")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 17
Pituitary Stalk Interruption Syndrome with Excessive Height Growth Combined with Congenital Absence of the Uterus and Ovaries: A Rare Case Report and Review of the Literature
Received 25 December 2023
Accepted for publication 3 April 2024
Published 16 April 2024 Volume 2024:17 Pages 1739—1747
DOI https://doi.org/10.2147/DMSO.S456678
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Rongqian Wu,1– 3 Jixiong Xu1– 3
1Department of Endocrinology and Metabolism, The 1st Affiliated Hospital, Jiangxi Medical College, Nanchang University, Nanchang, People’s Republic of China; 2Jiangxi Clinical Research Center for Endocrine and Metabolic Disease, Nanchang, People’s Republic of China; 3Jiangxi Branch of National Clinical Research Center for Metabolic Disease, Nanchang, People’s Republic of China
Correspondence: Jixiong Xu, Email [email protected]
Aim: Pituitary stalk interruption syndrome is a relatively rare disease. Patients with this disease usually have different degrees of short stature in adulthood. The purpose of this case report is to highlight a special case of unusually elongated limbs with excessive height growth and congenital absence of uterus and ovary, so as to improve clinicians understanding of the atypical manifestations of pituitary stalk interruption syndrome and provide reference for the clinical diagnosis and treatment of the disease.
Case Presentation: The 30-year-old female patient exhibited disproportionate growth in height, with a significant increase from 140 cm at the age of 16 to 180 cm currently. Physical examination revealed widened bilateral eye fissures, underdeveloped secondary sexual characteristics, and absence of menstruation. The patient ‘s parents are cousins, belonging to consanguineous marriage. The patient ‘s hypoglycemia provocation test suggested the lack of growth hormone and cortisol. Gonadorelin provocation test suggested hypogonadism, and thyroid function test showed hypothyroidism. Pituitary MRI plain scan and enhancement suggested pituitary stalk interruption syndrome, and abdominal and urinary color Doppler ultrasound suggested no echo of uterus and bilateral appendages in the pelvic cavity. The karyotype of peripheral blood was 45, X[3] / 46, XX [117]. The patient was diagnosed with pituitary stalk interruption syndrome, congenital uterine and ovarian deficiency, bone overgrowth, hypothyroidism and secondary osteoporosis. During hospitalization, the symptoms were improved and discharged after hormone replacement therapy such as physiological dose of glucocorticoid, estradiol valerate tablets and levothyroxine sodium tablets. Now the patient is still in our hospital endocrinology outpatient follow-up, no special discomfort.
Conclusion: The patient had special clinical manifestations and was clinically confirmed as pituitary stalk interruption syndrome. The patient ‘s height continues to grow in the absence of growth hormone in the body, and its mechanism remains to be further studied.
