")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 13
Polycythemia Vera: Barriers to and Strategies for Optimal Management
Authors Duminuco A , Harrington P, Harrison C, Curto-Garcia N
Received 27 September 2023
Accepted for publication 16 December 2023
Published 21 December 2023 Volume 2023:13 Pages 77—90
DOI https://doi.org/10.2147/BLCTT.S409443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wilson Gonsalves
Andrea Duminuco,1,2 Patrick Harrington,1 Claire Harrison,1 Natalia Curto-Garcia1
1Department of Haematology, Guy’s and St Thomas’ NHS Foundation Trust, London, UK; 2Haematology with BMT Unit, A.O.U. Policlinico “G.Rodolico-San Marco”, Catania, Italy
Correspondence: Claire Harrison, Guys’ and St Thomas’ Hospital, London, SE1 9RT, UK, Tel +207 188 2742, Email [email protected]
Abstract: Polycythemia vera (PV) is a subtype of myeloproliferative neoplasms characterized by impaired quality of life and severe complications. Despite the increasingly in-depth knowledge of this condition, it necessitates a multifaceted management approach to mitigate symptoms and prevent thrombotic and hemorrhagic events, ensuring prolonged survival. The therapeutic landscape has been revolutionized in recent years, where venesection and hydroxycarbamide associated with antiplatelet therapy have a central role and are now accompanied by other drugs, such as interferon and Janus kinase inhibitors. Ongoing research and advancements in targeted therapies hold promise for further enhancing the therapeutic choice for PV management.
Keywords: polycythemia vera, barriers to treatment, current approach, future perspectives
Introduction
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by the presence of erythrocytosis in peripheral blood due to an acquired mutation on the JAK2 gene (~95% with JAK2V617F in exon 14). Consequently, the JAK-STAT pathway is activated, promoting blood cell proliferation and inhibiting apoptosis. PV incidence is estimated at 22 per 100,000 population,1 and the average age of presentation is 65–74 years, although it has been described in younger patients.2,3
Excess erythrocytosis and panmyelosis characteristic of PV can lead to thrombotic events (TE – both arterial and venous), which may be the first presentation of PV or precede the diagnosis.4 Common symptoms in these patients are aquagenic pruritus, erythromelalgia (burning pain in the extremities), or hyperviscosity symptoms (headaches, blurry vision), and fatigue, among others.
Beyond the JAK2V617F mutation, other mutations have been associated with PV, such as those in exon 12 of JAK2, and other mutations involving epigenetic (ie, TET2, ASXL1) or splicing (ie, SRSF2, U2AF1); furthermore, abnormal cytogenetics such as del(20q), +8, and +9 are well described.5,6 The risk of disease progression to myelofibrosis (MF) and acute myeloid leukemia (AML) is estimated at 10 years at 4.9–6% and 2–5%, respectively, and at 20 years at 26% for MF and remains below 10% in the case of AML.7 The Mutation-Enhanced International Prognostic Scoring System for PV (MIPSS-PV) integrates genetic and clinical/demographic information to predict OS and risk of transformation to MF or AML, underlying the negative role of adverse mutations (among which spliceosome), age >67 years, leukocytosis ≥15 × 109/L, and thrombotic history.8
The current management of PV is based on risk stratification; hence, high-risk patients are defined as those aged ≥60–65 years old and/or the presence of PV-related TE. Other factors that have been considered are the presence of cardiovascular risk factors (CVRF – hypertension, hypercholesterolemia, diabetes); excess platelet levels ≥1500x109/L; need for venesection (VS) to keep hematocrit (Hct) <0.45, among others; increasingly genomic factors such as JAK2 VAF and additional mutations are also considered. Cytoreductive therapy with VS and antiplatelets is recommended for this high-risk population, while low-risk patients are managed with just venesection and antiplatelets.9,10
Here, we will focus on reviewing the classical and new therapies used in PV patients and the challenges in delivering care to this patient group, often not allowing optimal management.
Management of PV
At present, limited therapies are available to completely eradicate the neoplastic clone from which the MPN phenotype originates. Though recent studies have shown treatments are able to reduce the mutated allelic fraction, its disappearance is almost unprecedented, and the relevance of this reduction is not fully substantiated. The main goals of managing PV patients are to control hematocrit (Hct) and disease-related symptoms and reduce the risk of TE and disease progression. In this aspect, patients without a history of thrombosis and age ≤60/65 are stratified as low-risk disease, while, in case of a previous PV-related TE or age ≥60/65 years as high-risk.9,11 According to this stratification, the management approach ranges from a watch-and-wait to active treatment. Overall, VS and antiplatelet therapies are recommended for all PV patients to reduce the risk of thrombosis and would constitute watch and wait. Recent guidelines suggest cytoreductive treatment for low-risk patients with CVRF, uncontrolled Hct levels, elevated white blood cells (WBC), and extreme thrombocytosis,9 but these considerations can be challenging to implement, and, for example, guidelines are unclear as to what constitutes “elevation or extreme or uncontrolled”.
Prevention of Thrombotic Risk: General ApproachManagement of Cardiovascular Risk Factors and Lifestyle
It is well known that cardiovascular risk factors such as hypertension, hyperlipidemia, diabetes, or smoking habits can increase the risk of TE in the general population. In the PV patients, Cerquozzi et al described the correlation between the presence of hyperlipidemia (p = 0.03) and hypertension (p = 0.02) and arterial events.12 Equally, the survival of PV patients can be affected by the presence of cardiovascular risk factors.13 Therefore, it is highly recommended to manage these factors and promote a healthy lifestyle with an adequate diet, reduced smoking and alcohol consumption, and regular exercise. Moreover, some medications, such as ruxolitinib, can increase cholesterol levels; hence, a routine check of cholesterol levels is suggested in patients with this JAK inhibitor. Implementing routine cardiovascular risk factor screening for all patients and enhanced screening in some patients taking agents such as ruxolitinib is challenging, and which tools and thresholds for realization are unclear.
Few studies have focused on the role of diet in MPN in recent years. The Mediterranean diet has anti-inflammatory properties and could reduce the incidence of cardiovascular risk factors.14,15 The Nutrient Trial has explored the role of the Mediterranean diet in MPN patients, looking at reduction in inflammatory biomarkers and symptom burden, among others. Although no significant differences in the inflammatory markers were found, the adherence to this diet was good.16 Overall, a healthy and varied diet, together with regular exercise, should be encouraged for MPN patients, but, for the large part, these are not implemented in routine care.
Venesection
Venesection (VS) remains the cornerstone treatment for PV. Removing red blood cells from the peripheral blood is primarily aimed at reducing blood viscosity, thus preventing TE with associated complications and helping to alleviate symptoms common to PV, such as pruritus, headache, and fatigue.
In 1986, the PVSG-01 randomized trial (N = 431) prospectively evaluated the use of VS compared to other treatments. It demonstrated remarkable overall survival (OS) in the arm treated with VS (median 13.9 years) compared to patients treated with chlorambucil (8.9 years) or phosphorus-32 (11.8 years). Moreover, the study confirmed the hypothesis of a higher risk of transformation to AML in patients treated with alkylating or radioactive agents.17 Later, the randomized trial CYTO-PV defined the indications for VS and the Hct target to be achieved. The primary endpoint was the incidence of cerebral TE (stroke, transient ischemic attack), myocardial infarction, peripheral arterial occlusions, and venous thromboembolism were studied in two groups (target Hct <45% in one arm and between 45% and 50% in the other). The results reported a lower incidence of events (2.7%) in the group with a Hct target <45% (compared to 9.8% of the other group, p = 0.007). Moreover, considering the occurrence of superficial-vein thrombosis, the same differences were maintained (4.4% versus 10.9%, p = 0.02). No significant distinctions between the groups were reported for adverse bleeding events and/or myelofibrosis (MF)/AML/myelodysplasia progression.18
Thus, beyond the risk stratification, VS is indicated in PV patients with elevated Hct levels (>45%). In case of persistent symptomatic burden (erythromelalgia, amaurosis, persistent headache) or particular circumstances (pregnancy due to hemostatic changes, the greater thrombotic risk), the therapeutic target could also be lower (30–39%).19 Approximately 350/450 mL of blood is generally removed from the circulation during a procedure. The frequency of VS is individualized to maintain the Htc on target.
VS is generally well tolerated, but certain precautions and contraindications should be considered, and patients should be carefully evaluated before addressing the procedure in view of age and/or medical comorbidities such as hypertension or cardiac diseases.20 VS is also time-consuming and resource-intensive; it also results in a “see-saw” effect for patients, while some newer therapies (see below) aim to eradicate the need for this therapy.
