally, statins have been shown to reduce cholesterol levels and lesion areas in mice fed a high-fat “Western” diet. Therefore, it is hypothesized that the adjustment of triglycerides and the use of statins may assist in delaying the onset of VC in patients.
Microelement
The principal emphasis of this research pertains to the significance of magnesium and zinc in the prevention of VC. Numerous randomized studies have demonstrated that magnesium, via the antagonism of calcification-promoting protein (CPP) maturation, can inhibit the progression of VC.24 Magnesium has been observed to negatively regulate osteogenic transdifferentiation and VC25 by enhancing the activity of its transporter, transient receptor potential melastatin 7 (TRPM7), and its invasion into cells, resulting in the expression of anti-calcification proteins such as bone morphogenetic protein 7 (BMP-7), OPN, and MGP. Furthermore, magnesium has been shown to inhibit the WNT /β-catenin signalling pathway and repress Pit-1 expression26,27 within VSMCs. The anti-calcification influence of magnesium is mediated by its ability to impede the WNT /β-catenin pathway, thereby preventing calcium from penetrating VSMCs and osteogenic transdifferentiation.78 In addition, magnesium has the potential to prevent the formation of extracellular hydroxyapatite, thereby preventing VSMC calcification.79 Magnesium has also been shown to influence bone formation (Runx2, Smad1, and Osterix) by down-regulating microRNA expression (miR-133a, miR-30b, and miR-143a).28 Emerging evidence has highlighted the role of zinc in oxidative stress resistance and its potential in mitigating the progression of VC.29 Specifically, zinc may deter phosphate-induced arterial calcification by inhibiting the activation of NF-κB light chain enhancer. In individuals suffering from type 2 diabetes and CKD, serum zinc levels have been found to correlate positively with T50 (a shorter T50 indicates a heightened propensity to calcify).29 Thus, the judicious supplementation of magnesium, zinc, and various other trace elements may provide a promising approach to delaying the progression of VC.
AGE/RAGE Signal Cascade
AGEs refer to a class of proteins that are indirectly modified by the automatic oxidation of carbohydrates and reactive carbonyl compounds that are formed through the AGE process. The circulating AGEs in patients on dialysis, including pentanoside, are often found to be elevated.80 This class of molecules has been shown to act through a feedforward cycle, which results in the upregulation of RAGE expression and the exacerbation of oxidative stress.32,33 Upon interaction between AGEs and RAGE, RAGE initiates a downstream signaling cascade through PKC-ζ, which is mediated by p38 mitogen-activated protein kinase (MAPK), TGF-β and NF-κB, and PKC.30,31 Studies have shown that RAGE activation by AGE/RAGE signaling can stimulate the generation of reactive ROS through specific pathways such as TGF-β, NF-κB, and Nox-1.32 Evidence suggests that members of the AGE/RAGE signaling cascade, including p38 MAPK and ERK1/2, are phosphorylated following RAGE activation.34 Moreover, TGF-β signaling can lead to the phosphorylation of Smads, a family of transcriptional regulators.81 This implies that the AGE/RAGE cascade is also associated with Smad activation and TGF-β signaling.33–35 RAGE signaling has also been implicated in various mitotic pathways that regulate VSMC calcification. The p38 MAPK pathway is an essential component of AGE/RAGE-induced VSMC differentiation. The role of AGE/RAGE signaling in diabetes-induced VC is attributed to the calcification caused by oxidative stress and the AGE-induced transformation of VSMC phenotype.36,37 In addition, the AGE/RAGE signal has been proven by activating and lower SOD Nox-1 expression to increase the oxidative stress and promotes diabetes mediated VC.32,82
Furthermore, studies have demonstrated that AGE/RAGE signaling can directly affect ALP activity, where p38 MAPK plays a key role.83,84 Janda et al85 have found that elevated serum levels of fetal protein A (fetuin-A) are a positive indicator of increased AGE deposition in arteries, suggesting that fetuin-A may indirectly influence the AGE/RAGE pathway, particularly in the presence of inflammatory molecules. Studies have also linked AGEs to matrix alterations, specifically to the formation of irreversible crosslinks of stromal collagen and elastin, resulting in an increase in ECM stiffness and the development of arteriosclerosis. Non-enzymatic glycosylation of collagen renders it resistant to enzymatic degradation and contributes to its accumulation in the arterial wall.38,39 RAGE mediates PI-induced VSMC calcification86 in vitro and VC87 in enpp1 − / − mice. An example of AGE-induced matrix alterations is seen in enpp1 − / − mice, where AGEs alter the glomerular basement membrane, leading to increased albumin permeability, endothelial cell pathology, and the activation of pro-atherogenic and prothrombotic genes. Ultimately, this may lead to an impediment in the selective absorption of high-density lipoprotein (HDL) ester and HDL cholesterol from peripheral cells, thereby impeding the reverse transport of cholesterol.88
Extracellular Vesicles
Apart from the release of secretory vesicles by specialized cells, which carry, for example, hormones or neuro- transmitters, all cells are capable of secreting various types of membrane vesicles, known as extracellular vesicles, and this process is conserved throughout evo- solution from bacteria to humans and plants.89,90 Notably, these vesicles carry microRNAs (miRNAs, miRs), mRNAs, and proteins that undergo profound changes in protein composition as a result of elevated phosphate levels.91 From VSMCs, the apoptotic bodies of cells undergoing apoptosis (ABS) can also serve as a form of calcium crystal in the nuclear architecture, similar to matrix vesicles (MV).92 In physiological conditions, these vesicles remain free of crystals as they are equipped with potent mineralization inhibitors such as fetuin-A and MGP.93,94 However, an increase in phosphatemia and hypercalcemia can stimulate the secretion of matrix vesicles by human VSMCs, potentially triggering the mineralization of the extracellular matrix in bone.40 In addition, it has been found that apoptosis induced by high phosphate may be related to extracellular phosphate-induced VSMC calcification.40 Conversely, other research has concluded that phosphate-induced VSMC calcification is independent of apoptosis or extracellular vesicles.95 Therefore, whether extracellular vesicles can be novel targets for VC remains to be investigated.
Similarity of microRNA in Vascular Calcification
Recently, a study has found a number of miRNAs in diabetes, CKD, and coronary heart disease (CAD), which have a high prevalence of calcification: miR-21, and miR-146a are common in these diseases and have a regulatory role in the transdifferentiation of VCm and SMCs into osteoblast-like cells. Current evidence suggests that miRNAs are implicated in modulating various intracellular pathways that regulate osteogenic / chondrogenic phenotypic transitions in VSMCs, Including WNT/β-catenin pathway,96 PI3K signaling pathway,41 STAT3 signaling pathway42 or TGF-β1 /SMAD signaling pathway.43 These miRNAs exert a critical role in osteogenic/chondrogenic transdifferentiation of VSMCs by controlling various cellular operations under conditions of hyperphosphatemia, such as gene expression,44,45,97–99 inflammasome activation,100 apoptosis,98,101 senescence,42,98 or endoplasmic reticulum stress.101
A study by Zhou et al102 confirmed that the expression of miR-21 is stimulated by hemodynamic forces (such as oscillatory shear stress (OSS)) and facilitates the pro-inflammatory response in human umbilical vein endothelial cells (HUVECs) by augmenting the binding of c-Jun to the miR-21 promoter region. In endothelial cells, miRNAs suppress the expression of genes associated with NF-κB signalling, potentially playing a role in the progression of endothelial disease. Additionally, miR-146a-5p has been identified as a suppressor of NF-κB103 Diminished expression of miR-146a might result in diminished efficacy of suppression of target genes involved in NF-κB and other cytokine production and signalling pathways. Furthermore, interleukin-1 receptor-associated kinase (IRAK)1, TNF receptor-associated factor 6 (TRAF6), and IRAK2 have been identified as self-targets of miR-146a,104 further highlighting its role in mitigating inflammatory cytokine production and pro-inflammatory conditions. The role of epigenetics44 and the currently recognized microRNAs related to VC.45,105 This research sheds light on the potential role of miRNAs in VC and the complex regulatory network involving various miRNAs and their targets in the progression of cardiovascular disease.
Differences in Vascular Mechanisms Between the Two Environments
The development of VC is a complex and multifaceted process involving a number of mechanisms, such as chronic inflammation, oxidative stress, and dysregulation of bone-mineral metabolism. While both DM and CKD share certain similarities in their aetiologies, they also exhibit several significant differences in their respective mechanisms of development (Table 2). In the context of DM, hyperglycemia and hyperlipidemia serve as primary metabolic ecosystems that drive the progression of VC. In contrast, CKD is associated with a variety of metabolic disorders that contribute to the exacerbation of VC, such as disturbances in calcium and phosphorus metabolism, hormonal imbalances, and increased release of uremic toxins from the gastrointestinal tract. These disparities further underscore the distinct pathologies underlying VC in the two conditions.
 |
Table 2 Differences in Vascular Calcification in the Two Environments |
Differences in Oxidative Stress
The investigation of elevated blood sugar as a potent stimulus of oxidative stress has shown that it amplifies the circulatory flux of citric acid, stimulating NF-κB activation.133 On the converse, an excessive production of reactive ROS can repress insulin gene transcription, mediated by the activation of ATP-sensitive K+ channels, thereby inhibiting the generation and release of insulin. Alternatively, the augmented blood glucose level in diabetic patients facilitates the accumulation of advanced glycation end products (AGEs), which bind to the receptor for advanced glycation (RAGE), initiating inflammatory signalling and escalating oxidative stress.107,108 (Figure 1) In addition, within the context of CKD, phosphorus instigates oxidative stress in VSMCs by inciting an imbalance between antioxidant and reactive ROS production mechanisms,9,109,134 and phosphate absorption by PIT1 may lead to intracellular alkalization.135 Zhao et al9 have established for the first time that mitochondrial ROS-driven p65 nuclear translocation is implicated in the hyperphosphate-induced inflammation in VSMCs. This finding further supports the notion that phosphate-induced ROS production and oxidative stress are both significant contributors to this phenomenon.109,135 The precise mechanisms by which elevated blood sugar and phosphorus contribute to oxidative stress in CKD remain to be fully elucidated. Future research should focus on elucidating the complex interplay between these factors and their potential impact on the progression of CKD.