Keywords: pituitary stalk interruption syndrome, excessive height growth, congenital uterine absence, congenital ovarian absence, chromosome abnormality
Introduction
Pituitary stalk interruption syndrome, also known as pituitary stalk transection syndrome, is a relatively rare disease, which was first reported by Fujisawa in 1987.1 Pituitary stalk interruption syndrome is a clinical syndrome of one or more pituitary hormone deficiency due to the fact that the pituitary stalk is slender, shorter or absent due to various reasons, and the ectopic posterior lobe of the pituitary gland leads to the fact that the hormones secreted by the hypothalamus cannot be transported to the posterior lobe of the pituitary gland through the pituitary stalk, nor can they act on the anterior lobe of the pituitary gland through the pituitary portal system.2 The incidence of pituitary stalk interruption syndrome is approximately 5 per million, the ratio of male to female is 2.3:1, and most of them have no family history.3,4 The diagnosis of pituitary stalk interruption syndrome mainly depends on MRI examination, which is characterized by the characteristic triad of thin or disappearance of pituitary stalk, ectopic posterior pituitary and poor development of anterior pituitary.5 The pathogenesis of pituitary stalk interruption syndrome is still unclear. Relevant studies suggest that it may be closely related to gene mutation, chromosome abnormality and perinatal injury (such as postpartum asphyxia history, foot presentation, hip presentation, etc).6–8 However, only 5% of patients in the study found that the etiology is associated with HESX1, LHX4, OTX2, SOX3 