Adverse effects of VS are generally mild and transient, including fatigue, light-headedness, and iron deficiency. Rarely serious events are syncope, nerve injury, vasovagal reaction, and infections.21 Typically, one VS reduces the body’s iron amount by about 250 milligrams, and questions often arise about iron supplementation to restore deposits and minimize iron deficiency-associated side effects. However, iron replacement in PV patients should be carefully and individually assessed, and overall, it is not recommended as it can increase the Hct and hemoglobin levels.22
A different procedure to reduce Hct levels may be erythrocyte-apheresis (ECP), although its usage in PV is limited. This apheresis technique is an extracorporeal blood separation whereby blood is extracted from a patient, and through an external machine, the red blood cells are discarded, and the remaining blood is returned to circulation. The risks associated, in this case, are related to the use of a larger venous catheter (with risk of bleeding and infections) or to the citrate used as an anticoagulant, which can reduce serum ionized calcium levels causing dizziness, paresthesia, twitching, muscle cramps, and tetany.23
In summary, guidelines suggest VS should begin as soon as possible after diagnosis to keep Hct <45%. Fluid replacement with VS is based on local practice and the patient’s medical history. The use of cytoreductive therapy can be considered in case of a high number of VS requirements (>6–7 procedures per year) and/or if patients develop resistance/intolerance to the procedure24 or persistent symptoms related to iron deficiency.
Antiplatelet Therapy
The use of aspirin in PV has historically been debated. Initially, in a study conducted by the Polycythemia Vera Study Group (PVSG), the use of high doses of aspirin (900 mg daily) was associated with an elevated risk of gastrointestinal bleeding.25 In 2004, the European Collaborative Low-dose Aspirin (ECLAP) randomized, double-blind trial evaluated the efficacy of aspirin (100 mg daily) versus placebo in preventing TE in PV patients. The study demonstrated that this dosage was associated with a significant reduction in deaths from cardiovascular events and nonfatal thrombosis (eg, stroke, myocardial infarction, venous thrombosis and/or pulmonary embolisms) [hazard ratio (HR) 0.4; 95% confidence interval (CI) 0.18–0.91; p = 0.02] without a significant increase of major bleeding in the aspirin group [relative risk (RR), 1.62; 95% CI, 0.27 to 9.71].26 In the case of intolerance to aspirin, such as mild indigestion, gastric ulcers or bleeding events, among others, clopidogrel (an ADP-receptor antagonist) should be used, although limited data are available about its role in preventing TE in the PV population. Furthermore, it raises an important question in those patients with acute coronary syndrome where clopidogrel is recommended as prevention in the general population, as well as the combined usage with aspirin in indicated cases.27
Anticoagulation Therapy
Direct oral anticoagulation (DOAC) therapy usage has been raised in the last year to prevent and treat TE in cardiovascular diseases.28 DOACs are studied and evaluated in solid cancers, while data in hematological neoplasms are less explored.29 The SELECT-D study, including lymphoma (n = 23) and myeloma (n = 5) patients with deep vein thrombosis (DVT) and pulmonary embolism (PE) compared the efficacy of rivaroxaban versus LMWH (dalteparin). The cumulative incidence of recurrent TE rate at 6 months was lower (4%, [95% CI 2–9]) in rivaroxaban arm versus dalteparin arm (11%, [95% CI 7–16]), with HR 0.43, 95% CI 0.19–0.99, associated with a lower rate of major bleedings events in DOAC arm compared to LMWH [4%, 95% CI; 2–8; vs 6%, 95% CI 3–11; respectively, HR 1.83, 95% CI 0.68–4.96].30 Despite the small number of hematological patients included in these studies, the use of DOAC has been extending in recent years. Ianotto et al presented a study with 25 patients (8 PV and 17 ET) treated with DOAC for atrial fibrillation (AF) and TE, with only one case of TE with treatment and 3 major bleeding (1 due to traumatic injury and 2 post-surgery).31 In a recent small review with MPN patient and disease-associated TE (n = 102), DOAC were given in 32 with only 1 case of mesenteric ischemia without other cases of recurrent TE after 84.7 patients’ years cumulatively. Equally, no major bleeding events were described and only 3 cases of minor bleeding.32 Recently, two retrospective studies led by How et al and Barbui et al have demonstrated the efficacy and safety of DOAC. How et al studied 133 MPN patients who received DOAC for TE and AF, finding a recurrent thrombosis of 5.5% (1.5–9.5%) and bleeding of 12.3% (6.4–18.2%) after 1 year. Moreover, the multivariate analysis described a high-risk of thrombosis in patients with dabigatran and edoxaban, younger age and history of thrombosis, while high WBC was associated with an increased risk of bleeding.33 Barbui et al revised 442 MPN patients with DOAC indication for AF or VTE and described rates of recurrent thrombosis in 10 cases (2.1% patients’ year) and 22 cases (9.2% patients’ year), respectively.34 Based on these, a multicenter Phase 3 prospective, randomized, and open-label trial (AVAJAK, NCT05198960) has been designed to compare the efficacy of apixaban (2.5mg bd)/rivaroxaban (10mg od) versus aspirin 100mg daily in preventing the occurrence of TE. Within the second endpoints are the bleeding events, therapeutic adherence, overall survival, and/or quality of life, among others. This study’s results would help define the role of DOAC in the MPN population.
A brief summary of the described studies is reported in Table 1.
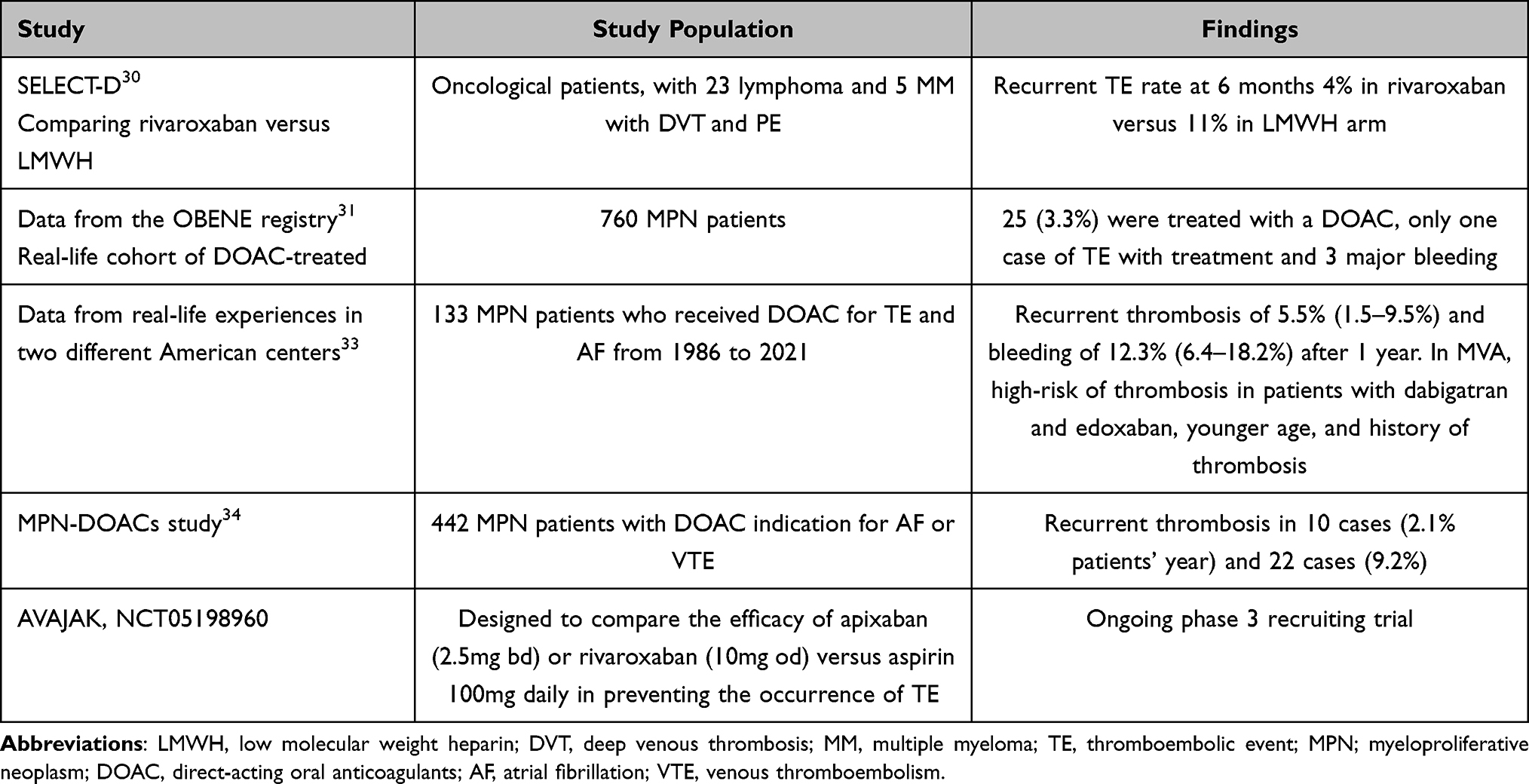 |
Table 1 Summary of Studies Reported in the Literature Regarding Anticoagulant Therapy in MPN |
Despite the current limited knowledge, overall, the use of DOAC has been extended and appears safe and efficacious for preventing and treating TE in PV.