Loss of Klotho Expression
It is well known that dysregulated mineral metabolism and exceptionally high levels of phosphorus and calcium are the most essential factors for the development and occurrence of calcification in CKD patients.106 The critical importance of minerals to human anatomy was recently underscored by an observational cohort study.136 Calcium homeostasis is primarily regulated by two major hormones, parathyroid hormone (PTH) and 1.25-dihydroxyvitamin D3 (1,25D3). In contrast, phosphorus homeostasis is predominantly controlled by the fibroblast growth factor-23 (FGF-23)/klotho axis (Klotho is a soluble anti-aging protein predominantly expressed in the kidney as a co-receptor for FGF23). Additionally, PTH and 1.25D3 mediate calcium homeostasis. In the context of CKD, the loss of Klotho expression can instigate calcification and osteoblast differentiation of VSMCs137 (Figure 2B). An investigation revealed that pretreating Klotho protein in human umbilical vein endothelial cells co-treated with inorganic sulfate (IS) decreased the expression of superoxide radicals and monocyte chemoattractant protein-1 (MCP-1)110 (Figure 2A). Additional research has also highlighted the role of the FGF-Klotho axis in VC in CKD patients, exacerbating calcium and phosphorus dysregulation138 (Figure 2C). Evidence suggests that calcimimetic agents can decrease calcium and phosphorus levels in the aorta139 and prevent the progression of VC in uremic mice.140
 |
Figure 2 Mechanism of calcium and phosphorus imbalance in chronic kidney disease. (A) In the environment of chronic kidney disease, the decline of renal function leads to the dysfunction of P excretion and the increase of serum Pi level, and the decrease of 1.25(OH)2D3 secretion leads to the decrease of serum Ca level. At the same time, renal injury changes the intestinal microenvironment and reduces the absorption of Ca. In addition, in the environment of chronic kidney disease, hyperparathyroidism and increased PTH levels can not only promote the activity of osteoblasts and osteoclasts, thereby promoting calcification, but also increase the serum Pi level and promote changes in the intestinal microenvironment. (B) PTH, FGF-23, Klotho, and 1,25D have feedback loops to maintain calcium and phosphorus homeostasis. (C) In the environment of chronic kidney disease, the hormone levels of PTH, 1,25D and FGF-23 are dysregulated, thereby increasing the osteogenic activity of osteoclasts and promoting vascular calcification. Graphics by Figdraw. |
The dependence of the sodium phosphate transporter on solute carrier membrane protein (SLC) group is crucial. Type II transporters (SLC34 family) consist of renal API-IIa (SLC34A1), intestinal API-IIb (SLC34A2),141 and NaPi-IIc (SLC34A3).142 The renal NaPi-IIa (SLC34A1) incorporates NaPi-IIaelectrogenic NaPi-IIa/b and electroneutral NaPi-IIc. These molecules are regulated by numerous factors, including parathyroid hormone (PTH) and FGF23, which are instrumental in mineral metabolism.143,144 The SLC20 family encompasses two proteins: Pit 1 (SLC20A1) and PiT-2 (SLC20A2). The phosphate signaling in osteoblasts and VSMCs may be orchestrated by the SLC20 transporter family. Pit 1 and PiT-2 are considered indispensable for Pi influx in systemic cells.145 A study indicated that phosphate uptake in VSMCs occurs through type III sodium-dependent P-cotransporters PiT-1 and PiT-2. These PiT transporters exhibit contrasting functions on VSMCs under hyperphosphatemic conditions.146 PiT-1 exerts a pathological effect by stimulating osteogenic differentiation of VSMC and the discharge of calcium phosphate-laden vesicles.111,112,146 Downstream effects of Pit-1 encompass Runx2/ core-binding factor alpha 1 (CBFA1) and osteopontin transcription.146 In contrast, PiT-2 deficient VSMC exhibited increased stromal calcification, which may be mediated by suppression of osteoclast inhibitor osteopontin.112
About Phosphate Dependency Bone Induction Signal Cascade
NF-kB Signaling Pathway
Within VSMCs, an increase in extracellular phosphate levels initiates the activation of NF-κB.113,147,148 The NF-κB signalling pathway partially stimulates the calcification of VSMCs by inducing the expression of MSX2 and elevating the levels of CBFA1, which subsequently augments the expression of ALP.148,149 TNF-α strengthens the expression of MSX2 in VSMCs via the NF-κB pathway, leading to the initiation of osteogenic/chondrogenic transdifferentiation of VSMCs.149 The receptor activator of NF-κB ligand (RANKL)/RANK system significantly contributes to VC through NF-κB activation.150
SGK1/NF-kB Signaling Pathway
Phosphate has been established as a cardiovascular toxin, particularly affecting VSMCs.151 It has been observed that phosphate can induce the expression and activation of serum and glucocorticoid-induced kinase (SGK1) in VSMCs.152 By interfering with the SGK1/NF-kB signaling pathway, phosphate could potentially maintain the anti-calcification environment of VSMCs, thereby improving VC during hyperphosphatemia.152 In a similar vein, IL-18 may activate the SGK1/NF-kB osteoinductive pathway,153 potentially contributing to the exacerbation of phosphate-induced calcification in VSMCs.153
WNT/β-Catenin Signaling Pathway
The WNT/β-catenin signaling pathway serves a critical role within the osteoinductive signaling cascade and also functions as a mediator of VC.26,154,155 Phosphate stimulation results in activation of the WNT/β-catenin pathway, leading to increased downstream effects of MSX2 and subsequent promotion of osteogenic transdifferentiation and calcification of VSMCs via direct upregulation of CBFA1156 and PIT1 gene expression.155 Moreover, the WNT/β-catenin pathway may also play a role in VC by inducing the expression of matrix metalloproteinase (MMP)2 and MMP9 in VSMCs.157 Similarly, RNA interference studies confirmed the role of the MSX gene in TNAP andexpression. Additionally, paracrine WNT signaling is integral for the refinement of osteogenic TNAP expression downstream of BMP2 and Sonic hedgehog (Shh).158
mTOR Signaling Pathway
In VSMC, klotho expression is down-regulated by phosphate,159 an effect associated with activated mTOR signalling that enhances VC.159,160
The protein kinase referred to as the mammalian target of rapamycin (mTOR) belongs to the family of PI3K-associated kinase (PIKK), a class of serine/threonine kinases. mTOR forms two distinct protein complexes, referred to as mTOR Complex 1 (mTORC1) and 2 (mTORC2), which play vital roles in various cellular processes, including protein synthesis, cell growth, and proliferation. Excessive activation of mTOR has been observed in various types of human cancers, as well as in age-related diseases such as atherosclerosis, obesity, diabetes, and Alzheimer’s disease. In a nutritionally adequate state, mTOR signalling is activated, and conversely, mTOR signalling is blocked, with it being reported that mTORC1 is specifically regulated by nutrient.161 The upregulation of mTOR by phosphate is particularly intriguing, as phosphates are essential nutrients involved in energy metabolism. Protein digestion leads to the production of amino acids and phosphates. It is likely that mTORC1 is involved in the signalling cascade activated by phosphate, which in turn activates the mTOR cascade in VSMCs, leading to a downregulation of Klotho expression. This downregulation can enhance the calcification of VSMCs.162 This upregulation of mTOR in a uremic environment is yet to be fully understood. It has been reported that mTOR inhibitors can be considered as an upregulation of Klotho. As CKD is associated with severe kidney and systemic Klotho deficiency, these drugs that upregulate Klotho have been shown to protect the kidney and vascular system. Additionally, there is substantial evidence that supports the broad-spectrum anti-atherosclerotic effects of mTOR inhibitors, and experimental studies have demonstrated that rapamycin can act as a stabilizer of atherosclerotic plaques and even promote plaque regression.163 In animal models (such as apolipoprotein E-deficient mice), rapamycin has been shown to effectively reduce inflammation, inhibit progression, and enhance the stability of atherosclerotic plaques,164 Thus, it can be anticipated that mTOR inhibitors could be used to prevent or delay the onset of atherosclerosis.
BMP-2 Signaling Pathway
TNF-α-induced VSMC calcification is associated with increased BMP2 signalling.165 Phosphate has been observed to stimulate BMP-2 expression in VSMCs Phosphate induces BMP-2 expression in VSMCS.166,167 Additionally, BMP-2 promotes VSMC calcification by stimulating the expression of PIT-1, which is upregulated and the activation of SMAD signalling pathways.165 Notably, phosphate has also been observed to promote TGFβ1 expression in VSMCs.168,169 Moreover, downstream osteoinductive signals of TGFβ1 include the transcription factor NFAT5 (also known as TonEBP), which mediates the upregulation of CBFA1 in VSMCs through SOX9. Interestingly.10,168,169 TGFβ1 may also contribute to VC by inducing cellular senescence, including the upregulation of plasminogen activator inhibitor PAI-1, which plays a pro-calcification role.10,168
Differences in Endothelial Dysfunction
Endothelial impairment has been established as a primary concern in individuals with CKD and has been found to persistently elevate throughout the course of the disease. Studies have consistently demonstrated that parathyroid hormone (PTH) and hyperphosphatemia significantly contribute to the progression of endothelial impairment.170,171 Specifically, PTH has been found to trigger osteoblast transdifferentiation in endothelial cells through the activation of the extracellular signal-regulated kinase (ERK)1/2 and NF-κB signaling cascades.127 Conversely, an experimental study has demonstrated that uremia, a significant complication of CKD, can lead to the amplification of fibroblast growth factor (FGF) signaling within endothelial cells, which exacerbates atherosclerosis.114 Additionally, recent studies have found that fluctuations in the functional properties of low-density lipoprotein-cholesterol (LDL-cholesterol) can further instigate endothelial impairment in CKD patients.115 Similarly, another in vitro experiment revealed that triggered by urinary toxin IS, injured endothelial cells generate excessive microvesicles, which modulate calcium accumulation, inflammation, and osteogenic transdifferentiation of SMCs.172 The same time, uremic toxins perturb endothelial function in CKD patients by competitively inhibiting eNOS.170 Recent studies have indicated that modifications in the functional properties of low-density lipoprotein-cholesterol may contribute to the development of endothelial impairment in patients with CKD. However, in the context of diabetes, hyperglycemia exacerbates hemodynamic damage, specifically shear stress, which can suppress NO production and induce alterations in endothelial phenotype. These changes can lead to alterations in vascular permeability and leukocyte recruitment during inflammation.173 It has been suggested that in the diabetic milieu, a decrease in the abundance of proteoglycans may provide protection against the development of atherosclerosis.116
Vitamin D and Vitamin K Deficiency
CKD is strongly associated with VC through Vitamin D deficiency. In fact, a vitamin D deficit often presents in individuals affected by CKD174 (Figure 2A). The potential mechanisms that link CKD and VC may involve the capacity of Vitamin D to inactivate RUNX2 regulation, as demonstrated in recent studies.175,176 Furthermore, individuals with CKD often experience elevated FGF23 concentrations due to a compromised Glomerular Filtration Rate (GFR). This elevation in FGF23 consequently impairs vitamin D levels.177 Moreover, 1.25(OH)2D3 suppresses NF-kB and TNFα, key signalling molecules involved in bone-induced signalling cascades.117,178 In a similar study involving patients with Type 1 Diabetes, vitamin D deficiency was associated with an increased risk of Coronary Artery Calcification (CAC).118 Therefore, vitamin D supplements have shown beneficial effects on VC in CKD.
Despite a correlation between vitamin K deficiency and enhanced VC severity in CKD patients, this correlation did not consistently lead to increased cardiovascular disease or mortality.Vitamin K deficiency was associated with increased severity of VC in patients with CKD, but, paradoxically, vitamin K deficiency was not consistently associated with cardiovascular disease and mortality.120 Potential mechanisms for vitamin K’s ability to reduce calcification propensity include an increase in the activity of vitamin K-dependent calcification inhibitors or a reduction in inflammation by inhibiting innate immune system components.179
MicroRNAs and Epigenetic Modifications at Different Points in Vascular Calcification
There were different epigenetic modifications to VC in the two environments. During hyperphosphatemia, different epigenetic modifications, including DNA methylation, histone modifications, and dysregulation of microRNAs, contribute to osteoinduced cell signaling.119,180 It has been confirmed that methylation in the SM22α promoter region can induce VC at higher pi levels. It has been reported that the downregulation of miR-29b and activation of WNT/β-catenin signalling may be involved in VC in IS-induced CKD.181 A potential lncRNA H19/miR-138/TLR3 axis121 was predicted, and its downstream microRNAs were enriched in the NF-κB signalling pathway, which may be related to VC in CKD. Interestingly, microRNAs play a role in the diabetic environment by affecting vascular dysfunction and inflammatory status. As endothelial miR-126 affects vascular dysfunction and inflammatory status,122 it is thought that activation of miR-21 may be a key event associated with the development of diabetes and atherosclerosis. It may trigger endothelial activation, affecting foam cell formation, circulating monocyte adhesion, macrophage apoptosis, and phagocyte clearance.123 In the diabetic environment, miR-21 functions again appear in inhibitory mechanisms of antioxidant response, regulating ROS homeostasis through Krev interaction trapping protein (KRIT)-1 and silencing the major mitochondrial antioxidant enzyme SOD2.182 Therefore, miRNAs could serve as potential therapeutic targets for the treatment of VC.