and PROKR2 gene mutations.9,10 For most patients, the etiology of their genetic factors is still unclear. The clinical manifestations of pituitary stalk interruption syndrome are mainly caused by a variety of hormone deficiencies, such as growth hormone deficiency leading to growth retardation and dwarfism, gonadotropin deficiency leading to gonadal and secondary sexual dysplasia, and thyroid stimulating hormone deficiency leading to hypothyroidism. Therefore, many patients often see a doctor due to short stature and short penis, and some of them show hyponatremia and hypoglycemia. Because of its complex and diverse clinical manifestations, it is easy to cause misdiagnosis and missed diagnosis. Therefore, this case report can improve people ‘s understanding of the atypical clinical manifestations of pituitary stalk interruption syndrome, and has certain clinical significance for the diagnosis and treatment of this disease.
Case Presentation
On May 6, 2023, a 30-year-old woman was admitted to our hospital. The patient self-reported that there was no obvious inducement for slender limbs and abnormal height growth rate 15 years ago. Before the age of 16, the height was about 140 cm, and then the height began to grow so far. It has been in a state of growth, and the annual growth rate is unknown. The patient ‘s limbs were slender, bilateral eye fissures widened, secondary sexual characteristics were not developed, and there was no history of menstruation. The patient was treated in the outpatient department of endocrinology of our hospital in 2016. At that time, the results of color Doppler ultrasound examination of uterus and appendages