Cytoreductive Treatment
Hydroxycarbamide (HC) has been the gold standard treatment in PV patients for years. It is a DNA synthesis inhibitor due to the activity of the hydroxylamine group (-NHOH) that interferes with essential enzymes for DNA synthesis, such as ribonucleotide-diphosphate reductase.35
The PVSG and the French Polycythemia Study Group reported the first evidence of HC’s role in PV in the 1970s. PSVG compared 51 patients treated with HC and 194 only with VS, evidencing an inferior thrombotic risk in the HC arm (9.8% vs 32.8% in the VS group; p = 0.009).36 Conversely, in a randomized trial, the French Polycythemia Study Group compared HC treatment to pipobroman in 292 patients <65 years. During a median follow-up of 9 years, no differences in terms of TE were reported; however, an increased risk of leukemic transformation was observed.37 Furthermore, the extended follow-up (16 years) confirmed a higher risk of AML/myelodysplastic syndrome transformation during pipobroman therapy (cumulative incidence of 52% at 20 years vs 24% with HC)38 that was suggested in previous studies.
A subgroup analysis of the ECLAP cohort compared 1042 patients with PV who received only VS or HC to maintain the Hct level <45%. The occurrence of TE was statistically higher in the VS group than in the HC arm (5.8 vs 3.0 per 100 person-years, p = 0.002) during a comparable observation period (29.9 months for VS and 34.7 for HC). MF progression was reported only in patients not treated with HC (n = 8, 2.3%) and AML transformation in 3 cases (n = 2 in the VS arm and n = 1 in the HC arm). Furthermore, the study demonstrated that the rate of TE and excess mortality was significantly higher in VS-cohort patients with high-risk disease and those who did not achieve target Hct <45% (p = 0.000).39
Recent trials have also reviewed the efficacy of HC compared to interferon-α as described by the randomized Myeloproliferative Disorders Research Consortium (MPD-RC 112) study.40 This trial demonstrated no difference between the overall response rates at 12 months between arms (69.8% for HC and 78% for IFN, p = 0.22). The PROUD-PV phase 3 clinical trial has recently published the results of monopegylated-IFN-α-2b (ropeg-IFN) use in both naïve and previously HC-treated high-risk PV patients. PROUD-PV was designed as a non-inferiority study with HC, and the study reached its primary objective of achieving complete hematological and spleen volume response by 12 months. The CONTI-PV study compared ropeg-IFN with the best available therapy as a continuation of the PROUD-PV study. Surprisingly, in the PROUD-PV study, at 12 months, the complete hematological response (CHR) favored the HC-treated cohort [75% vs 62.1%, p = 0.12],41 although in the CONTI-PV study, as ailed below, these results reverted in favor of IFN arm.42
Despite the good efficacy of HC, various side effects have been reported, as well as the onset of resistance/intolerance leading to treatment suspension. Hematological/oncological toxicities are manifested by myelosuppression, with anemia, leukopenia (the more common side effect), and thrombocytopenia, which leads to increasing infection and bleeding risk. Due to drug interactions, several gastrointestinal side effects (ie, gastritis, mucositis, and oral mucosa ulcers), such as liver toxicity and fatal and nonfatal pancreatitis, have been described. As a chemotherapy drug, it has toxicity on the reproductive system by reducing the count and motility of spermatozoa, with related effects responsible for malformations or fetal-neonatal toxicity, so it is contraindicated in pregnancy and while breastfeeding.43
On the other hand, HC has been associated with increased skin toxicities such as skin ulcers, which is a common cause of treatment interruption, as demonstrated by Antonioli et al among others. The authors showed that between 5% and 10% of MPN patients on HC stopped treatment due to these side effects.44 The development of non-melanoma skin cancers in HC-treated patients has been described in several different studies,45 even if the study MPN-K showed that patients exposed to HC had a risk of skin cancer similar to unexposed patients during a median period of 3 years.46 Patient education and skin monitoring for this toxicity are recommended.
Thus, the central challenge in managing PV patients with HC is the development of intolerance and resistance to the medication, which could lead to the interruption of therapy.47
The 2017 European Leukaemia Network (ELN) guidelines defined resistance as those who required VS to attain the established target of Hct <0.45, with platelets >400 × 109/L, and with failure to reduce splenomegaly by 50%, or to relieve symptoms, despite the use of HC dose of 2 g/day for 3 consecutive months (maximum dosage). Larran et al evaluated in a retrospective study 261 PV patients who received HC for a median of 4.4 years and found that 11.5% (30 patients) were/became refractory, as defined by ELN criteria, while complete response was achieved by 24% of patients. HC resistance was associated with a higher risk of death (HR 5.6, p < 0.001) and progression to AML or MF (HR 6.8, p < 0.001), with a median survival of 1.2 years after resistance was identified.48
Aiming to provide clear and standardized indications, the 2021 ELN guidelines respecified the refractory/intolerance to HC and subsequent treatment change recommendations (detailed in Box 1). While these criteria exist, operationalizing them in practice is challenging and delayed, or lack of recognition of resistance/intolerance has been well described. A further problem in current practice is a lack of definitive data regarding the long-term toxicity of HC, even if the studies available in sickle cell disease confirm its safety in patients treated for many years.49
 |
Box 1 ELN 2021 Criteria to Define Intolerance/Resistance to Hydroxycarbamide |
Interferon
In younger patients, in whom HC use could lead to critical long-term toxicities, or in those refractories to previous therapy, interferon-α (IFN) represents the treatment of choice and, more recently, is considered perhaps the optimum treatment. Several trials aimed to establish the role of IFN and have demonstrated high rates of both hematological and molecular responses through the stimulation of an immune response directed towards neoplastic cells and the antiproliferative effect on hematopoietic precursor cells.19,50 Recently, the new formulation ropeg-IFN has been tested in the PROUD-PV trial (1:1 randomized phase 3 open-label), as introduced above. The study assessed the efficacy and safety of ropeg-IFN versus HC in 254 PV patients, stratified by prior HC exposure, age at enrolment (≤60 or >60 years), and the occurrence of previous thromboembolic events. The CHR (defined as Hct <45% at least 3 months after the last VS, accompanied by platelets <400 × 109/L and white blood cells <10 × 109/L at the 12-month treatment timepoint) was achieved by 43.1% (53 of 123 patients) in the ropeg-IFN arm, confirming thus a non-inferiority versus HC. Normalization of spleen size was not demonstrated. On the other side, the ongoing CONTINUATION-PV study (extension of PROUD-PV trial) evaluated the CHR, normalization of spleen size and improvement in disease burden (such as splenomegaly, microvascular disturbances, pruritus, and headache) in subsets of patients continuing from PROUD-PV. An interim analysis at 36 months reported CHR associated with improved disease burden (including spleen reduction) in 50 (53%) of 95 patients in the ropeg-IFN group, higher than 28 (38%) of 74 patients in the HC group [1.42 (1.01 to 2.00), p = 0.044], thus demonstrating a treatment alternative for patients diagnosed with PV showing advantages compared with HC therapy beyond the second year of treatment.42
In a new recent re-evaluation after overall 5 years of treatment, 53/95 patients (55.8%) in the ropeg-IFN arm and 33/75 (44.0%) in the HC arm confirmed CHR. The occurrence of PV progression among ropeg-IFN-treated patients was 0.2%-patient-years (MF = 1), while this was 1%-patient-years in the control treatment arm (n = 4 cases, MF = 2 and AML = 2). Regarding TE, no clear differences were reported with 5 events (1%-patient-year, n = 2 in the same patient) in the ropeg-IFN arm and 5 events (1.2%-patient-year).51 The ongoing ECLIPSE PV, phase 3b, open-label, multicenter study, aims to evaluate the efficacy (in terms of hematologic response at 24 weeks), safety, and tolerability of ropeg-IFN utilizing higher dose (from 250 to 500mcg) compared to the currently labeled dosing (Q2W starting at 100 up to 500, with 50mcg increases) (NCT05481151). Finally, peginterferon importantly reduced the JAK2 V617F allelic burden or VAF significantly by 35% (starting from a median of 41%) by week 18 of treatment (p = 0.479), and to 25% by week 50 (p < 0.001).52
Concerning side effects, the use of interferon-α (IFN) is limited by the wide range of side effects. Commonly, there are flu-like symptoms, fatigue and neuropsychiatric manifestations, such as depression and anxiety, that generally lead to treatment discontinuation in approximately 24–40% of patients within 1–2 years. Neutropenia is a frequent hematological toxicity, noted in 20% of patients in some studies, as well as autoimmune disease (mainly thyroid disorders), hepatotoxicity and retinopathies.53,54 Hence, we recommend completing a baseline liver, thyroid and autoimmune profile and retinal screening before starting IFN. During the treatment period, it is essential to closely monitor for these toxicities and adjust the dose of pegylated interferon (peg-IFN) to reduce the possible side effects. Problematically, no standardized criteria exist for IFN intolerance or resistance, and the role of JAK2 VAF monitoring requires guidance, as discussed later.