Intestinal Microbiota and Uremic Toxins
As a source of uremic toxins, the gut microbiome represents a significant contributor to cardiovascular disease in CKD. It executes a pivotal role in human health and the onset of kidney disease.124,183 It is hypothesized that the altered gut microbiome and its metabolic byproducts in CKD may be implicated in the VC process.184 A number of studies have shown that uremic toxins such as indoxyl sulfate (IS), p-cresol (PC), p-cresol sulfate (PCS), uric acid, etc., released by the gut microbiome, stimulate oxidative stress and augment the inflammatory state by upregulating proinflammatory cytokines such as TNF-α and IL-6.185 In addition, endotoxin (LPS) and short-chain fatty acids (SCFA) are specific categories of gut microbiome-derived metabolites that are capable of causing systemic inflammation if the intestinal barrier is compromised. (Figure 3). Uric acid may be synthesized in endothelial cells, leading to local vascular smooth muscle cell proliferation and migration through the release of platelet derived growth factor, thereby promoting inflammation through the release of monocyte chemoattractant protein-1, and intensifying oxidized low-density lipoprotein.125 Furthermore, urate deposits serve as a trigger for local intravascular inflammation, which can stimulate the progression of atherosclerosis and the formation of adjacent inflammatory plaques.126,186,187 As a result, maintaining the balance of the gut flora is crucial for the therapeutic management of CKD patients, as well as patients with end-stage renal disease (ESRD).
 |
Figure 3 Uremic toxin and intestinal flora disorder. Lipopolysaccharide and short chain fatty acid imbalance, promote intestinal flora disorder, promote intestinal microorganisms and uremic toxins to destroy intestinal barrier. Uremic toxins such as IS increase TNF-α and IL-6, leading to an increased inflammatory state by promoting oxidative stress. Graphics by Figdraw. |
Hormone Levels
The hormone release rates in the respective environments are not consistent, which in turn results in variations in the mechanisms associated with VC. The experimental findings from both in vivo and in vitro models propose that the regulation of calcification is mediated by a combination of hormones: angiotensin II,188 aldosterone,189,190 and glucocorticoid,191 such as estrogen, thyroid, adiponectin, parathyroid, FGF23, calcitriol, testosterone, insulin, etc., exert protective effects against VC. Recent animal investigations have suggested that the increase of PTH triggers the transformation of aortic endothelial cells into chondrocytes171 and the PTH receptor 1 (PTHR1) signaling of endothelial cells plays a pivotal role in PTH-induced osteogenesis and is correlated to angiogenesis.192 It is acknowledged that insulin resistance is closely associated with diabetes, and insulin stimulates the release of endothelial NO, which in turn oxidizes lipoproteins, decelerates the rate of intimal calcification, and suppresses vasa vasorum formation.128 Upon the occurrence of insulin resistance (IR), the higher concentration of free fatty acids in the liver prompts the body to amplify the absorption and very low-density lipoprotein (VLDL) triglyceride production and secretion to compensate; therefore, the susceptibility to blood vessel calcification escalates. Iguchi et al have demonstrated that individuals with higher IR levels are predisposed to develop thin cap fibroatheroma (TCFA) and exhibit notably thinner caps.193 Elevated insulinemia reduces OPG and MGP levels, activates BMP2, and enhances carboxy-terminal propeptide of type I collagen (CBFPa-1) and ALP levels, culminating in the bone-like transformation of stromal cells.129 As such, hormone levels are equally indispensable for the progression of VC.
Persistent Activation of the Renin-Angiotensin-Aldosterone System (RAAS)
The role of RAAS in the pathophysiology of hypertension, cardiovascular and renal diseases is well known.CKD complications such as RAAS activation can induce endothelial dysfunction and atherosclerosis. These pathologies coexist during the progression of CKD and exacerbate VC. VSMCs express the mineralocorticoid receptor, and the activation of mineralocorticoid promotes VC.130 As renal function declines, the standard defence mechanism, phosphorus (Pi) and calcium (Ca) endocrine axis, becomes overwhelmed and disrupted. Pi can directly induce the expression of aldosterone synthase in the adrenal gland and vascular tissues, which may be related to FGF-23-mediated activation of RAAS.131 Increased levels of angiotensin II increase the production of aldosterone that activates sodium-phosphate cotransporter (Pit-1), leading Pi to enter the VSMC. In addition, aldosterone promotes inflammatory processes by inducing TNF-α.194 Furthermore, vascular injury caused by excessive activation of mineral corticosteroid receptor (MR) can be manifested as inappropriate vascular remodelling, endothelial dysfunction and increased vascular stiffness.132
Therapeutic Strategies for Vascular Calcification in Two Environments
Research on the treatment of VC targets still has important clinical significance, the next (Table 3) summed up in two kinds of environments drugs for the treatment of VC.
 |
Table 3 Comparison of the Treatment of Vascular Calcification in Two Environments |
Metformin
Currently, metformin is the first-line medication to treat T2DM in most guidelines and is used daily by >200 million patients. Surprisingly, the mechanisms underlying its therapeutic action are complex and are still not fully understood. Metformin activates protein kinase (AMPK) through phosphorylated AMP, and by activating AMPK signalling in VSMC, the RANKL gene is an essential inducer of VC. Metformin can down-regulate the overexpression of the receptor activator of RANKL gene.224 Metformin has been shown to alleviate VC through the AMPK-eNOS-NO pathway.196 In the rat model of adenine-induced CKD, Metformin has shown that the expression of inflammatory cytokines TNF-a and IL-1β and to decrease serum PTH and FGF-23 hormone levels significantly are reduced in the kidneys of rats treated with Metformin.195 Metformin can improve macrophage dysfunction, reduce NLRP3 inflammatory vesicles, and inhibits ROS production.225 Therefore, Therefore, metformin is currently one of the most promising therapies to reduce VC. However, Interestingly, More insights into the benefits of metformin on healthspan and lifespan in humans will hopefully be provided by the MILES (Metformin in Longevity Study) and TAME (Targeting Aging with Metformin) clinical trials.197 The ability of metformin to promote health or increase lifespan in healthy populations remains to be determined.
Thiazolidinediones (TZD)
TZDs exhibit their therapeutic effects by stimulating the activity of nuclear peroxisome proliferator-activated receptor gamma (PPARγ). This stimulation of PPARγ signalling has been implicated in the regression of VC. Previous research indicates that PPARγ is capable of regulating the proliferation of VSMCs induced by platelet-derived growth factor-induced protein (PI)226 Further studies have shown that the administration of a PPAR-γ inducer was able to suppress NF-kB-dependent secretion of inflammatory cytokines in a rodent model.198 Similarly, pioglitazone, a thiazolidinedione-like drug, has been shown to mitigate TNF-α-induced endothelial dysfunction in patients with diabetes.199 A retrospective study involving 156 patients with T2DM demonstrated that rosiglitazone significantly reduced TNF-α, interleukin-6, high-sensitivity C-reactive protein (hs-CRP), and malondialdehyde (MDA) levels.200 Based on these findings, thiazolidinedione (TZD) drugs can be utilized as the primary treatment for VC. TZDs are commonly utilized as second-line oral hypoglycemic drugs. However, there is emerging evidence suggesting that long-term use of TZDs may exacerbate cardiac dysfunction.227 Therefore, it is crucial for healthcare professionals to consider multiple factors when administering TZD drugs.
Sulfonylurea
Sulfonylureas, such as glimepiride, elicit insulin release by virtue of their ability to inhibit ATP-sensitive K+ channel receptors located in pancreatic β cells.228 This phenomenon is underscored by a study demonstrating that treatment with glimepiride decreased renal NLRP-3 expression in a rodent model of adenine-induced CKD.201,204 In addition, in diabetic mice treated with glibenclamide, it was demonstrated that VC development was delayed by down-regulating IL-10, IL-18, TGF-β, and mRNA levels.202 A study on whether diabetes treatment has a beneficial effect on calcification markers showed that anti-diabetic treatment with glibenclamide for 18 weeks resulted in a significant reduction in circulating OPG, suggesting that this may slow the progression of VC.203 This finding suggests that sulfonylureas may exert anti-calcifying effects through the manipulation of inflammatory and oxidative pathways.
Dipeptidyl Peptidase-4 (DPP-4) Inhibitor
The dipeptidyl peptidase-4 (DPP4) is a crucial enzyme in the regulation of the incretin system. This system, which is responsible for regulating blood glucose levels, functions by suppressing the hormone glucagon-like peptide 1 (GLP-1). By blocking the activity of DPP4, DPP4-inhibitors (DPP4-i) can enhance GLP-1 concentration, thereby controlling blood glucose levels.229 A study conducted on high-fat diet-induced obesity-related glomerulopathy (ORG) rat model demonstrated the ability of ghrelin to suppress local inflammation by reducing the presence of mast cells and macrophages, and by decreasing the expression of proinflammatory cytokines such as TNF-a and IL-6.230 Interestingly, a random clinical study on T2D patients suggested that linagliptin treatment for 4 weeks prevented impairment of renal endothelial functions205 However, DPP4-I is currently controversial, and recent studies have shown that DPP-4 inhibition can induce cancer metastasis.206 However, this controversy in the carcinogenic mechanism of diabetes has been shown to be complex, including excessive ROS formation, destruction of essential biomolecules, chronic inflammation and impaired healing phenomena, which together lead to carcinogenic effects in diabetic patients. A recent study by Zendehdel et al231 found that patients with type 1 diabetes had a 19% increased risk of developing cancer, particularly gastric, cervical, and endometrial cancers. This risk is higher compared to the risk observed in patients with CKD. Compared to CKD, although it can promote kidney cancer itself, some other anti-tumour drugs (such as cisbo) can themselves have a harmful effect on kidney function, leading to renal cell carcinoma. The combination of cancer with impaired renal function worsens patients’ outcomes and complicates their management and treatment.
Selective Sodium-Glucose Transporter-2 Inhibitors (SSGT −2)
SGLT-2 inhibitors, such as empagliflozin, function by inhibiting glucose reuptake in the proximal tubules of the nephron.232 In an ApoE−/− mouse model, studies have indicated that chronic empagliflozin administration for eight weeks resulted in a decrease in atherosclerotic lesions and a reduction in TNF-α and IL-6 levels.233 Furthermore, clinical trials have provided support for the anti-inflammatory effects of empagliflozin in reducing atherosclerosis in individuals with T2DM.207 A subsequent analysis of the therapeutic efficacy of empagliflozin on the Kidney Disease Improving Global Outcomes (KDIGO) classification of CKD suggested that SGLT-2 inhibitors may exhibit cardioprotective and nephroprotective effects.234 A recent comprehensive meta-analysis of 11 cardiovascular outcome trials (CVOTs) examining the effects of SGLT-2 inhibitors on cardiorenal outcomes in individuals with type 2 diabetes agreed with these findings.235
GLP-1 Agonists
GLP-1 primarily modulates postprandial glucose surge through augmenting insulin production and suppressing glucagon release.236 Several recent studies have further elucidated the effects of GLP-1 and its receptor agonists on cardiac and vascular physiology, inflammatory responses, and platelet function.237 For instance, Liraglutide, a GLP-1 agonist, has been shown to significantly reduce pro-inflammatory cytokines, such as TNF-α and IL-6, thus attenuating local inflammation.230 Similarly, Exenatide, another GLP-1 receptor agonist, has demonstrated the ability to mitigate VC by downregulating RANKL expression.208 Thus, these data suggest that GLP-1 agonists may have therapeutic potential in the management of VC. Furthermore, it has been observed that co-administration of liraglutide with GLP-1 agonists has reduced the risk of cardiovascular events in patients with diabetes.238 The cardiovascular safety profile of liraglutide in patients with type 2 diabetes appears to be relatively reassuring.
Alpha-Glucosidase Inhibitors (AGI)
AGI exhibits a promising role in the treatment of VC by virtue of its ability to suppress postprandial hyperglycemia through the restraint of α-glucosidase in the brush border of the small intestinal mucosa. Studies have demonstrated the efficacy of acarbose, an α-glucosidase inhibitor, in the management of type 2 diabetes (DM) in rodent models. Acarbose treatment was observed to diminish oxidative stress and reduce reactive ROS levels.239 Additionally, it was shown to have an anti-inflammatory effect, thereby dampening pro-inflammatory factors.209,210 It is noteworthy that AGI displays the same therapeutic efficacy as other anti-diabetic medications in controlling glycosylated haemoglobin and blood glucose levels, and its effect is comparable to metformin. Despite its promising potential, AGI is currently underutilized in clinical practice, particularly in patients with advanced CKD, due to its limited safety profile in these patients. However, further research on AGI’s safety in severe renal impairment is warranted to determine its therapeutic potential in this patient population.
Antioxidants
Oxidative stress is considered to play a pivotal role in the progression of VC, which has been established as a standard mechanism for the development of VC in conditions such as DM and CKD. Research indicates that various nutrients and dietary plant metabolites exhibit antioxidant activities, such as α- and γ-tocopherols, retinol, β-cryptophanes, ascorbic acid, α- and β-carotene, lutein and zeaxanthin, and lycopene.211,212 For example, Vitamin E has been shown to act as an antioxidant and has been identified as a compound that can decelerate VC progression.212 Furthermore, Vitamin C, possessing vital antioxidant functions, is capable of reducing reactive ROS levels and maintaining vascular and endothelial functionality.213 Other dietary plant metabolites, such as quercetin,240 curcumin,184 resveratrol,241 ursolic acid and others, can also function as antioxidants and exert protective effects against VC.In a systematic review and meta-analysis of the effects of age and dose on the glycemic impact of resveratrol in managing type 2 diabetes, it was demonstrated that resveratrol significantly reduced blood glucose, insulin, and Glycosylated Hemoglobin, Type A1C (HbA1c) levels in patients aged 60 years.242 Therefore, the positive and significant role of antioxidant compounds in CKD is undeniable, highlighting the significant role they play in disease prevention and intervention through the regulation of free radicals.The consumption of these antioxidant compounds through diet, including fruits, vegetables, and grains, plays a crucial role in disease prevention and intervention.