in other hospitals showed no echo of uterus and appendages in pelvic cavity, and the results of magnetic resonance imaging of pituitary saddle area suggested pituitary stalk interruption syndrome. The patient ‘s parents are cousins and belong to consanguineous marriage.
The patient was 180 cm in height and 72 kg in weight at admission. The patient was admitted to the hospital to improve the relevant test, and the test results are shown in Table 1. Pituitary MRI plain scan and enhanced scan at admission suggested pituitary stalk interruption syndrome (Figure 1). The bone age of the right wrist showed that the patient was not fully mature (Figure 2A). Pelvic magnetic resonance plain scan showed absence of uterus and bilateral ovaries (Figure 2B). Chest CT scan showed a few small nodules in both lungs, and bone mineral density examination showed low bone mass. Thyroid ultrasound showed diffuse thyroid lesions. Conventional electrocardiogram showed sinus rhythm, left chest lead QRS low voltage.
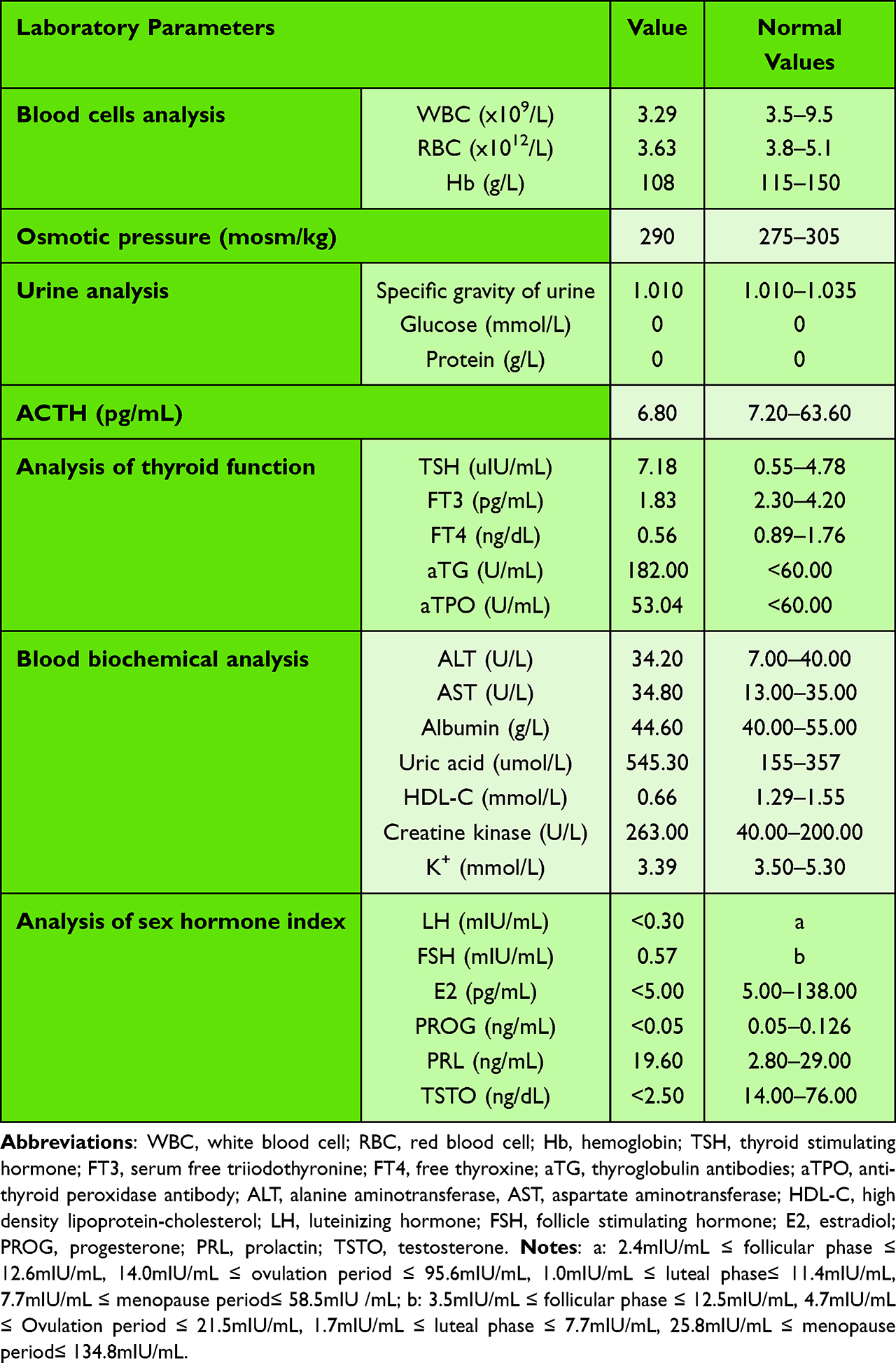 |
Table 1 Preliminary Summary of Laboratory Test Results |
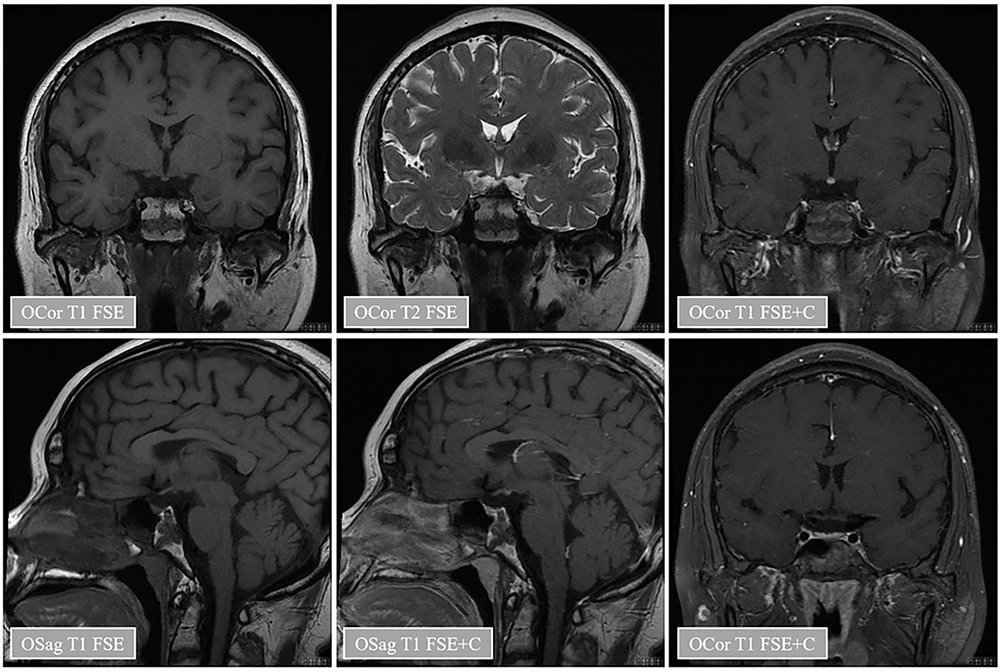 |
Figure 1 Pituitary magnetic resonance plain scan and enhanced images of patients. Notes: The results of pituitary magnetic resonance plain scan combined with enhancement in patients showed that the pituitary volume was reduced, the pituitary stalk was absent, and there was no T1 high signal shadow in the normal neurohypophysis, which seemed to move upward. The enhanced scan showed uniform enhancement of the pituitary gland, shallow sella, and no abnormal signal shadow near the sella. The epiphyseal plate