Janus Kinase Inhibitor
The Janus kinase inhibitors (JAKi) widely impacted the treatment of MF, above all in the patients with worse prognosis according to the standard prognostic models,55 where over the years, several JAKi were tested and are currently used with significant results in terms of symptoms control, improvement in quality of life, and aiming to modify the disease outcome, alone or combined to other molecules with a different mechanism of action.56,57 In PV, ruxolitinib is the only JAKi licensed and widely used in intolerance or resistance to HC cases. A multicenter phase 3 (RESPONSE) trial compared ruxolitinib to other physician choices in a second-line therapy setting following HC in PV patients with splenomegaly, stratifying patients according to inadequate response or unacceptable side effects. The primary endpoints were Hct control (less than 45% in the absence of VS) and a 35% reduction of spleen size, evaluating as secondary aims the duration of response, the symptoms control and the safety. After 81 weeks in the ruxolitinib and 34 in the control arms (the median exposure to therapy assumed as data cut-off), 84.5% and 3.6% continued treatment, respectively, in the two groups, and the composite primary efficacy endpoints (Hct and spleen reduction taken together) were significantly more achieved by patients treated with JAKi (20.9% vs 0.9% in control arm, p < 0.001), without any difference according to whether patients were refractory or intolerant to HC. Moreover, at week 32, 36 of 74 patients in the ruxolitinib group (49%) reported at least a 50% reduction in the MPN-SAF total symptom score, higher than the standard-therapy group (4 of 81 patients, 5%) and a CHR in 24%.58 The RESPONSE-2 trial (enrolling HC-resistant or intolerant PV patients without splenomegaly) confirmed the efficacy of ruxolitinib compared to the best available treatment (HC, IFN, pipobroman, lenalidomide, or no therapy). Hct control (as primary endpoint) was achieved in 46 (62%) of ruxolitinib-treated patients, significantly higher than 14 (19%) of those who received the best available therapy (OR 7.28 [95% CI 3.43–15.45]; p < 0.0001).59 These results have allowed authorization of the use of ruxolitinib by different regulatory agencies such as FSA and EMA. Recently, the results of the Phase 2 trial MAJIC-PV confirmed the extended safety of ruxolitinib as a second-line treatment versus BAT in a UK cohort, assessing the complete response within 1 year, duration of response, event-free survival (EFS), control of symptoms, and the molecular response. CHR was achieved in 43% of patients on ruxolitinib compared to 26% on BAT, associated with improved thromboembolic-event-free survival (HR 0.56, 95% CI 0.32, 1.00, p = 0.05). The molecular response, evaluated as >50% reduction in JAK2 VAF, was more frequent with ruxolitinib and was correlated with reduced progression-free-survival, EFS and overall survival (all p < 0.01).60
Despite the well-assessed efficacy of ruxolitinib in controlling Hct levels, reducing spleen size, and improving symptom burden, several occurrences and/or adverse effects may limit ruxolitinib use, and its efficiency in reducing thrombosis and later events was unclear. Hematological toxicity is mainly represented by anemia and thrombocytopenia.61 Furthermore, not being a specific inhibitor of mutated JAK2, it may be related to impaired immune function with an increased risk of new infections or reactivation of latent infections,62,63 or impaired response to immunological stimulation (eg, in cases of vaccines both in MF and PV-treated patients).64,65
A further important and severe effect is the appearance of non-melanoma skin cancers, for which patients should be routinely screened for suspicious skin lesions.66 Also, in this case, MPN-K study, demonstrated how ruxolitinib shows a high risk (OR = 3.87, 95% CI 1.18–12.75), without excess risk of carcinoma and hematological second cancer compared with unexposed patients.46 Focusing on the cohort of MF patients in the COMFORT-1 trial, ruxolitinib-treated patients reported different metabolic side effects, such as weight gain and increased total cholesterol.67
Regardless of the potential adverse effects widely reported, the long-term data from the MAJIC-PV study demonstrated an acceptable safety with significative benefits, without any death related to infections, and not confirming metabolic side effects.60
The routine use of ruxolitinib as a second-line therapy is limited by cost, recognition of resistance or intolerance to HC and a lack of guidance for IFN failure. In addition, identifying early predictive parameters for an acceptable response to ruxolitinib represents a research target, such as the RR6 score for myelofibrosis.68,69 Currently, ruxolitinib is tested as a first-line PV therapy in the MITHRIDATE trial, compared to HC or IFN (any formulation) to evaluate the event-free survival (in terms of first major thrombosis/hemorrhage, death, progression/transformation), and, as second endpoints, the impact in quality-of-life, JAK2 V617 VAF reduction, and the adverse events (NCT04116502) during a follow-up of 3 years.
Pipobroman, Busulfan and Phosphorus-32
As mentioned above, pipobroman, busulfan and radioactive phosphorus were in the past widely used in the management of these conditions. They are currently recommended by the ELN only in subsequent lines of treatment after intolerance/loss of response to therapies such as either HC or IFN.70 In a Swedish cohort study, two or more lines of cytoreductive treatment (including an alkylating agent) were correlated with a 2.9-fold increase in transformation in AML/myelodysplastic syndrome rate.71 Because of the potential leukemogenicity, these agents should be reserved for elderly patients (>80 years) with a low risk of progression or those with a disease where the risk of thrombosis is superior to the risk of transformation.
A brief explanation of the available strategies to control PV clinical manifestations and reduce thrombotic risk, with their common side effects, is reported in Table 2.