Angiotensin-Converting Enzyme II Receptor Antagonist (MRA)
An experimental study was performed, in which angiotensin di-receptor (AT2) knockout rats were utilized. Results indicated that these rats demonstrated diminished aortic ring calcification in comparison to controls, indicating that AT2 stimulation can inhibit phosphate-induced VC.243 Finenone, an MR antagonist, can inhibit reactive ROS production, decrease oxidative stress, and prevent endothelial dysfunction, thereby providing control over VC.214,244 It is worth noting that the expression of pro-inflammatory molecules and macrophages plays a crucial role in the development and manifestation of VC. A study conducted on rats with CKD demonstrated that finenone therapy reduced the activity of matrix metalloproteinase-2,9 (MMP-2,9) in plasma, thereby curtailing VC and oxidative stress.215 In addition, MR antagonists can also block the pro-inflammatory effects of IL-6.216 Further, MRAs are considered a promising therapeutic strategy in diabetic kidney disease. Additionally, other non-steroidal MRAs have shown anti-albuminuric effects in diabetic kidney disease. Whether the concurrent administration of MRAs with other nephroprotective drugs, such as sodium-glucose cotransporter-2 (SGLT2) inhibitors, can provide synergistic protective effects requires further investigation.
Combating Mineral Disorders
It has been documented that phosphate-binding agents augment P excretion and diminish VC by reducing FGF-23 in clinical trials pertaining to phosphate disturbances.245
In a rat model, Mg alleviated inflammatory damage associated with oxidative stress.246 Zinc has been shown to inhibit ROS-induced LDL-C oxidation.48 Similarly, inadequate Zn intake was associated with increased ROS production,29 which is essential in reducing the burden of VC.219 Cinacalcet is a second-generation calcifying agent, and a clinical meta-analysis of Cinacalcet showed that Cinacalcet reduced PTH synthesis and secretion, decreased serum calcium, and was often accompanied by a decrease in phosphate, thereby reducing VC.247 Klotho is a critical protein that protects the kidney. Its deficiency is implicated in the pathogenesis and progression of CKD. Conversely, an increase in Klotho levels results in improved kidney function and delays CKD progression. Therefore, mineral supplementation may become a therapeutic agent for VC in the future.
Vitamin Supplement and New Therapies
Vitamin K supplementation is essential for VC.220 I In previously publicized studies, vitamin K-dependent carboxylation of glutamate residues activates MGP in hemodialysis patients, and VC is regulated and, to a certain extent, inhibited by MGP.248 Moreover, preliminary results from clinical trials imply that vitamin K supplementation reduces VC.249 Additionally, paricalcitol and lower clinical doses of paricalcitol may thwart VC.218 However, a clinical randomized controlled trial revealed no significant distinction in the progression of coronary artery calcification (CAC) or valvular calcification between treatment with osteopontin and paricalcitol in patients with CKD over 48 weeks.250 A more advanced drug now available in clinical studies for the treatment of VC is an inositol hexaphosphate (IP6) formulation called Sanifit’s SNF472.251 Recent studies have identified an OEG2-derived inositol phosphate (OEG2-IP4) that may have clinical implications in the prevention and treatment of VC.252 Novel therapies may help prevent VC progression in CKD and DM. Further clinical studies are needed to determine clinical efficacy.
Statins
Statins promote low-density lipoprotein metabolism and increase high-density lipoprotein concentration by inhibiting hydroxymethylglutaryl-CoA (HMG-CoA) reductase in the body’s cholesterol synthesis process.217 This has been evaluated in several randomized controlled clinical trials, each showing that statins have a significant LDL-lowering effect.221,253 However, the effect of statins on VC is still controversial. A prospective, multi-country study using continuously computed tomography angiography (CT) scans at 2-year intervals to detect atherosclerotic plaque progression showed that patients taking statins had a faster rate of lesion calcification, a lower rate of atherosclerosis progression and a lower occurrence of high-risk plaque features compared to controls.222 This means that statins do not affect progression in the percentage of coronary artery disease stenosis severity but induce plaque phenotypic transformation. Another prospective study suggests a possible mechanism by which statins promote calcium accumulation in arterial walls by inhibiting vitamin K-dependent proteins and participating in vascular protection.254 In contrast, Doran et al223 recently suggested that statin therapy promotes the reversal of significant microcalcification in mice. Another study found that treating coronary heart disease patients treated with statin with PCSK9 inhibitor evolocumab can significantly reduce LDL cholesterol and plaque regression. Therefore, whether for patients with CKD or DM, long-term statin use can significantly prolong the survival of patients by reducing apolipoprotein a, but there is also an impact on VC. Generally speaking, the benefits outweigh the disadvantages.
Conclusions and Perspectives
Due to meticulous investigation into their mechanisms, significant breakthroughs have been achieved in the pharmacotherapy of two diseases. Studies have established a link between diabetes and CKD with VC. Recent data suggest the existence of multiple, convergent mechanisms accelerating VC within the framework of these diseases. These mechanisms involve mitochondrial oxidative stress, pro-inflammatory cytokine release, endothelial dysfunction, lipid metabolic disorders, hypoxia, and common signalling cascades. Additionally, extracellular vesicles and miRnas are also implicated in VC. Although these factors share some similarities, there are also distinct differences. For example, metabolic disorders are the primary factors for VC in diabetes, while excessive calcification in CKD is intensified by calcium and phosphorus imbalances, hormone dysregulation, and the release of massive amounts of uremic toxins from the intestine. Despite significant advances in the research of VC within diabetic and CKD milieus, including the elucidation of mechanistic aspects and the initiation of clinical pharmacological trials, the complex and heterogeneous pathophysiological mechanisms underlying VC within these contexts have yet to be fully understood. This complexity has posed significant challenges for the identification of optimal therapeutic targets and the advancement of drug development. Furthermore, it is important to emphasize the need for increased research on the pathophysiological mechanisms and preventative measures of VC. By addressing these aspects of the disease, we can develop more effective strategies to mitigate the negative consequences of VC and ultimately improve patient outcomes.
Acknowledgment
Graphical abstract and figures created using Figdraw.
Funding
This work was supported as follows: the National Natural Science Foundation of China (82070455); the related Foundation of Jiangsu Province (BK20201225, BE2022780).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Demer LL. Effect of calcification on in vivo mechanical response of rabbit arteries to balloon dilation. Circulation. 1991;83(6):2083–2093. doi:10.1161/01.cir.83.6.2083
2. Proudfoot D, Shanahan CM. Biology of calcification in vascular cells: intima versus media. Herz. 2001;26(4):245–251. doi:10.1007/pl00002027
3. Rubin MR, Silverberg SJ. Vascular calcification and osteoporosis--The nature of the nexus. J Clin Endocrinol Metab. 2004;89(9):4243–4245. doi:10.1210/jc.2004-1324
4. Aghagolzadeh P, Bachtler M, Bijarnia R, et al. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis. 2016;251:404–414. doi:10.1016/j.atherosclerosis.2016.05.044
5. Awan Z, Denis M, Roubtsova A, et al. Reducing vascular calcification by Anti-IL-1β monoclonal antibody in a mouse model of familial hypercholesterolemia. Angiology. 2016;67(2):157–167. doi:10.1177/0003319715583205
6. Bhatnagar S, Panguluri SK, Gupta SK, Dahiya S, Lundy RF, Kumar A. Tumor necrosis factor-α regulates distinct molecular pathways and gene networks in cultured skeletal muscle cells. PLoS One. 2010;5(10):e13262. doi:10.1371/journal.pone.0013262
7. Hénaut L, Massy ZA. New insights into the key role of interleukin 6 in vascular calcification of chronic kidney disease. Nephrol Dial Transplant. 2018;33(4):543–548. doi:10.1093/ndt/gfx379
8. Byon CH, Javed A, Dai Q, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283(22):15319–15327. doi:10.1074/jbc.M800021200
9. Zhao MM, Xu MJ, Cai Y, et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79(10):1071–1079. doi:10.1038/ki.2011.18
10. Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovas Res. 2008;77(1):221–230. doi:10.1093/cvr/cvm049
11. Larivière R, Gauthier-Bastien A, Ung RV, et al. Endothelin type A receptor blockade reduces vascular calcification and inflammation in rats with chronic kidney disease. J Hypert. 2017;35(2):376–384. doi:10.1097/hjh.0000000000001161
12. Sánchez-Duffhues G, García de Vinuesa A, van de Pol V, et al. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol. 2019;247(3):333–346. doi:10.1002/path.5193
13. Archuleta TL, Lemieux AM, Saengsirisuwan V, et al. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: role of p38 MAPK. Free Radic Biol Med. 2009;47(10):1486–1493. doi:10.1016/j.freeradbiomed.2009.08.014
14. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668. doi:10.1126/science.271.5249.665
15. Van Belle E, Rioufol G, Pouillot C, et al. Outcome impact of coronary revascularization strategy reclassification with fractional flow reserve at time of diagnostic angiography: insights from a large French multicenter fractional flow reserve registry. Circulation. 2014;129(2):173–185. doi:10.1161/CIRCULATIONAHA.113.006646
16. You H, Yang H, Zhu Q, et al. Advanced oxidation protein products induce vascular calcification by promoting osteoblastic trans-differentiation of smooth muscle cells via oxidative stress and ERK pathway. Renal Failure. 2009;31(4):313–319. doi:10.1080/08860220902875182
17. Blanc A, Pandey NR, Srivastava AK. Distinct roles of Ca2+, calmodulin, and protein kinase C in H2O2-induced activation of ERK1/2, p38 MAPK, and protein kinase B signaling in vascular smooth muscle cells. Antioxid Redox Signal. 2004;6(2):353–366. doi:10.1089/152308604322899422
18. Erickson JR, Joiner ML, Guan X, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–474. doi:10.1016/j.cell.2008.02.048
19. Liberman M, Bassi E, Martinatti MK, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28(3):463–470. doi:10.1161/atvbaha.107.156745
20. Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007(407):cm8. doi:10.1126/stke.4072007cm8
21. Feng H, Wang JY, Zheng M, et al. CTRP3 promotes energy production by inducing mitochondrial ROS and up-expression of PGC-1α in vascular smooth muscle cells. Exp Cell Res. 2016;341(2):177–186. doi:10.1016/j.yexcr.2016.02.001
22. Ribeiro S, Faria Mdo S, Silva G, et al. Oxidized low-density lipoprotein and lipoprotein(a) levels in chronic kidney disease patients under hemodialysis: influence of adiponectin and of a polymorphism in the apolipoprotein(a) gene. Hemodial Int Symp Home Hemodial. 2012;16(4):481–490. doi:10.1111/j.1542-4758.2012.00687.x