can be seen on the slope, and the surrounding bone signal is not uniform, showing T1 slightly longer T2 signal similar to the epiphyseal plate, and there is no obvious enhancement. Long T2 signal shadow was seen in the left sphenoid sinus and parapharyngeal space, and there was no obvious enhancement. |
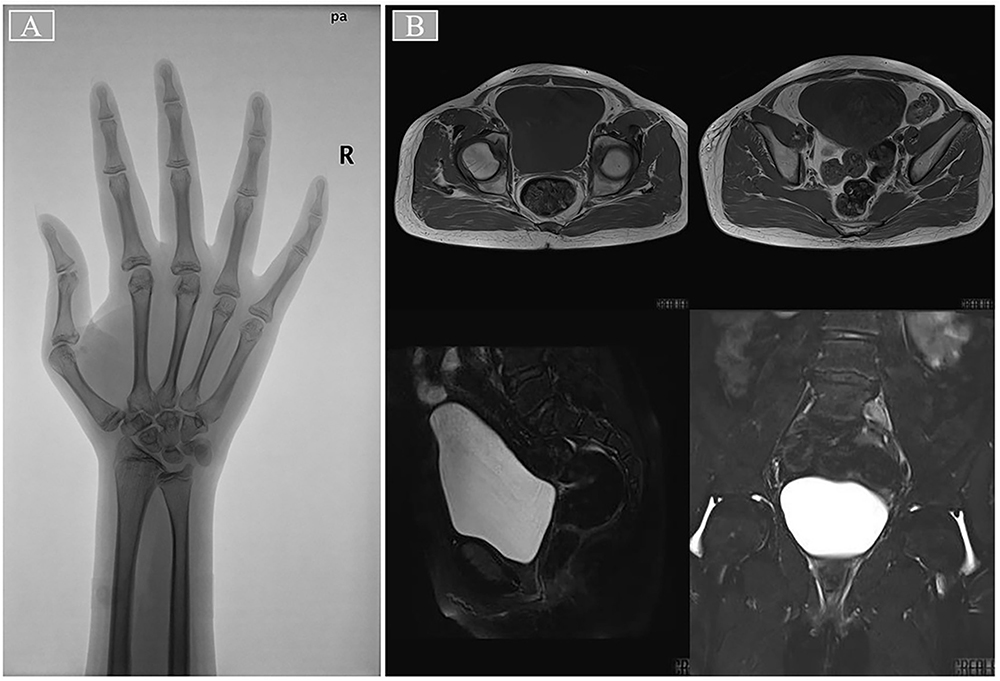 |
Figure 2 The patient ‘s right wrist bone age film and pelvic magnetic resonance image. Notes: (A) The epiphyseal line of the right distal ulna and distal radius was not closed, and the ulnar styloid process appeared. There were 8 ossification centers in the wrist, and the uncinate process of the hook bone was mature. The scaphoid bone appeared dense white line, and the pea bone appeared. The epiphysis of the metacarpal bone was not completely closed, and the medial sesamoid of the thumb appeared. (B) The uterus and bilateral ovaries were not shown, the pelvic wall structure was normal, and the fat space around the rectum was normal. |
In order to clarify the etiology of the patient, the patient underwent peripheral blood chromosome karyotype examination and analysis. The karyotypes of 120 cells were analyzed. The results showed that one was a homologous chimeric karyotype, and the cell line had only one X chromosome and three cells were found. Another cell line was a normal female karyotype, and 117 cells were found. The karyotype of the patient was 45, X[3] / 46, XX [117].
In order to further guide hormone replacement therapy, the patient underwent hypoglycemia provocation test and Gonadorelin test. Hypoglycemia provocation test showed growth hormone and cortisol deficiency in patients (Table 2). The patient ‘s Gonadorelin test suggested hypogonadism (Table 3).