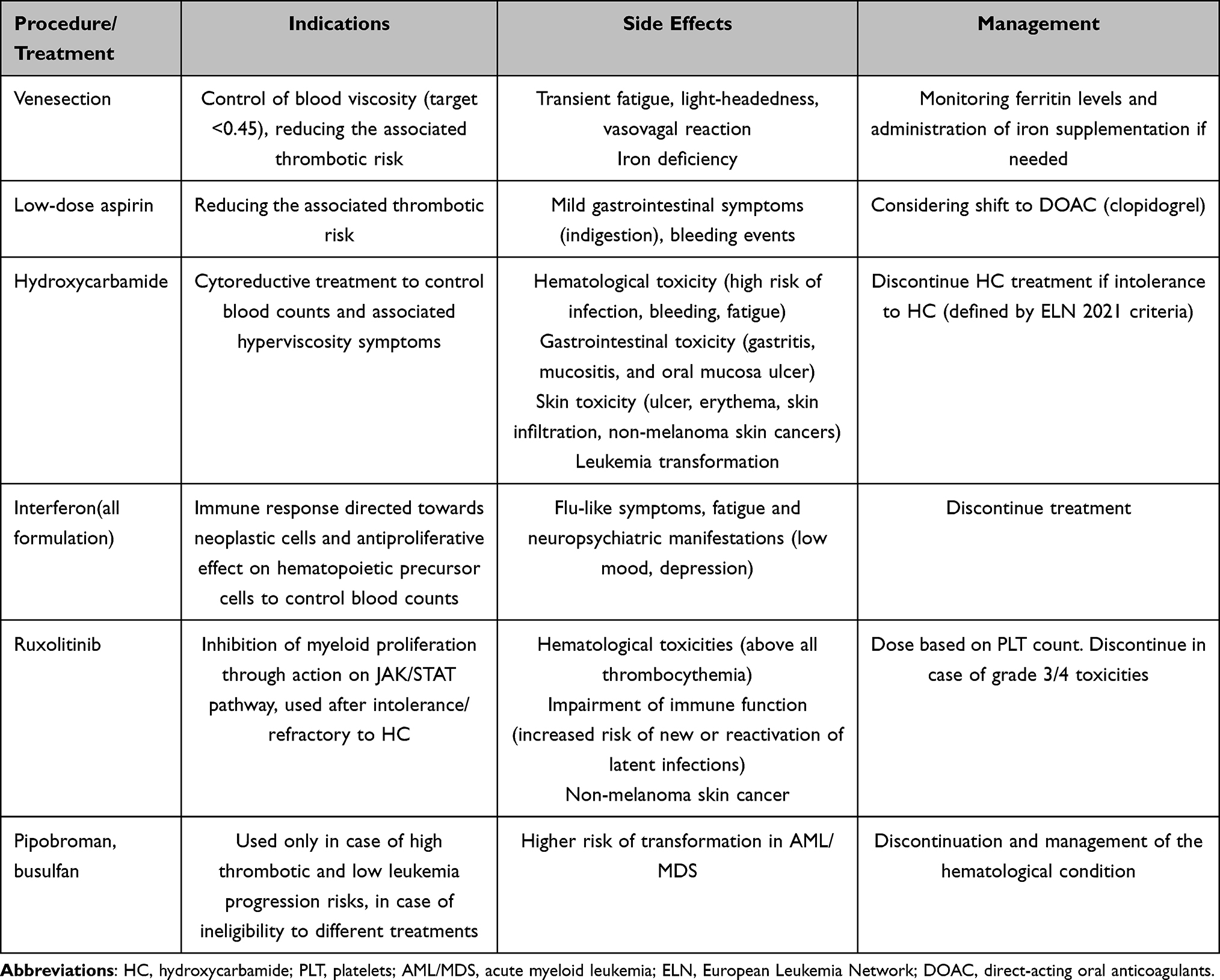 |
Table 2 Available Strategies for Polycythemia Vera Management and Commonly Reported Side Effects with Their Management |
Management of Symptoms
Symptomatic management of PV can be problematic. As already described, among the symptoms commonly present in patients with PV, pruritus is the most frequent (up to 70–85% of cases), with a spontaneous occurrence or evoked by water or changes in temperature (pruritus aquagenic) and causes a significant negative impact on quality of life.72 It is usually treated simultaneously with cytoreductive therapy/Hct control but can persist even in patients with well-controlled blood counts.73 Other purely symptomatic approaches are antihistamines (both H1 and H2 receptor antagonists),74,75 selective serotonin reuptake inhibitors (SSRIs),76 or anticonvulsant drugs. The referral to a dermatologist could be considered for refractory patients trying to treat with narrow-band-ultraviolet (UVB) or ultraviolet A (UVA), associated or not with oral psoralen to UV light (PUVA).77 These options are preferred in patients with only the presence of these symptoms but not with a thrombotic risk that requires cytoreductive therapy. In any case, persistent pruritus resistant to the above options can be an indication to start an active treatment just to control the symptom burden.
Regarding fatigue, no specific medicine can effectively treat this condition secondary to MPN or to side effects of treatment. Anxiety and depression related to cancer can increase it. Lifestyle modifications are central in the management, with adequate sleep with scheduled breaks, abstention from smoking and alcohol, hydration, and a balanced diet, associated with a referral to a psychologist to discuss one’s condition and receive support. The management of fatigue is a significant challenge in the modern management of PV.
Management of Thrombotic Events
The occurrence of a thrombotic event (TE) represents, as previously described, the factor that fixes patients as being at high risk of further thrombosis. Acute events should be managed according to current guidelines. TEs are divided into arterial (such as stroke or transient ischemic attack, acute myocardial infarction, and peripheral arterial thrombosis) and venous (deep vein thrombosis, superficial thrombophlebitis, and pulmonary embolism). Splanchnic venous thrombosis deserves special mention as, when necessary, it presents behind an MPN in a variable number of cases between 10% and 50%,78 where the specific cytoreductive therapy does not show effectiveness in reducing the risk of recurrence.79 For acute events involving the venous system, the first choice is low molecular weight heparin (LMWH), followed by indefinite secondary prophylaxis with vitamin K antagonists (VKA), as per guidelines.80 Novel direct oral anticoagulants (DOACs) are increasingly used as secondary prophylaxis with efficacy, although data on MPNs are still limited,31 as discussed earlier. In any case, every patient should be screened for CVRF, such as hypertension, hyperlipidemia, diabetes mellitus, and smoking history, as well as for prothrombotic-associated conditions (ie, antiphospholipid antibody syndrome, coagulation factor V and II mutations).
Conversely, in the case of an arterial event, the acute aim is the reperfusion of the affected extremity through thrombolysis or recanalization of the occluded vessel. It could be via catheter-directed thrombolysis (CDT, as interventional radiology treatment), surgical treatment (open bypass surgery, or the more common endovascular therapy, such as stenting, balloon angioplasty, or atherectomy) or use of thrombolytic drugs (serine proteases that cleave plasminogen into active plasmin such as streptokinase, alteplase, reteplase).81
Special conditions may require a particular approach. In the case of cerebral venous thrombosis, patients are stratified based on risk factors for poor outcome (malignancy, coma, deep venous thrombosis, mental status disturbance, male sex, intracranial hemorrhage) and treated with LMWH in case of lower risk, or endovascular procedures (endovascular thrombolysis or thrombectomy) or neurosurgery (decompressive craniotomy) in case of risk of high risk, followed by oral anticoagulation lifelong in case of the simultaneous severe prothrombotic condition, such as PV.82
Management of Hemorrhagic Events
Hemorrhagic events have been documented in individuals with PV, primarily affecting the integumentary system, mucous membranes, and gastrointestinal tract (particularly notable in cases with existing oesophageal varices). These occurrences are commonly attributed to persistent anticoagulation usage or platelet counts exceeding 1500 × 109/L, which increase the risk for acquired von Willebrand syndrome.83 The bleeding episodes are generally less frequent and severe events than the thrombotic counterpart and require management with tranexamic acid and/or platelet transfusion, associated with a subsequent cease/reduce aspirin shifting to DOACs (as mentioned above), and optimizing of cytoreductive treatment.80
Future Directions
In addition to the classic and established therapies currently available, there are several molecules (newly or used for different conditions) that are being tested in phase 2/3 trials and which, in the years to come, could represent one more possibility in the management of patients with PV in need of further treatment.
Concerning already available drugs, the efficacy and safety of peg-IFNα-2b in combination with ruxolitinib versus peg-IFNα-2b alone for treating HC-resistant/intolerant PV are currently being tested in a multicenter phase 2 study. The results, expected in 2028, could open a new window in the combination therapy between two already widely used molecules (NCT05870475).
Among the new molecules, rusfertide is a hepcidin-mimetic that binds ferroportin, causing a reduced availability of iron in the bone marrow, a fundamental element for erythrocytosis.84 It was tested in two different phase 2 trials. REVIVE trial (NCT04057040) enrolled patients with uncontrolled Hct despite VS (≥3 in the 6 months before study entry) associated with a cytoreductive therapy. This trial was divided into a 28-week open-label dose-finding phase (starting from rusfertide 20 mg subcutaneously, with subsequent dose titration, and associated with any prior cytoreductive treatment), followed by a 3-month double-blind randomization (compared to placebo), and a 3-year open-label extension, with rusfertide for all the patients enrolled. The results showed that the average clinically effective dose is approximately 40 mg weekly, without an increase in toxicity, whether rusfertide was administered with concurrent VS or cytoreductive therapy.85 The second trial (PACIFIC) enrolled patients with high Hct (>48%) for an induction period with 40mg twice weekly until the control of Hct value (<45%) and maintenance, with a schedule that may be adjusted every 2 to 4 weeks to maintain Hct, with a target of <43% (NCT04767802).
Based on these results, the phase 3 VERIFY trial is ongoing, recruiting PV patients who required ≥3 VS in the previous 6 months or ≥5 in the last 12 months due to inadequate Hct control. Patients with a stable cytoreductive regimen must be in a well-controlled scheme, with no need for dose modification, while patients managed only with VS must have interrupted cytoreductive therapy 2–6 months before screening. Patients are double-blinding randomized to 32 weeks of rusfertide or placebo added to each subject’s ongoing treatment.86 The results will definitively indicate the role of rusfertide in the PV setting.