23. Chu M, Wang AY, Chan IH, Chui SH, Lam CW. Serum small-dense LDL abnormalities in chronic renal disease patients. Br J Biomed Sci. 2012;69(3):99–102. doi:10.1080/09674845.2012.12069133
24. Bressendorff I, Hansen D, Schou M, et al. Oral magnesium supplementation in chronic kidney disease stages 3 and 4: efficacy, safety, and effect on serum calcification propensity-a prospective randomized double-blinded placebo-controlled clinical trial. Kidney Int Rep. 2017;2(3):380–389. doi:10.1016/j.ekir.2016.12.008
25. Montezano AC, Zimmerman D, Yusuf H, et al. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension. 2010;56(3):453–462. doi:10.1161/hypertensionaha.110.152058
26. de Oca A M, Guerrero F, Martinez-Moreno JM, et al. Magnesium inhibits Wnt/β-catenin activity and reverses the osteogenic transformation of vascular smooth muscle cells. PLoS One. 2014;9(2):e89525. doi:10.1371/journal.pone.0089525
27. Sonou T, Ohya M, Yashiro M, et al. Magnesium prevents phosphate-induced vascular calcification via TRPM7 and Pit-1 in an aortic tissue culture model. Hypertens Res. 2017;40(6):562–567. doi:10.1038/hr.2016.188
28. Louvet L, Metzinger L, Büchel J, Steppan S, Massy ZA. Magnesium attenuates phosphate-induced deregulation of a MicroRNA signature and prevents modulation of smad1 and osterix during the course of vascular calcification. Biomed Res Int. 2016;2016:7419524. doi:10.1155/2016/7419524
29. Nakatani S, Mori K, Shoji T, Emoto M. Association of zinc deficiency with development of CVD events in patients with CKD. Nutrients. 2021;13(5):1680. doi:10.3390/nu13051680
30. Hegazi RA, Sutton-Tyrrell K, Evans RW, et al. Relationship of adiposity to subclinical atherosclerosis in obese patients with type 2 diabetes. Obesity Res. 2003;11(12):1597–1605. doi:10.1038/oby.2003.212
31. Nakanishi R, Rajani R, Cheng VY, et al. Increase in epicardial fat volume is associated with greater coronary artery calcification progression in subjects at intermediate risk by coronary calcium score: a serial study using non-contrast cardiac CT. Atherosclerosis. 2011;218(2):363–368. doi:10.1016/j.atherosclerosis.2011.07.093
32. Daffu G, Del Pozo CH, O’Shea KM, Ananthakrishnan R, Ramasamy R, Schmidt AM. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int J Mol Sci. 2013;14(10):19891–19910. doi:10.3390/ijms141019891
33. Hutchinson KR, Lord CK, West TA, Stewart JA Jr. Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS One. 2013;8(8):e72080. doi:10.1371/journal.pone.0072080
34. Li JH, Huang XR, Zhu HJ, et al. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 2004;18(1):176–178. doi:10.1096/fj.02-1117fje
35. Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med. 2010;48(1):1–15. doi:10.1016/j.freeradbiomed.2009.09.026
36. Steitz SA, Speer MY, Curinga G, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89(12):1147–1154. doi:10.1161/hh2401.101070
37. Suga T, Iso T, Shimizu T, et al. Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J Atheroscler Thromb. 2011;18(8):670–683. doi:10.5551/jat.7120
38. Verzijl N, DeGroot J, Thorpe SR, et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275(50):39027–39031. doi:10.1074/jbc.M006700200
39. Konova E, Baydanoff S, Atanasova M, Velkova A. Age-related changes in the glycation of human aortic elastin. Exp Gerontol. 2004;39(2):249–254. doi:10.1016/j.exger.2003.10.003
40. West SL, Swan VJ, Jamal SA. Effects of calcium on cardiovascular events in patients with kidney disease and in a healthy population. Clin J Am Soc Nephrol. 2010;5 Suppl 1:S41–7. doi:10.2215/cjn.05860809
41. Liu J, Xiao X, Shen Y, et al. MicroRNA-32 promotes calcification in vascular smooth muscle cells: implications as a novel marker for coronary artery calcification. PLoS One. 2017;12(3):e0174138. doi:10.1371/journal.pone.0174138
42. Lin L, He Y, Xi BL, et al. MiR-135a suppresses calcification in senescent VSMCs by regulating KLF4/STAT3 pathway. Curr Vasc Pharmacol. 2016;14(2):211–218. doi:10.2174/1570161113666150722151817
43. Song R, Fullerton DA, Ao L, Zhao KS, Meng X. An epigenetic regulatory loop controls pro-osteogenic activation by TGF-β1 or bone morphogenetic protein 2 in human aortic valve interstitial cells. J Biol Chem. 2017;292(21):8657–8666. doi:10.1074/jbc.M117.783308
44. Wu SS, Lin X, Yuan LQ, Liao EY. The role of epigenetics in arterial calcification. Biomed Res Int. 2015;2015:320849. doi:10.1155/2015/320849
45. Nanoudis S, Pikilidou M, Yavropoulou M, Zebekakis P. The Role of MicroRNAs in arterial stiffness and arterial calcification. an update and review of the literature. Front Genetics. 2017;8:209. doi:10.3389/fgene.2017.00209
46. Son DJ, Jung YY, Seo YS, et al. Interleukin-32alpha inhibits endothelial inflammation, vascular smooth muscle cell activation, and atherosclerosis by upregulating timp3 and reck through suppressing microRNA-205 biogenesis. Theranostics. 2017;7(8):2186–2203. doi:10.7150/thno.18407
47. Yao L, Chandra S, Toque HA, et al. Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovas Res. 2013;97(3):509–519. doi:10.1093/cvr/cvs371
48. Heath JM, Sun Y, Yuan K, et al. Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ Res. 2014;114(7):1094–1102. doi:10.1161/CIRCRESAHA.114.302968
49. Azpiazu D, Gonzalo S, Villa-Bellosta R. Tissue non-specific alkaline phosphatase and vascular calcification: a potential therapeutic target. Current Cardiol Rev. 2019;15(2):91–95. doi:10.2174/1573403x14666181031141226
50. Zickler D, Luecht C, Willy K, et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol Dial Transplant. 2018;33(4):574–585. doi:10.1093/ndt/gfx316
51. Pietrowski E, Bender B, Huppert J, White R, Luhmann HJ, Kuhlmann CRW. Pro-inflammatory effects of interleukin-17A on vascular smooth muscle cells involve NAD(P)H- oxidase derived reactive oxygen species. J Vascu Res. 2011;48(1):52–58. doi:10.1159/000317400
52. Barrera-Chimal J, Estrela GR, Lechner SM, et al. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. 2018;93(6):1344–1355. doi:10.1016/j.kint.2017.12.016
53. Canto C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev. 2012;64(1):166–187. doi:10.1124/pr.110.003905
54. Liu X, Chen A, Liang Q, et al. Spermidine inhibits vascular calcification in chronic kidney disease through modulation of SIRT1 signaling pathway. Aging Cell. 2021;20(6):e13377. doi:10.1111/acel.13377
55. Massy ZA, Ivanovski O, Nguyen-Khoa T, et al. Uremia accelerates both atherosclerosis and arterial calcification in apolipoprotein E knockout mice. J Am Soc Nephrol. 2005;16(1):109–116. doi:10.1681/asn.2004060495
56. Sun H, Zhang F, Xu Y, et al. Salusin-β promotes vascular calcification via nicotinamide adenine dinucleotide phosphate/reactive oxygen species-mediated klotho downregulation. Antioxid Redox Signal. 2019;31(18):1352–1370. doi:10.1089/ars.2019.7723
57. Sutra T, Morena M, Bargnoux AS, Caporiccio B, Canaud B, Cristol JP. Superoxide production: a procalcifying cell signalling event in osteoblastic differentiation of vascular smooth muscle cells exposed to calcification media. Free Radic Res. 2008;42(9):789–797. doi:10.1080/10715760802400766
58. Liu H, Li X, Qin F, Huang K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J Bio Inorg Chem. 2014;19(3):375–388. doi:10.1007/s00775-013-1078-1
59. Gomez C, Martinez L, Mesa A, et al. Oxidative stress induces early-onset apoptosis of vascular smooth muscle cells and neointima formation in response to injury. Biosci Rep. 2015;35(4). doi:10.1042/bsr20140122
60. Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol. 2014;307(8):F891–900. doi:10.1152/ajprenal.00163.2014
61. Erickson JR. Mechanisms of CaMKII activation in the heart. Front Pharmacol. 2014;5:59. doi:10.3389/fphar.2014.00059
62. Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E. NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovas Res. 2013;100(3):392–401. doi:10.1093/cvr/cvt201
63. Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256(5060):1199–1202. doi:10.1126/science.256.5060.1199
64. Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM. S-Nitrosylation induces both autonomous activation and inhibition of calcium/calmodulin-dependent protein kinase II δ. J Biol Chem. 2015;290(42):25646–25656. doi:10.1074/jbc.M115.650234
65. Anderson ME. Oxidant stress promotes disease by activating CaMKII. J Mol Cell Cardiol. 2015;89(Pt B):160–167. doi:10.1016/j.yjmcc.2015.10.014
66. Feng H, Wang JY, Yu B, et al. Peroxisome proliferator-activated receptor-γ coactivator-1α inhibits vascular calcification through sirtuin 3-mediated reduction of mitochondrial oxidative stress. Antioxid Redox Signal. 2019;31(1):75–91. doi:10.1089/ars.2018.7620
67. Jelic MD, Mandic AD, Maricic SM, Srdjenovic BU. Oxidative stress and its role in cancer. J Cancer Res Ther. 2021;17(1):22–28. doi:10.4103/jcrt.JCRT_862_16
68. Zheng L, Shen X, Ye J, Xie Y, Yan S. Metformin alleviates hyperglycemia-induced apoptosis and differentiation suppression in osteoblasts through inhibiting the TLR4 signaling pathway. Life Sci. 2019;216:29–38. doi:10.1016/j.lfs.2018.11.008
69. Li T, Ni L, Liu X, Wang Z, Liu C. High glucose induces the expression of osteopontin in blood vessels in vitro and in vivo. Biochem Biophys Res Commun. 2016;480(2):201–207. doi:10.1016/j.bbrc.2016.10.027
70. Zhang X, Xiao J, Li R, et al. Metformin alleviates vascular calcification induced by vitamin D3 plus nicotine in rats via the AMPK pathway. Vascul Pharmacol. 2016;81:83–90. doi:10.1016/j.vph.2016.01.002
71. Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: a focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016;9:244–253. doi:10.1016/j.redox.2016.08.015
72. Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, Orekhov AN. The diabetes mellitus-atherosclerosis connection: the role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci. 2020;21(5):1835. doi:10.3390/ijms21051835
73. Duvillard L, Florentin E, Pont F, et al. Endogenous chronic hyperinsulinemia does not increase the production rate of VLDL apolipoprotein B: proof from a kinetic study in patients with insulinoma. J Clin Endocrinol Metab. 2011;96(7):2163–2170. doi:10.1210/jc.2010-2946
74. Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci USA. 2004;101(44):15597–15602. doi:10.1073/pnas.0405238101
75. Chen NX, O’Neill K, Akl NK, Moe SM. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449(1):151–156. doi:10.1016/j.bbrc.2014.05.005
76. Yu B, Huo L, Liu Y, et al. PGC-1α controls skeletal stem cell fate and bone-fat balance in osteoporosis and skeletal aging by inducing TAZ. Cell Stem Cell. 2018;23(2):193–209.e5. doi:10.1016/j.stem.2018.06.009
77. Patten IS, Arany Z. PGC-1 coactivators in the cardiovascular system. Trend Endocrinol Metabol. 2012;23(2):90–97. doi:10.1016/j.tem.2011.09.007
78. Altura BM, Altura BT, Carella A, Gebrewold A, Murakawa T, Nishio A. Mg2+-Ca2+ interaction in contractility of vascular smooth muscle: mg2+ versus organic calcium channel blockers on myogenic tone and agonist-induced responsiveness of blood vessels. Can J Physiol Pharmacol. 1987;65(4):729–745. doi:10.1139/y87-120
79. Ter Braake AD, Tinnemans PT, Shanahan CM, Hoenderop JGJ, de Baaij JHF. Magnesium prevents vascular calcification in vitro by inhibition of hydroxyapatite crystal formation. Sci Rep. 2018;8(1):2069. doi:10.1038/s41598-018-20241-3
80. Miyata T, Sprague SM. Advanced glycation of beta 2-microglobulin in the pathogenesis of bone lesions in dialysis-associated amyloidosis. Nephrol Dial Transplant. 1996;11 Suppl 3:86–90. doi:10.1093/ndt/11.supp3.86
81. Zimmerman CM, Padgett RW. Transforming growth factor β signaling mediators and modulators. Gene. 2000;249(1–2):17–30. doi:10.1016/s0378-1119(00)00162-1
82. Stefan N, Fritsche A, Weikert C, et al. Plasma fetuin-A levels and the risk of type 2 diabetes. Diabetes. 2008;57(10):2762–2767. doi:10.2337/db08-0538
83. Tanikawa T, Okada Y, Tanikawa R, Tanaka Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J Vascu Res. 2009;46(6):572–580. doi:10.1159/000226225
84. Hu Y, Chan E, Wang SX, Li B. Activation of p38 mitogen-activated protein kinase is required for osteoblast differentiation. Endocrinology. 2003;144(5):2068–2074. doi:10.1210/en.2002-220863
85. Janda K, Krzanowski M, Gajda M, et al. Vascular effects of advanced glycation end-products: content of immunohistochemically detected AGEs in radial artery samples as a predictor for arterial calcification and cardiovascular risk in asymptomatic patients with chronic kidney disease. Dis. Markers. 2015;2015:153978. doi:10.1155/2015/153978
86. Belmokhtar K, Ortillon J, Jaisson S, et al. Receptor for advanced glycation end products: a key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter PIT-1 expression. Nephrol Dial Transplant. 2019;34(12):2018–2030. doi:10.1093/ndt/gfz012
87. Cecil DL, Terkeltaub RA. Arterial calcification is driven by RAGE in Enpp1-/- mice. J Vascu Res. 2011;48(3):227–235. doi:10.1159/000318805
88. La Sala L, Micheloni S, De Nigris V, Prattichizzo F, Ceriello A. Novel insights into the regulation of miRNA transcriptional control: implications for T2D and related complications. Acta diabetologica. 2018;55(10):989–998. doi:10.1007/s00592-018-1149-4
89. Schorey JS, Cheng Y, Singh PP, Smith VL. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015;16(1):24–43. doi:10.15252/embr.201439363
90. Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 2012;80(6):1948–1957. doi:10.1128/iai.06014-11
91. Chaudhary SC, Khalid S, Smethurst V, et al. Proteomic profiling of extracellular vesicles released from vascular smooth muscle cells during initiation of phosphate-induced mineralization. Connect Tissue Res. 2018;53:55–61. doi:10.1080/03008207.2018.1444759
92. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87(11):1055–1062. doi:10.1161/01.res.87.11.1055
93. Proudfoot D, Skepper JN, Shanahan CM, Weissberg PL. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler Thromb Vasc Biol. 1998;18(3):379–388. doi:10.1161/01.atv.18.3.379
94. Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15(11):2857–2867. doi:10.1097/01.Asn.0000141960.01035.28
95. Tuttle KR, Short RA. Longitudinal relationships among coronary artery calcification, serum phosphorus, and kidney function. Clin J Am Soc Nephrol. 2009;4(12):1968–1973. doi:10.2215/cjn.01250209
96. Zhang H, Chen J, Shen Z, et al. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/β-catenin signaling. Toxicol Lett. 2018;284:29–36. doi:10.1016/j.toxlet.2017.11.033
97. Alkagiet S, Tziomalos K. Vascular calcification: the role of microRNAs. Biomol Conc. 2017;8(2):119–123. doi:10.1515/bmc-2017-0001
98. Badi I, Mancinelli L, Polizzotto A, et al. miR-34a promotes vascular smooth muscle cell calcification by downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler Thromb Vasc Biol. 2018;38(9):2079–2090. doi:10.1161/atvbaha.118.311298
99. Jiang W, Zhang Z, Yang H, Lin Q, Han C, Qin X. The Involvement of miR-29b-3p in arterial calcification by targeting matrix metalloproteinase-2. Biomed Res Int. 2017;2017:6713606. doi:10.1155/2017/6713606
100. Wen C, Yang X, Yan Z, et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol. 2013;168(3):2242–2247. doi:10.1016/j.ijcard.2013.01.211
101. Duan X, Zhou Y, Teng X, Tang C, Qi Y. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem Biophys Res Commun. 2009;387(4):694–699. doi:10.1016/j.bbrc.2009.07.085
102. Zhou J, Wang KC, Wu W, et al. MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci USA. 2011;108(25):10355–10360. doi:10.1073/pnas.1107052108
103. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103(33):12481–12486. doi:10.1073/pnas.0605298103
104. Hou J, Wang P, Lin L, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183(3):2150–2158. doi:10.4049/jimmunol.0900707
105. Leopold JA. MicroRNAs Regulate Vascular Medial Calcification. Cells. 2014;3(4):963–980. doi:10.3390/cells3040963
106. Dusing P, Zietzer A, Goody PR, et al. Vascular pathologies in chronic kidney disease: pathophysiological mechanisms and novel therapeutic approaches. J Mol Med. 2021;99(3):335–348. doi:10.1007/s00109-021-02037-7
107. Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013;34(31):2436–2443. doi:10.1093/eurheartj/eht149
108. Yamagishi S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp Gerontol. 2011;46(4):217–224. doi:10.1016/j.exger.2010.11.007
109. Luong TTD, Schelski N, Boehme B, et al. Fibulin-3 attenuates phosphate-induced vascular smooth muscle cell calcification by inhibition of oxidative stress. Cell Physiol Biochem. 2018;46(4):1305–1316. doi:10.1159/000489144
110. Yang K, Nie L, Huang Y, et al. Amelioration of uremic toxin indoxyl sulfate-induced endothelial cell dysfunction by Klotho protein. Toxicol Lett. 2012;215(2):77–83. doi:10.1016/j.toxlet.2012.10.004
111. Chavkin NW, Chia JJ, Crouthamel MH, Giachelli CM. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp Cell Res. 2015;333(1):39–48. doi:10.1016/j.yexcr.2015.02.002
112. Yamada S, Leaf EM, Chia JJ, Cox TC, Speer MY, Giachelli CM. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int. 2018;94(4):716–727. doi:10.1016/j.kint.2018.05.015
113. Voelkl J, Tuffaha R, Luong TTD, et al. Zinc Inhibits phosphate-induced vascular calcification through TNFAIP3-mediated suppression of NF-κB. J Am Soc Nephrol. 2018;29(6):1636–1648. doi:10.1681/asn.2017050492
114. Ding H, Li D, Zhang Y, et al. Luteolin inhibits smooth muscle cell migration and proliferation by attenuating the production of Nox4, p-Akt and VEGF in endothelial cells. Current Pharm Biotechnol. 2014;14(12):1009–1015. doi:10.2174/1389201015666140113113843
115. Speer T, Owala FO, Holy EW, et al. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur Heart J. 2014;35(43):3021–3032. doi:10.1093/eurheartj/ehu111
116. Wasty F, Alavi MZ, Moore S. Distribution of glycosaminoglycans in the intima of human aortas: changes in atherosclerosis and diabetes mellitus. Diabetologia. 1993;36(4):316–322. doi:10.1007/bf00400234
117. Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3(2):109–121. doi:10.4161/oxim.3.2.11354
118. Young KA, Snell-Bergeon JK, Naik RG, et al. Vitamin D deficiency and coronary artery calcification in subjects with type 1 diabetes. Diabetes Care. 2011;34(2):454–458. doi:10.2337/dc10-0757
119. Azechi T, Sato F, Sudo R, Wachi H. 5-aza-2’-Deoxycytidine, a DNA methyltransferase inhibitor, facilitates the inorganic phosphorus-induced mineralization of vascular smooth muscle cells. J Atheroscler Thromb. 2014;21(5):463–476. doi:10.5551/jat.20818
120. Villa JKD, Diaz MAN, Pizziolo VR, Martino HSD. Effect of vitamin K in bone metabolism and vascular calcification: a review of mechanisms of action and evidences. Crit Rev Food Sci Nutr. 2017;57(18):3959–3970. doi:10.1080/10408398.2016.1211616
121. Liu Q, Qi H, Yao L. A long non-coding RNA H19/microRNA-138/TLR3 network is involved in high phosphorus-mediated vascular calcification and chronic kidney disease. Cell Cycle. 2022;21(16):1667–1683. doi:10.1080/15384101.2022.2064957
122. Zhang X, Chen J, Meng Q, et al. The protective effects of long non-coding RNA-ANCR on arterial calcification. J Bone Mineral Metabol. 2020;38(4):421–431. doi:10.1007/s00774-019-01076-y
123. Wang D, Deuse T, Stubbendorff M, et al. Local MicroRNA modulation using a novel anti-mir-21-eluting stent effectively prevents experimental in-stent restenosis. Arterioscler Thromb Vasc Biol. 2015;35(9):1945–1953. doi:10.1161/atvbaha.115.305597
124. Ramezani A, Massy ZA, Meijers B, Evenepoel P, Vanholder R, Raj DS. Role of the gut microbiome in uremia: a potential therapeutic target. Am J Kidney Dis. 2016;67(3):483–498. doi:10.1053/j.ajkd.2015.09.027
125. Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. 2000;46(4):493–499. doi:10.1136/gut.46.4.493
126. Felici C, Ciari I, Terzuoli L, et al. Purine catabolism in advanced carotid artery plaque. Nucleosides Nucleotides Nucleic Acids. 2006;25(9–11):1291–1294. doi:10.1080/15257770600890772
127. Cheng ZY, Ye T, Ling QY, Wu T, Wu GY, Zong GJ. Parathyroid hormone promotes osteoblastic differentiation of endothelial cells via the extracellular signal-regulated protein kinase 1/2 and nuclear factor-κB signaling pathways. Exp Ther Med. 2018;15(2):1754–1760. doi:10.3892/etm.2017.5545
128. Yao H, Sun Z, Zang G, et al. Epidemiological research advances in vascular calcification in diabetes. J Diabetes Res. 2021;2021:4461311. doi:10.1155/2021/4461311
129. Walrand S, Guillet C, Boirie Y, Vasson MP. In vivo evidences that insulin regulates human polymorphonuclear neutrophil functions. J Leukocy Biol. 2004;76(6):1104–1110. doi:10.1189/jlb.0104050
130. Jaffe IZ, Tintut Y, Newfell BG, Demer LL, Mendelsohn ME. Mineralocorticoid receptor activation promotes vascular cell calcification. Arteriosclerosis Thrombosis Vasc Biol. 2007;27(4):799–805. doi:10.1161/01.ATV.0000258414.59393.89
131. de Seigneux S, Martin PY. Phosphate and FGF23 in the renoprotective benefit of RAAS inhibition. Pharmacol Res. 2016;106:87–91. doi:10.1016/j.phrs.2016.02.015
132. Kolkhof P, Lawatscheck R, Filippatos G, Bakris GL. Nonsteroidal mineralocorticoid receptor antagonism by finerenone-translational aspects and clinical perspectives across multiple organ systems. Int J Mol Sci. 2022;23(16):9243. doi:10.3390/ijms23169243
133. Kawakami R, Katsuki S, Travers R, et al. S100A9-RAGE axis accelerates formation of macrophage-mediated extracellular vesicle microcalcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2020;40(8):1838–1853. doi:10.1161/atvbaha.118.314087
134. Agharazii M, St-Louis R, Gautier-Bastien A, et al. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am J Hypertens. 2015;28(6):746–755. doi:10.1093/ajh/hpu225
135. Nguyen TT, Quan X, Xu S, et al. Intracellular alkalinization by phosphate uptake via type III sodium-phosphate cotransporter participates in high-phosphate-induced mitochondrial oxidative stress and defective insulin secretion. FASEB J. 2016;30(12):3979–3988. doi:10.1096/fj.201600455RR
136. Al-Qaridhi A, Ghosh S, Luo D, Huang H. Magnesium and zinc intake ratio mediates the increase of coronary artery calcification through upregulating interleukin 6. Libyan J Med. 2022;17(1):2028997. doi:10.1080/19932820.2022.2028997
137. Viegas C, Araújo N, Marreiros C, Simes D. The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): challenging old concepts with new facts. Aging. 2019;11(12):4274–4299. doi:10.18632/aging.102046
138. Kuro OM. The Klotho proteins in health and disease. Nat Rev Nephrol. 2019;15(1):27–44. doi:10.1038/s41581-018-0078-3
139. Ivanovski O, Nikolov IG, Joki N, et al. The calcimimetic R-568 retards uremia-enhanced vascular calcification and atherosclerosis in apolipoprotein E deficient (apoE-/-) mice. Atherosclerosis. 2009;205(1):55–62. doi:10.1016/j.atherosclerosis.2008.10.043
140. Kawata T, Nagano N, Obi M, et al. Cinacalcet suppresses calcification of the aorta and heart in uremic rats. Kidney Int. 2008;74(10):1270–1277. doi:10.1038/ki.2008.407