Another class of targeted agents is histone deacetylase (HDAC) inhibitors. Givinostat, an HDAC inhibitor specific for JAK2 V617F-mutated cells, was tested in a phase 2 trial. Givinostat was administered in MPNs patients (including 12 with PV) at a starting dose of 50 mg twice daily. Apart from 2 patients who discontinued treatment, 7 out of 10 PV patients (70%) reached VS independence, and splenomegaly and pruritus were resolved in 70% and 90% of patients, respectively, with a reported reduction of JAK2 V617F allele burden. It was well tolerated, with no grade 4 toxicities.87
Subsequently, in a multicenter clinical trial, givinostat long-term efficacy and safety were tested. Of 45 patients enrolled and receiving treatment for a median of 4 years, complete/partial hematological remissions were described in 11% and 89%, respectively, with a Hct control (<45%) without VS and standard spleen size both observed in 56% of patients. The incidence of thrombosis was 2.3% of patients per year. Only three grade 3 and no grade 4 toxicities were observed.88 During an extended follow-up, the excellent safety profile was confirmed, with an overall response rate always more significant than 80%,89 laying the foundations for future phase 3 trials.
Vorinostat, another HDAC inhibitor, was tested in a phase 2 trial. Most patients who completed 24 weeks of treatment (24/33; 72%) reported a response, although 44% of patients discontinued the therapy due to adverse events, of which 25% of cases were severe (deep vein thrombosis, diarrhea, headache, progression to AML, palpitations, neuropathy, fatigue, and renal impairment).90 Using lower dosages of vorinostat combined with other drugs (eg, JAKi) will probably represent the therapy capable of conferring good responses without excessive toxicities.
In addition, several other molecules are currently being studied in phase 2 studies, which may represent potential mechanisms in the future. Summaries of these trials are shown in Table 3.
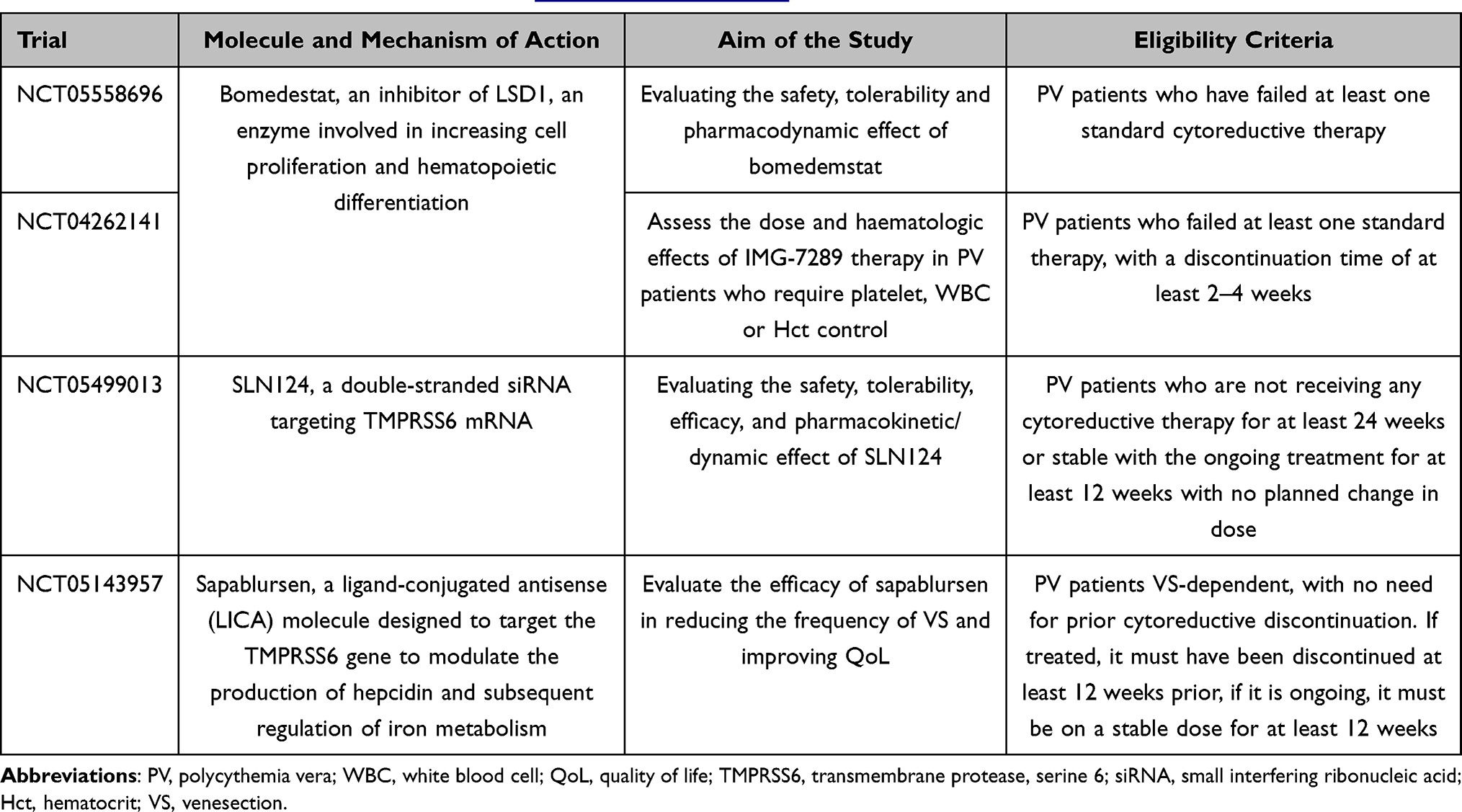 |
Table 3 Summary of Ongoing Clinical Phase 1/2 Trial Testing New Molecules in the Setting of PV Patients Intolerant/Refractory to Standard Cytoreductive Treatment Available on https://clinicaltrials.gov/.91 |
Conclusion
In the last decade, considerable advancements have transpired within the therapeutic landscape of PV, predominantly inspired by the revelation of the disease’s fundamental molecular driver mutations. However, in order to deliver effective care for patients, attention to details such as vascular risk, optimum control, defining how to utilize molecular assays, and recognizing when patients fail to continue to be a challenge. Notably, optimal management of debilitating symptoms, particularly fatigue, and cases with an associated complex clinical background necessitate the provision of an enhanced approach.
Disclosure
Andrea Duminuco reports personal fees from BMS Celgene, personal fees from Incyte, personal fees from EusaPharma, outside the submitted work. Patrick Harrington reports grants from Bristol Myers Squibb, personal fees from Incyte, outside the submitted work. Claire Harrison reports grants, personal fees and travel support from Novartis, grants, personal fees from Constellation, grants, personal fees from Celgene/BMS, personal fees, non-financial support from AbbVie, grants, personal fees from AOP, grants, personal fees from CTI/Sobi, grants, personal fees from IMAGO/MSD, personal fees from Galecto, personal fees from Geron, personal fees from Keros, grants, personal fees, and writing from GSK, outside the submitted work; and Work with European Haematology Association Work with patient advocacy – Blood Cancer UK, MPN Voice. The authors report no other conflicts of interest in this work.