141. Hilfiker H, Hattenhauer O, Traebert M, Forster I, Murer H, Biber J. Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc Natl Acad Sci USA. 1998;95(24):14564–14569. doi:10.1073/pnas.95.24.14564
142. Segawa H, Kaneko I, Takahashi A, et al. Growth-related renal type II Na/Pi cotransporter. J Biol Chem. 2002;277(22):19665–19672. doi:10.1074/jbc.M200943200
143. Tatsumi S, Miyagawa A, Kaneko I, Shiozaki Y, Segawa H, Miyamoto K. Regulation of renal phosphate handling: inter-organ communication in health and disease. J Bone Mineral Metabol. 2016;34(1):1–10. doi:10.1007/s00774-015-0705-z
144. Hernando N, Gagnon K, Lederer E. Phosphate Transport in Epithelial and Nonepithelial Tissue. Physiol Rev. 2021;101(1):1–35. doi:10.1152/physrev.00008.2019
145. Forster IC, Hernando N, Biber J, Murer H. Phosphate transporters of the SLC20 and SLC34 families. Mol Aspect Med. 2013;34(2–3):386–395. doi:10.1016/j.mam.2012.07.007
146. Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98(7):905–912. doi:10.1161/01.RES.0000216409.20863.e7
147. Zhao G, Xu MJ, Zhao MM, et al. Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. 2012;82(1):34–44. doi:10.1038/ki.2012.40
148. Yoshida T, Yamashita M, Horimai C, Hayashi M. Smooth muscle-selective nuclear factor-κB inhibition reduces phosphate-induced arterial medial calcification in mice with chronic kidney disease. J Am Heart Assoc. 2017;6(11). doi:10.1161/jaha.117.007248
149. Lee HL, Woo KM, Ryoo HM, Baek JH. Tumor necrosis factor-alpha increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem Biophys Res Commun. 2010;391(1):1087–1092. doi:10.1016/j.bbrc.2009.12.027
150. Panizo S, Cardus A, Encinas M, et al. RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ Res. 2009;104(9):1041–1048. doi:10.1161/circresaha.108.189001
151. Lang F, Voelkl J. Therapeutic potential of serum and glucocorticoid inducible kinase inhibition. Expert Opin Invest Drugs. 2013;22(6):701–714. doi:10.1517/13543784.2013.778971
152. Voelkl J, Luong TT, Tuffaha R, et al. SGK1 induces vascular smooth muscle cell calcification through NF-κB signaling. J Clin Invest. 2018;128(7):3024–3040. doi:10.1172/jci96477
153. Schelski N, Luong TTD, Lang F, Pieske B, Voelkl J, Alesutan I. SGK1-dependent stimulation of vascular smooth muscle cell osteo-/chondrogenic transdifferentiation by interleukin-18. Pflugers Archiv. 2019;471(6):889–899. doi:10.1007/s00424-019-02256-5
154. Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest. 2005;115(5):1210–1220. doi:10.1172/jci24140
155. Yao L, Sun YT, Sun W, et al. High phosphorus level leads to aortic calcification via β-catenin in chronic kidney disease. Am J Nephrol. 2015;41(1):28–36. doi:10.1159/000370250
156. Cai T, Sun D, Duan Y, et al. WNT/β-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. 2016;345(2):206–217. doi:10.1016/j.yexcr.2016.06.007
157. Freise C, Kretzschmar N, Querfeld U. Wnt signaling contributes to vascular calcification by induction of matrix metalloproteinases. BMC Cardiovascu Disord. 2016;16(1):185. doi:10.1186/s12872-016-0362-8
158. Rawadi G, Vayssière B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res. 2003;18(10):1842–1853. doi:10.1359/jbmr.2003.18.10.1842
159. Lim K, T-S L, Molostvov G, et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012;125(18):2243–2255. doi:10.1161/CIRCULATIONAHA.111.053405
160. Zhao Y, Zhao MM, Cai Y, et al. Mammalian target of rapamycin signaling inhibition ameliorates vascular calcification via Klotho upregulation. Kidney Int. 2015;88(4):711–721. doi:10.1038/ki.2015.160
161. Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci. 2013;38(5):233–242. doi:10.1016/j.tibs.2013.01.004
162. Vervloet MG, Adema AY, Larsson TE, Massy ZA. The role of klotho on vascular calcification and endothelial function in chronic kidney disease. Semin Nephrol. 2014;34(6):578–585. doi:10.1016/j.semnephrol.2014.09.003
163. Martinet W, De Loof H, De Meyer GRY. mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis. 2014;233(2):601–607. doi:10.1016/j.atherosclerosis.2014.01.040
164. Elloso MM, Azrolan N, Sehgal SN, et al. Protective effect of the immunosuppressant sirolimus against aortic atherosclerosis in apo E-deficient mice. Am J Transplant. 2003;3(5):562–569. doi:10.1034/j.1600-6143.2003.00094.x
165. Liberman M, Johnson RC, Handy DE, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun. 2011;413(3):436–441. doi:10.1016/j.bbrc.2011.08.114
166. Zhang D, Bi X, Liu Y, et al. High phosphate-induced calcification of vascular smooth muscle cells is associated with the TLR4/NF-κb signaling pathway. Kidney Blood Press Res. 2017;42(6):1205–1215. doi:10.1159/000485874
167. Sage AP, Lu J, Tintut Y, Demer LL. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;79(4):414–422. doi:10.1038/ki.2010.390
168. Leibrock CB, Alesutan I, Voelkl J, et al. NH4Cl treatment prevents tissue calcification in klotho deficiency. J Am Soc Nephrol. 2015;26(10):2423–2433. doi:10.1681/asn.2014030230
169. Alesutan I, Musculus K, Castor T, Alzoubi K, Voelkl J, Lang F. Inhibition of phosphate-induced vascular smooth muscle cell osteo-/chondrogenic signaling and calcification by bafilomycin A1 and methylamine. Kidney Blood Press Res. 2015;40(5):490–499. doi:10.1159/000368524
170. Abbasian N, Burton JO, Herbert KE, et al. Hyperphosphatemia, phosphoprotein phosphatases, and microparticle release in vascular endothelial cells. J Am Soc Nephrol. 2015;26(9):2152–2162. doi:10.1681/asn.2014070642
171. Soriano S, Carmona A, Triviño F, et al. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am J Physiol Renal Physiol. 2014;307(11):F1302–F1311. doi:10.1152/ajprenal.00114.2014
172. Alique M, Bodega G, Corchete E, et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification in vitro. Comput Struct Biotechnol J. 2020;18:953–966. doi:10.1016/j.csbj.2020.04.006
173. Sulistyowati E, Permatasari N, Aris Widodo M. Combined effects of shear stress and glucose on the morphology, actin filaments, and VE-cadherin of endothelial cells in vitro. Int J Cardiol Heart Vasculature. 2017;15:31–35. doi:10.1016/j.ijcha.2017.03.004
174. Pilz S, Verheyen N, Grübler MR, Tomaschitz A, März W. Vitamin D and cardiovascular disease prevention. Nat Rev Cardiol. 2016;13(7):404–417. doi:10.1038/nrcardio.2016.73
175. Cianciolo G, La Manna G, Della Bella E, et al. Effect of vitamin D receptor activator therapy on vitamin D receptor and osteocalcin expression in circulating endothelial progenitor cells of hemodialysis patients. Blood Purif. 2013;35(1–3):187–195. doi:10.1159/000347102
176. Jiang YJ, Teichert AE, Fong F, Oda Y, Bikle DD. 1α,25(OH)2-dihydroxyvitamin D3/VDR protects the skin from UVB-induced tumor formation by interacting with the β-catenin pathway. J Steroid Biochem Mol Biol. 2013;136:229–232. doi:10.1016/j.jsbmb.2012.09.024
177. Prié D, Friedlander G. Reciprocal control of 1,25-dihydroxyvitamin D and FGF23 formation involving the FGF23/Klotho system. Clin J Am Soc Nephrol. 2010;5(9):1717–1722. doi:10.2215/CJN.02680310
178. Cohen-Lahav M, Shany S, Tobvin D, Chaimovitz C, Douvdevani A. Vitamin D decreases NFkappaB activity by increasing IkappaBalpha levels. Nephrol Dial Transplant. 2006;21(4):889–897. doi:10.1093/ndt/gfi254
179. Vissers LET, Dalmeijer GW, Boer JMA, Verschuren WMM, van der Schouw YT, Beulens JWJ. The relationship between vitamin K and peripheral arterial disease. Atherosclerosis. 2016;252:15–20. doi:10.1016/j.atherosclerosis.2016.07.915
180. Xie SA, Zhang T, Wang J, et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: role of DNA methyltransferase 1. Biomaterials. 2018;155:203–216. doi:10.1016/j.biomaterials.2017.11.033
181. Kétszeri M, Kirsch A, Frauscher B, et al. MicroRNA-142-3p improves vascular relaxation in uremia. Atherosclerosis. 2019;280:28–36. doi:10.1016/j.atherosclerosis.2018.11.024
182. Zhan Q, Song R, Zeng Q, et al. Activation of TLR3 induces osteogenic responses in human aortic valve interstitial cells through the NF-κB and ERK1/2 pathways. Int J Bio Sci. 2015;11(4):482–493. doi:10.7150/ijbs.10905
183. Mahmoodpoor F, Rahbar Saadat Y, Barzegari A, Ardalan M, Zununi Vahed S. The impact of gut microbiota on kidney function and pathogenesis. Biomed Pharmacother. 2017;93:412–419. doi:10.1016/j.biopha.2017.06.066
184. Rapa SF, Di Iorio BR, Campiglia P, Heidland A, Marzocco S. Inflammation and oxidative stress in chronic kidney disease-potential therapeutic role of minerals, vitamins and plant-derived metabolites. Int J Mol Sci. 2019;21(1):263. doi:10.3390/ijms21010263
185. Pieniazek A, Bernasinska-Slomczewska J, Gwozdzinski L. Uremic toxins and their relation with oxidative stress induced in patients with CKD. Int J Mol Sci. 2021;22(12):6196. doi:10.3390/ijms22126196
186. Suarna C, Dean RT, May J, Stocker R. Human atherosclerotic plaque contains both oxidized lipids and relatively large amounts of alpha-tocopherol and ascorbate. Arterioscler Thromb Vasc Biol. 1995;15(10):1616–1624. doi:10.1161/01.atv.15.10.1616
187. Patetsios P, Song M, Shutze WP, et al. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am J Cardiol. 2001;88(2):188–91, a6. doi:10.1016/s0002-9149(01)01621-6
188. Armstrong ZB, Boughner DR, Drangova M, Rogers KA. Angiotensin II type 1 receptor blocker inhibits arterial calcification in a pre-clinical model. Cardiovas Res. 2011;90(1):165–170. doi:10.1093/cvr/cvq391
189. Alesutan I, Voelkl J, Feger M, et al. Involvement of vascular aldosterone synthase in phosphate-induced osteogenic transformation of vascular smooth muscle cells. Sci Rep. 2017;7(1):2059. doi:10.1038/s41598-017-01882-2
190. Lang F, Ritz E, Voelkl J, Alesutan I. Vascular calcification--is aldosterone a culprit? Nephrol Dial Transplant. 2013;28(5):1080–1084. doi:10.1093/ndt/gft041
191. Mori K, Shioi A, Jono S, Nishizawa Y, Morii H. Dexamethasone enhances In vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19(9):2112–2118. doi:10.1161/01.atv.19.9.2112
192. Cohn-Schwartz D, Schary Y, Yalon E, et al. PTH-induced bone regeneration and vascular modulation are both dependent on endothelial signaling. Cells. 2022;11(5):897. doi:10.3390/cells11050897
193. Iguchi T, Hasegawa T, Otsuka K, et al. Insulin resistance is associated with coronary plaque vulnerability: insight from optical coherence tomography analysis. Europ Heart J Cardiovas Imag. 2014;15(3):284–291. doi:10.1093/ehjci/jet158
194. Verma A, Vaidya A, Subudhi S, Waikar SS. Aldosterone in chronic kidney disease and renal outcomes. Eur Heart J. 2022;43(38):3781–3791. doi:10.1093/eurheartj/ehac352
195. Neven E, Vervaet B, Brand K, et al. Metformin prevents the development of severe chronic kidney disease and its associated mineral and bone disorder. Kidney Int. 2018;94(1):102–113. doi:10.1016/j.kint.2018.01.027
196. Cao X, Li H, Tao H, et al. Metformin inhibits vascular calcification in female rat aortic smooth muscle cells via the AMPK-eNOS-NO pathway. Endocrinology. 2013;154(10):3680–3689. doi:10.1210/en.2013-1002
197. Mohammed I, Hollenberg MD, Ding H, Triggle CR. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front Endocrinol. 2021;12:718942. doi:10.3389/fendo.2021.718942
198. Scirpo R, Fiorotto R, Villani A, Amenduni M, Spirli C, Strazzabosco M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-γ limits NF-κB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology. 2015;62(5):1551–1562. doi:10.1002/hep.28000
199. Martens FMAC, Rabelink TJ, op ‘t Roodt J, de Koning EJP, Visseren FLJ. TNF-alpha induces endothelial dysfunction in diabetic adults, an effect reversible by the PPAR-gamma agonist pioglitazone. Eur Heart J. 2006;27(13):1605–1609. doi:10.1093/eurheartj/ehl079
200. Li J, Shen X. Effect of rosiglitazone on inflammatory cytokines and oxidative stress after intensive insulin therapy in patients with newly diagnosed type 2 diabetes. Diabetol Metab Syndr. 2019;11:35. doi:10.1186/s13098-019-0432-z
201. Diwan V, Gobe G, Brown L. Glibenclamide improves kidney and heart structure and function in the adenine-diet model of chronic kidney disease. Pharmacol Res. 2014;79:104–110. doi:10.1016/j.phrs.2013.11.007
202. Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes. 2014;63(3):1103–1114. doi:10.2337/db13-0927
203. Schinzari F, Tesauro M, Bertoli A, et al. Calcification biomarkers and vascular dysfunction in obesity and type 2 diabetes: influence of oral hypoglycemic agents. Am J Physiol Endocrinol Metab. 2019;317(4):E658–E666. doi:10.1152/ajpendo.00204.2019
204. Dowling JK, O’Neill LA. Biochemical regulation of the inflammasome. Crit Rev Biochem Mol Biol. 2012;47(5):424–443. doi:10.3109/10409238.2012.694844
205. Ott C, Kistner I, Keller M, et al. Effects of linagliptin on renal endothelial function in patients with type 2 diabetes: a randomised clinical trial. Diabetologia. 2016;59(12):2579–2587. doi:10.1007/s00125-016-4083-4
206. Busek P, Duke-Cohan JS, Sedo A. Does DPP-IV inhibition offer new avenues for therapeutic intervention in malignant disease? Cancers. 2022;14(9). doi:10.3390/cancers14092072
207. Bosch A, Ott C, Jung S, et al. How does empagliflozin improve arterial stiffness in patients with type 2 diabetes mellitus? Sub analysis of a clinical trial. Cardiov Diabetol. 2019;18(1):44. doi:10.1186/s12933-019-0839-8
208. Zhan J-K, Tan P, Wang Y-J, et al. Exenatide can inhibit calcification of human VSMCs through the NF-kappaB/RANKL signaling pathway. Cardiov Diabetol. 2014;13:153. doi:10.1186/s12933-014-0153-4
209. Lin Y-C, Chen Y-C, Hsiao H-P, et al. The effects of acarbose on chemokine and cytokine production in human monocytic THP-1 cells. Hormones. 2019;18(2):179–187. doi:10.1007/s42000-019-00101-z
210. Mo D, Liu S, Ma H, et al. Effects of acarbose and metformin on the inflammatory state in newly diagnosed type 2 diabetes patients: a one-year randomized clinical study. Drug Des Devel Ther. 2019;13:2769–2776. doi:10.2147/DDDT.S208327
211. Darenskaya MA, Kolesnikova LI, Kolesnikov SI. Oxidative stress: pathogenetic role in diabetes mellitus and its complications and therapeutic approaches to correction. Bull Exp Biol Med. 2021;171(2):179–189. doi:10.1007/s10517-021-05191-7
212. Rafael MS, Cavaco S, Viegas CSB, et al. Insights into the association of Gla-rich protein and osteoarthritis, novel splice variants and γ-carboxylation status. Mol Nutr Food Res. 2014;58(8):1636–1646. doi:10.1002/mnfr.201300941
213. Stock M, Menges S, Eitzinger N, et al. A dual role of upper zone of growth plate and cartilage matrix-associated protein in human and mouse osteoarthritic cartilage: inhibition of aggrecanases and promotion of bone turnover. Arthritis Rheumatol. 2017;69(6):1233–1245. doi:10.1002/art.40042
214. Lattenist L, Lechner SM, Messaoudi S, et al. Nonsteroidal mineralocorticoid receptor antagonist finerenone protects against acute kidney injury-mediated chronic kidney disease: role of oxidative stress. Hypertension. 2017;69(5):870–878. doi:10.1161/HYPERTENSIONAHA.116.08526
215. Gil-Ortega M, Vega-Martín E, Martín-Ramos M, et al. Finerenone reduces intrinsic arterial stiffness in Munich Wistar frömter rats, a genetic model of chronic kidney disease. Am J Nephrol. 2020;51(4):294–303. doi:10.1159/000506275
216. Luther JM, Gainer JV, Murphey LJ, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension. 2006;48(6):1050–1057. doi:10.1161/01.HYP.0000248135.97380.76
217. Sirtori CR. The pharmacology of statins. Pharmacol Res. 2014;88:3–11. doi:10.1016/j.phrs.2014.03.002
218. Cheng S, Coyne D. Oral paricalcitol for the treatment of secondary hyperparathyroidism in chronic kidney disease. Therap Clin Risk Manag. 2006;2(3):297–301. doi:10.2147/tcrm.2006.2.3.297
219. Strauss HW, Nakahara T, Narula N, Narula J. Vascular calcification: the evolving relationship of vascular calcification to major acute coronary events. J Nucl Med. 2019;60(9):1207–1212. doi:10.2967/jnumed.119.230276
220. Shea MK, Holden RM. Vitamin K status and vascular calcification: evidence from observational and clinical studies. Advan Nutr. 2012;3(2):158–165. doi:10.3945/an.111.001644
221. Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692–711. doi:10.1016/j.jacc.2016.11.042
222. Lee SE, Chang HJ, Sung JM, et al. Effects of statins on coronary atherosclerotic plaques: the PARADIGM study. JACC Cardiovasc Imaging. 2018;11(10):1475–1484. doi:10.1016/j.jcmg.2018.04.015
223. Handelsman Y, Lepor NE. PCSK9 inhibitors in lipid management of patients with diabetes mellitus and high cardiovascular risk: a review. J Am Heart Assoc. 2018;7(13). doi:10.1161/JAHA.118.008953
224. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20(6):953–966. doi:10.1016/j.cmet.2014.09.018
225. Feng X, Chen W, Ni X, et al. Metformin, macrophage dysfunction and atherosclerosis. Front Immunol. 2021;12:682853. doi:10.3389/fimmu.2021.682853
226. Reinhold S, Blankesteijn WM, Foulquier S. The Interplay of WNT and PPARγ signaling in vascular calcification. Cells. 2020;9(12):2658. doi:10.3390/cells9122658
227. Chen YH, Chiang MH, Liu JS, et al. Thiazolidinediones and risk of long-term dialysis in diabetic patients with advanced chronic kidney disease: a nationwide cohort study. PLoS One. 2015;10(6):e0129922. doi:10.1371/journal.pone.0129922
228. Usuda R. スルホニル尿素 [Sulfonylurea]. Nihon Rinsho. 2015;73(3):409–415. Japanese.
229. Barchetta I, Ciccarelli G, Barone E, et al. Greater circulating DPP4 activity is associated with impaired flow-mediated dilatation in adults with type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis. 2019;29(10):1087–1094. doi:10.1016/j.numecd.2019.07.010
230. He J, Yuan G, Cheng F, Zhang J, Guo X. Mast cell and M1 macrophage infiltration and local pro-inflammatory factors were attenuated with incretin-based therapies in obesity-related glomerulopathy. Metab Syndr Relat Disord. 2017;15(7):344–353. doi:10.1089/met.2017.0057
231. Swerdlow AJ, Laing SP, Qiao Z, et al. Cancer incidence and mortality in patients with insulin-treated diabetes: a UK cohort study. Br J Cancer. 2005;92(11):2070–2075. doi:10.1038/sj.bjc.6602611
232. Loutradis C, Papadopoulou E, Angeloudi E, Karagiannis A, Sarafidis P. The beneficial hemodynamic actions of SGLT-2 inhibitors beyond the management of hyperglycemia. Curr Med Chem. 2020;27(39):6682–6702. doi:10.2174/0929867326666191029111713
233. Han JH, Oh TJ, Lee G, et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE (-/-) mice fed a western diet. Diabetologia. 2017;60(2):364–376. doi:10.1007/s00125-016-4158-2
234. Levin A, Perkovic V, Wheeler DC, et al. Empagliflozin and cardiovascular and kidney outcomes across KDIGO risk categories: post hoc analysis of a randomized, double-blind, placebo-controlled, multinational trial. Clin J Am Soc Nephrol. 2020;15(10):1433–1444. doi:10.2215/CJN.14901219
235. Giugliano D, Longo M, Scappaticcio L, Bellastella G, Maiorino MI, Esposito K. SGLT-2 inhibitors and cardiorenal outcomes in patients with or without type 2 diabetes: a meta-analysis of 11 CVOTs. Cardiovasc Diabetol. 2021;20(1):236. doi:10.1186/s12933-021-01430-3
236. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018;27(4):740–756. doi:10.1016/j.cmet.2018.03.001
237. Helmstädter J, Keppeler K, Küster L, Münzel T, Daiber A, Steven S. Glucagon-like peptide-1 (GLP-1) receptor agonists and their cardiovascular benefits-The role of the GLP-1 receptor. Br J Pharmacol. 2022;179(4):659–676. doi:10.1111/bph.15462
238. Buse JB, Bain SC, Mann JFE, et al. Cardiovascular risk reduction with liraglutide: an exploratory mediation analysis of the LEADER trial. Diabetes Care. 2020;43(7):1546–1552. doi:10.2337/dc19-2251
239. Rosen P, Osmers A. Oxidative stress in young Zucker rats with impaired glucose tolerance is diminished by acarbose. Horm Metab Res. 2006;38(9):575–586. doi:10.1055/s-2006-950397
240. Yang H, Song Y, Liang YN, Li R. Quercetin treatment improves renal function and protects the kidney in a rat model of adenine-induced chronic kidney disease. Med Sci Monit. 2018;24:4760–4766. doi:10.12659/MSM.909259
241. Huang DD, Shi G, Jiang Y, Yao C, Zhu C. A review on the potential of Resveratrol in prevention and therapy of diabetes and diabetic complications. Biomed Pharmacother. 2020;125:109767. doi:10.1016/j.biopha.2019.109767
242. García-Martínez BI, Ruiz-Ramos M, Pedraza-Chaverri J, Santiago-Osorio E, Mendoza-Núñez VM. Hypoglycemic effect of resveratrol: a systematic review and meta-analysis. Antioxidants. 2021;10(1):69. doi:10.3390/antiox10010069
243. Kukida M, Mogi M, Kan-No H, et al. AT2 receptor stimulation inhibits phosphate-induced vascular calcification. Kidney Int. 2019;95(1):138–148. doi:10.1016/j.kint.2018.07.028
244. Barrera-Chimal J, André-Grégoire G, Nguyen Dinh Cat A, et al. Benefit of mineralocorticoid receptor antagonism in AKI: role of vascular smooth muscle rac1. J Am Soc Nephrol. 2017;28(4):1216–1226. doi:10.1681/ASN.2016040477
245. Schantl AE, Ivarsson ME, Leroux J-C. Investigational pharmacological treatments for vascular calcification. Adv Ther. 2019;2(1):1800094. doi:10.1002/adtp.201800094
246. Yang YZ, Liu ZH, Wang SC, et al. Magnesium isoglycyrrhizinate alleviates fructose-induced liver oxidative stress and inflammatory injury through suppressing NOXs. Eur J Pharmacol. 2020;883:173314. doi:10.1016/j.ejphar.2020.173314
247. Zu Y, Lu X, Song J, Yu L, Li H, Wang S. Cinacalcet treatment significantly improves all-cause and cardiovascular survival in dialysis patients: results from a meta-analysis. Kidney Blood Press Res. 2019;44(6):1327–1338. doi:10.1159/000504139
248. De Vriese AS, Caluwé R, Pyfferoen L, et al. Multicenter randomized controlled trial of vitamin K antagonist replacement by rivaroxaban with or without vitamin K2 in hemodialysis patients with atrial fibrillation: the valkyrie study. J Am Soc Nephrol. 2020;31(1):186–196. doi:10.1681/asn.2019060579
249. Haroon SW, Tai BC, Ling LH, et al. Treatment to reduce vascular calcification in hemodialysis patients using vitamin K (Trevasc-HDK): a study protocol for a randomized controlled trial. Medicine. 2020;99(36):e21906. doi:10.1097/md.0000000000021906
250. Anis KH, Pober D, Rosas SE. Vitamin D analogues and coronary calcification in CKD Stages 3 and 4: a randomized controlled trial of calcitriol versus paricalcitol. Kidn Med. 2020;2(4):450–458. doi:10.1016/j.xkme.2020.05.009
251. Perelló J, Ferrer MD, Del Mar Pérez M, et al. Mechanism of action of SNF472, a novel calcification inhibitor to treat vascular calcification and calciphylaxis. Br J Pharmacol. 2020;177(19):4400–4415. doi:10.1111/bph.15163
252. Grases F, Sanchis P, Perello J, et al. Phytate (Myo-inositol hexakisphosphate) inhibits cardiovascular calcifications in rats. Front Biosci. 2006;11:136–142. doi:10.2741/1786
253. Doran AC, Terry JG, Carr JJ, Linton MF. Statins and atherosclerotic lesion microcalcification: a new mechanism for plaque stability? Arterioscler Thromb Vasc Biol Apr. 2021;41(4):1306–1308. doi:10.1161/atvbaha.121.315949
254. Zhelyazkova-Savova MD, Yotov YT, Nikolova MN, et al. Statins, vascular calcification, and vitamin K-dependent proteins: is there a relation? Kaohs J Med Sci. 2021;37(7):624–631. doi:10.1002/kjm2.12373
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.