References
1. Ma X, Vanasse G, Cartmel B, Wang Y, Selinger HA. Prevalence of polycythemia vera and essential thrombocythemia. Am J Hematol. 2008;83(5):359–362. doi:10.1002/ajh.21129
2. Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol. 2014;92(4):289–297. doi:10.1111/ejh.12256
3. Ianotto JC, Curto-Garcia N, Lauermannova M, Radia D, Kiladjian JJ, Harrison C. Characteristics and outcomes of patients with essential thrombocythemia or polycythemia vera diagnosed before 20 years of age, a systematic review. Haematologica. 2019;2018:200832. doi:10.3324/haematol.2018.200832
4. Falanga A, Marchetti M. Thrombotic disease in the myeloproliferative neoplasms. Hematology. 2012;2012(1):571–581. doi:10.1182/asheducation.V2012.1.571.3798557
5. Tefferi A, Lasho TL, Guglielmelli P, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21–30. doi:10.1182/bloodadvances.2016000216
6. Tang G, Hidalgo Lopez JE, Wang SA, et al. Characteristics and clinical significance of cytogenetic abnormalities in polycythemia vera. Haematologica. 2017;102(9):1511–1518. doi:10.3324/haematol.2017.165795
7. Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5(11):e366–10. doi:10.1038/bcj.2015.95
8. Tefferi A, Guglielmelli P, Lasho TL, et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2020;189(2):291–302. doi:10.1111/BJH.16380
9. McMullin MF, Harrison CN, Ali S, et al. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Br J Haematol. 2019;184(2):176–191. doi:10.1111/bjh.15648
10. Tefferi A, Vannucchi AM, Barbui T. Polycythemia vera: historical oversights, diagnostic details, and therapeutic views. Leukemia. 2021;35(12):3339–3351. doi:10.1038/s41375-021-01401-3
11. Tefferi A, Barbui T. Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023;98(9):1465–1487. doi:10.1002/ajh.27002
12. Cerquozzi S, Barraco D, Lasho T, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 2017;7(12):662. doi:10.1038/s41408-017-0035-6
13. Mancuso S, Santoro M, Accurso V, et al. Cardiovascular risk in polycythemia vera: thrombotic risk and survival: can cytoreductive therapy be useful in patients with low-risk polycythemia vera with cardiovascular risk factors? Oncol Res Treat. 2020;43(10):526–530. doi:10.1159/000509376
14. Estruch R, Ros E, Salas-Salvadó J, et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med. 2013;368(14):1279–1290. doi:10.1056/NEJMoa1200303
15. Casas R, Urpi-Sardà M, Sacanella E, et al. Anti-inflammatory effects of the Mediterranean diet in the early and late stages of atheroma plaque development. Mediators Inflamm. 2017;2017:3674390. doi:10.1155/2017/3674390
16. Mendez LF, Nguyen H, Nguyen J, et al. The Nutrient Trial (NUTRitional Intervention among myEloproliferative Neoplasms): feasibility Phase. Blood. 2019;134:5380. doi:10.1182/blood-2019-130851
17. Berk PD, Goldberg JD, Donovan PB, Fruchtman S, Berlin NI, Wasserman LR. Therapeutic recommendations in polycythemia vera based on polycythemia vera study group protocols. Semin Hematol. 1986;23:132–143.
18. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2012;368(1):22–33. doi:10.1056/NEJMoa1208500
19. Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018;32(5):1057–1069. doi:10.1038/s41375-018-0077-1
20. Assi TB, Baz E. Current applications of therapeutic phlebotomy. Blood Transfus. 2014;12(Suppl 1):s75–83. doi:10.2450/2013.0299-12
21. Ohnishi H. Side effects of phlebotomy: pathophysiology, diagnosis, treatment and prophylaxis. Rinsho Byori. 2005;53(10):904–910.
22. Barton JC, Bottomley SS. Iron deficiency due to excessive therapeutic phlebotomy in hemochromatosis. Am J Hematol. 2000;65(3):223–226. doi:10.1002/1096-8652(200011)65:3<223::aid-ajh8>3.0.co;2-9
23. Padmanabhan A, Connelly-Smith L, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice – evidence-based approach from the writing committee of the American society for apheresis: the eighth special issue. J Clin Apher. 2019;34(3):171–354. doi:10.1002/jca.21705
24. Barbui T, Passamonti F, Accorsi P, et al. Evidence- and consensus-based recommendations for phlebotomy in polycythemia vera. Leukemia. 2018;32(9):2077–2081. doi:10.1038/s41375-018-0199-5
25. Tartaglia AP, Goldberg JD, Berk PD, Wasserman LR. Adverse effects of antiaggregating platelet therapy in the treatment of polycythemia vera. Semin Hematol. 1986;23(3):172–176.
26. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–124. doi:10.1056/NEJMoa035572
27. Landolfi R, Di Gennaro L. Prevention of thrombosis in polycythemia vera and essential thrombocythemia. Haematologica. 2008;93(3 SE–Editorials):331–335. doi:10.3324/haematol.12604
28. Ashley C, Eric S, Bruce AW. Direct oral anticoagulant use: a practical guide to common clinical challenges. J Am Heart Assoc. 2020;9(13):e017559. doi:10.1161/JAHA.120.017559
29. Raskob GE, van Es N, Verhamme P, et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N Engl J Med. 2017;378(7):615–624. doi:10.1056/NEJMoa1711948
30. Young AM, Marshall A, Thirlwall J, et al. Comparison of an oral factor xa inhibitor with low molecular weight heparin in patients with cancer with venous thromboembolism: results of a randomized trial (SELECT-D). J Clin Oncol. 2018;36(20):2017–2023. doi:10.1200/JCO.2018.78.8034
31. Ianotto JC, Couturier MA, Galinat H, et al. Administration of direct oral anticoagulants in patients with myeloproliferative neoplasms. Int J Hematol. 2017;106(4):517–521. doi:10.1007/S12185-017-2282-5
32. Curto-Garcia N, Doyle AJ, Breen KA, et al. Outcomes of patients receiving direct oral anticoagulants for myeloproliferative neoplasm-associated venous thromboembolism in a large tertiary centre in the UK. Br J Haematol. 2020;189. doi:10.1111/bjh.16485
33. How J, Story C, Ren S, et al. Practice patterns and outcomes of direct oral anticoagulant use in myeloproliferative neoplasm patients. Blood Cancer J. 2021;11(11). doi:10.1038/S41408-021-00566-5
34. Barbui T, De Stefano V, Carobbio A, et al. Direct oral anticoagulants for myeloproliferative neoplasms: results from an international study on 442 patients. Leukemia. 2021;35(10):2989. doi:10.1038/S41375-021-01279-1
35. Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol. 1992;19:3).
36. Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, Wasserman LR. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol. 1997;34(1):17–23.
37. Najean Y, Rain JD. Treatment of polycythemia vera: the use of hydroxyurea and pipobroman in 292 patients under the age of 65 years. Blood. 1997;90(9):3370–3377. doi:10.1182/blood.V90.9.3370
38. Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29(29):3907–3913. doi:10.1200/JCO.2011.36.0792
39. Barbui T, Vannucchi AM, Finazzi G, et al. A reappraisal of the benefit-risk profile of hydroxyurea in polycythemia vera: a propensity-matched study. Am J Hematol. 2017;92(11):1131–1136. doi:10.1002/ajh.24851
40. Mascarenhas J, Kosiorek HE, Prchal JT, et al. A randomized phase 3 trial of interferon-α vs hydroxyurea in polycythemia vera and essential thrombocythemia. Blood. 2022;139(19):2931–2941. doi:10.1182/blood.2021012743
41. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients; 2016.
42. Gisslinger H, Klade C, Georgiev P, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020;7(3):e196–e208. doi:10.1016/S2352-3026(19)30236-4
43. Berthaut I, Guignedoux G, Kirsch-Noir F, et al. Influence of sickle cell disease and treatment with hydroxyurea on sperm parameters and fertility of human males. Haematologica. 2008;93(7):988–993. doi:10.3324/haematol.11515
44. Antonioli E, Guglielmelli P, Pieri L, et al. Hydroxyurea-related toxicity in 3411 patients with Ph’-negative MPN. Am J Hematol. 2012;87(5):552–554. doi:10.1002/ajh.23160
45. Gavini DR, Salvi DJ, Shah PH, Uma D, Lee JH, Hamid P. Non-melanoma skin cancers in patients on hydroxyurea for Philadelphia chromosome-negative myeloproliferative neoplasms: a systematic review. Cureus. 2021;13(8):e16978. doi:10.7759/cureus.16978
46. Barbui T, Ghirardi A, Masciulli A, et al. Second cancer in Philadelphia negative myeloproliferative neoplasms (MPN-K). A nested case-control study. Leukemia. 2019;33(8):1996–2005. doi:10.1038/S41375-019-0487-8
47. Alvarez-Larrán A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119(6):1363–1369. doi:10.1182/blood-2011-10-387787
48. Barosi G, Birgegard G, Finazzi G, et al. Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood. 2009;113(20):4829–4833. doi:10.1182/blood-2008-09-176818
49. Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. 2010;85(6):403. doi:10.1002/AJH.21699
50. Kiladjian JJ, Giraudier S, Cassinat B. Interferon-alpha for the therapy of myeloproliferative neoplasms: targeting the malignant clone. Leukemia. 2016;30(4):776–781. doi:10.1038/leu.2015.326
51. Kiladjian JJ, Klade C, Georgiev P, et al. Long-term outcomes of polycythemia vera patients treated with ropeginterferon Alfa-2b. Leukemia. 2022;36(5):1408–1411. doi:10.1038/s41375-022-01528-x
52. Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, et al. Ropeginterferon alfa-2b, a novel IFNα-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015;126(15):1762–1769. doi:10.1182/blood-2015-04-637280
53. Kiladjian JJ, Chomienne C, Fenaux P. Interferon-alpha therapy in bcr-abl-negative myeloproliferative neoplasms. Leukemia. 2008;22(11):1990–1998. doi:10.1038/leu.2008.280
54. Quintás-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27(32):5418–5424. doi:10.1200/JCO.2009.23.6075
55. Duminuco A, Nardo A, Giuffrida G, et al. Myelofibrosis and survival prognostic models: a journey between past and future. J Clin Med. 2023;12(6):2188. doi:10.3390/JCM12062188
56. Duminuco A, Vetro C, Giallongo C, Palumbo GA. The pharmacotherapeutic management of patients with myelofibrosis: looking beyond JAK inhibitors. Expert Opin Pharmacother. 2023;24:1449–1461. doi:10.1080/14656566.2023.2228695
57. Duminuco A, Torre E, Palumbo GA, Harrison C. A Journey Through JAK inhibitors for the treatment of myeloproliferative diseases. Curr Hematol Malignancy Rep. 2023;18:176–189. doi:10.1007/S11899-023-00702-X
58. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. doi:10.1056/NEJMOA1409002/SUPPL_FILE/NEJMOA1409002_DISCLOSURES.PDF
59. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 2017;18(1):88–99. doi:10.1016/S1470-2045(16)30558-7
60. Harrison CN, Nangalia J, Boucher R, et al. Ruxolitinib versus best available therapy for polycythemia vera intolerant or resistant to hydroxycarbamide in a randomized trial. J Clin Oncol. 2023;41(19):3534–3544. doi:10.1200/jco.22.01935
61. Vannucchi AM, Te Boekhorst PAW, Harrison CN, et al. EXPAND, a dose-finding study of ruxolitinib in patients with myelofibrosis and low platelet counts: 48-week follow-up analysis. Haematologica. 2019;104(5):947–954. doi:10.3324/HAEMATOL.2018.204602
62. Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib-associated infections: a systematic review and meta-analysis. Am J Hematol. 2018;93(3):339–347. doi:10.1002/AJH.24976
63. Duminuco A, Scarso S, Cupri A, et al. Leishmania infection during ruxolitinib treatment: the cytokines-based immune response in the setting of immunocompromised patients. J Clin Med. 2023;12(2):578. doi:10.3390/JCM12020578
64. Palumbo GA, Cambria D, La Spina E, et al. Ruxolitinib treatment in myelofibrosis and polycythemia vera causes suboptimal humoral immune response following standard and booster vaccination with BNT162b2 mRNA COVID-19 vaccine. Front Oncol. 2023:13. doi:10.3389/FONC.2023.1117815
65. Duminuco A, Nardo A, Orofino A, et al. Efficacy and safety of tixagevimab-cilgavimab versus SARS-CoV-2 breakthrough infection in the hematological conditions. Cancer. 2023. doi:10.1002/CNCR.35005
66. Lin JQ, Li SQ, Li S, et al. A 10-year retrospective cohort study of ruxolitinib and association with nonmelanoma skin cancer in patients with polycythemia vera and myelofibrosis. J Am Acad Dermatol. 2022;86(2):339–344. doi:10.1016/J.JAAD.2021.10.004
67. Mesa RA, Verstovsek S, Gupta V, et al. Effects of ruxolitinib treatment on metabolic and nutritional parameters in patients with myelofibrosis from COMFORT-I. Clin Lymphoma Myeloma Leuk. 2015;15(4):214–221.e1. doi:10.1016/J.CLML.2014.12.008
68. Maffioli M, Mora B, Ball S, et al. A prognostic model to predict survival after 6 months of ruxolitinib in patients with myelofibrosis. Blood Adv. 2022;6(6):1855–1864. doi:10.1182/BLOODADVANCES.2021006889
69. Duminuco A, Nardo A, Garibaldi B, et al. Prediction of survival and prognosis migration from gold-standard scores in myelofibrosis patients treated with ruxolitinib applying the rr6 prognostic model in a monocentric real-life setting. J Clin Med. 2022;11(24):7418. doi:10.3390/JCM11247418
70. Marchetti M, Vannucchi AM, Griesshammer M, et al. Appropriate management of polycythaemia vera with cytoreductive drug therapy: European LeukemiaNet 2021 recommendations. Lancet Haematol. 2022;9(4):e301–e311. doi:10.1016/S2352-3026(22)00046-1
71. Björkholm M, Derolf ÅR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29(17):2410. doi:10.1200/JCO.2011.34.7542
72. Siegel FP, Tauscher J, Petrides PE. Aquagenic pruritus in polycythemia vera: characteristics and influence on quality of life in 441 patients. Am J Hematol. 2013;88(8):665–669. doi:10.1002/AJH.23474
73. Gangat N, Strand JJ, Lasho TL, Li C-Y, Pardanani A, Tefferi A. Pruritus in polycythemia vera is associated with a lower risk of arterial thrombosis. Am J Hematol. 2008;83(6):451–453. doi:10.1002/AJH.21156
74. Weick JK, Donovan PB, Najean Y, et al. The use of cimetidine for the treatment of pruritus in polycythemia vera. Arch Intern Med. 1982;142(2):241–242. doi:10.1001/ARCHINTE.1982.003401500410010
75. Steinman HK, Greaves MW. Aquagenic pruritus. J Am Acad Dermatol. 1985;13(1):91–96. doi:10.1016/S0190-9622(85)70149-1
76. Tefferi A, Fonseca R. Selective serotonin reuptake inhibitors are effective in the treatment of polycythemia vera-associated pruritus. Blood. 2002;99(7):2627. doi:10.1182/BLOOD.V99.7.2627
77. Rivard J, Lim HW. Ultraviolet phototherapy for pruritus. Dermatol Ther. 2005;18(4):344–354. doi:10.1111/J.1529-8019.2005.00032.X
78. Finazzi G, De Stefano V, Barbui T. Splanchnic vein thrombosis in myeloproliferative neoplasms: treatment algorithm 2018. Blood Cancer J. 2018;8(7):1–6. doi:10.1038/s41408-018-0100-9
79. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica. 2008;93(3):372–380. doi:10.3324/HAEMATOL.12053
80. McMullin MFF, Mead AJ, Ali S, et al. A guideline for the management of specific situations in polycythaemia vera and secondary erythrocytosis. Br J Haematol. 2019;184(2):161–175. doi:10.1111/BJH.15647
81. Smith DA, Lilie CJ. Acute arterial occlusion. StatPearls; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441851/.
82. Ulivi L, Squitieri M, Cohen H, Cowley P, Werring DJ. Cerebral venous thrombosis: a practical guide. Pract Neurol. 2020;20(5):356–367. doi:10.1136/PRACTNEUROL-2019-002415
83. Wille K, Huenerbein K, Jagenberg E, et al. Bleeding complications in bcr-abl-negative myeloproliferative neoplasms (MPN): a retrospective single-center study of 829 MPN patients. Eur J Haematol. 2022;108(2):154–162. doi:10.1111/EJH.13721
84. Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. 2018;131(16):1790. doi:10.1182/BLOOD-2017-11-737411
85. Gerds AT, Gotlib J, Palmer JM, et al. Rusfertide for polycythemia vera: similar dosing in patients receiving therapeutic phlebotomy alone or in combination with cytoreductive treatment. Blood. 2022;140(Supplement 1):12241–12243. doi:10.1182/BLOOD-2022-163847
86. Verstovsek S, Kuykendall A, Hoffman R, et al. Verify: a phase 3 study of the hepcidin mimetic rusfertide (PTG-300) in patients with polycythemia vera. Blood. 2022;140(Supplement 1):3929–3931. doi:10.1182/BLOOD-2022-163755
87. Rambaldi A, Dellacasa CM, Finazzi G, et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150(4):446–455. doi:10.1111/J.1365-2141.2010.08266.X
88. Finazzi G, Iurlo A, Martino B, et al. A long-term safety and efficacy study of givinostat in patients with polycythemia vera: the first 4 years of treatment. Blood. 2017;130(Supplement 1):1648. doi:10.1182/BLOOD.V130.SUPPL_1.1648.1648
89. Rambaldi A, Iurlo A, Vannucchi AM, et al. Long-term safety and efficacy of givinostat in polycythemia vera: 4-year mean follow up of three phase 1/2 studies and a compassionate use program. Blood Cancer J. 2021;11(3):1–7. doi:10.1038/s41408-021-00445-z
90. Andersen CL, Mcmullin MF, Ejerblad E, et al. A Phase II study of vorinostat (MK-0683) in patients with polycythaemia vera and essential thrombocythaemia. Br J Haematol. 2013;162(4):498–508. doi:10.1111/BJH.12416
91. ClinicalTrials.gov. Home. Available from: https://clinicaltrials.gov/.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.