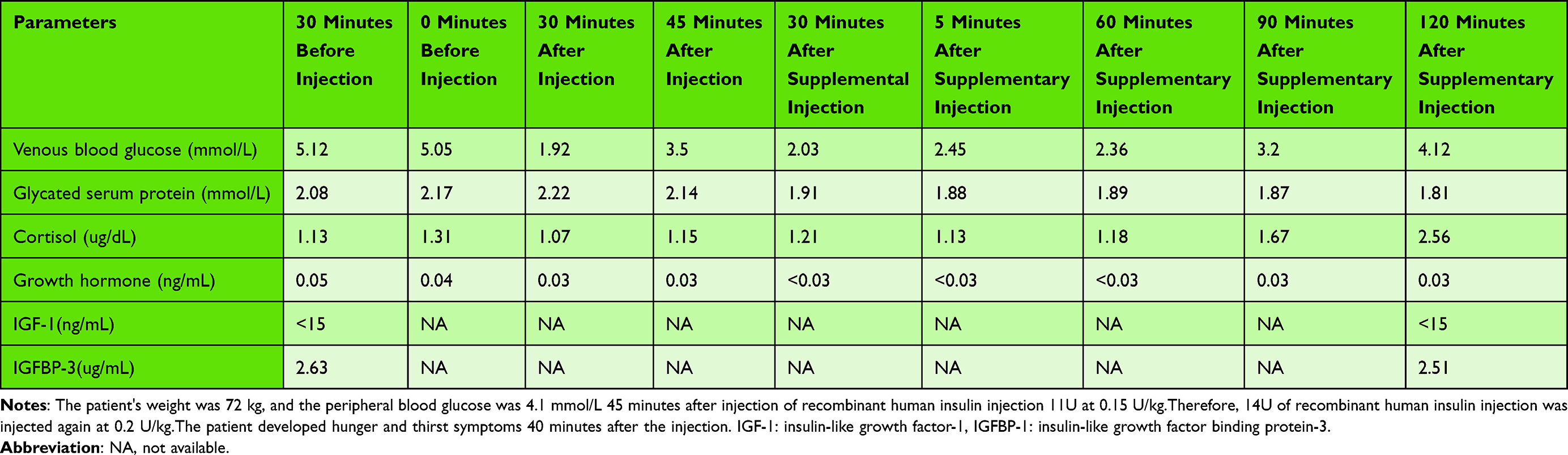 |
Table 2 Insulin-Induced Hypoglycemia Provocation Test |
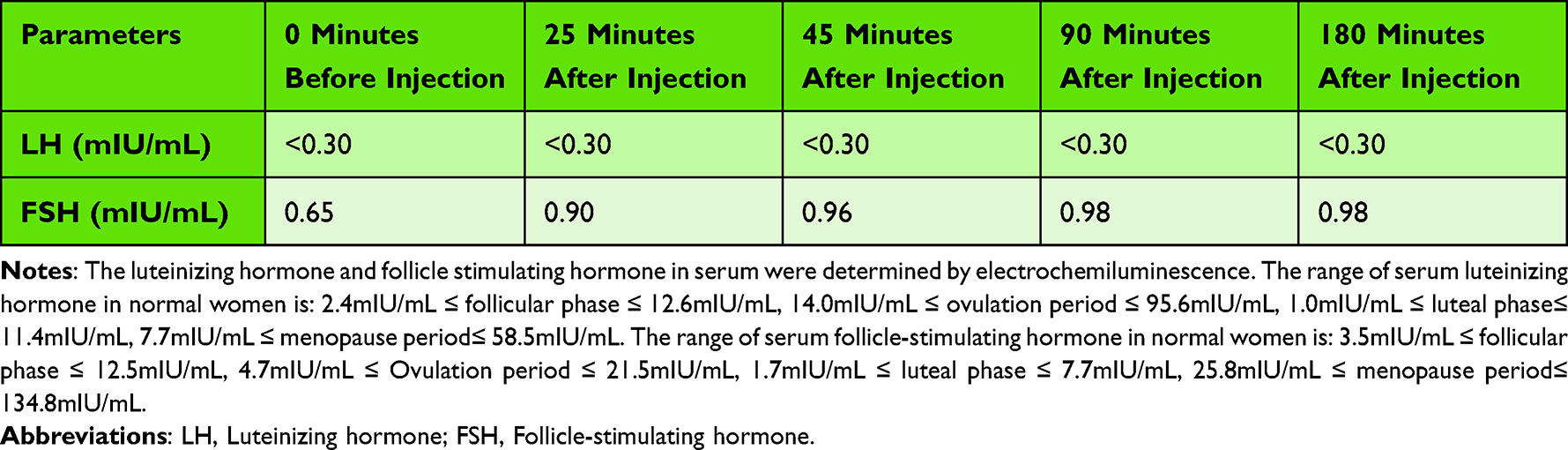 |
Table 3 Gonadorelin Stimulation Test (Gonadorelin 100ug) |
Discussion
Pituitary stalk interruption syndrome was first reported in 1987, with an extremely rare incidence of only 0.005 ‰, and it is more common in men. Most of its cases are sporadic, and only about 5% have familial inheritance.1,3 The pathogenesis of pituitary stalk interruption syndrome is that hormones secreted by the hypothalamus cannot be transported to the pituitary gland through the pituitary stalk, which in turn impairs the pituitary-gonadal axis, pituitary-adrenal cortex axis, pituitary-thyroid axis and growth hormone synthesis, and ultimately leads to the clinical manifestations of hypopituitarism in patients. Because the blood supply of the posterior pituitary is from the branch of the internal carotid artery and is less affected by the pituitary portal system, there is generally no hormone deficiency in the posterior pituitary.
The etiology of pituitary stalk interruption syndrome is still unclear, and it cannot be found during prenatal examination. Many studies suggest that gene mutations, chromosomal abnormalities and perinatal damage may be its pathogenesis. It has been reported that there is a correlation between chromosome abnormalities of 18p deletion, 2p25 duplication, 2q37 deletion and 17q21.31 microdeletion and pituitary stalk interruption syndrome.7,11,12 In this study, the patient ‘s chromosome was found to have two karyotypes. One is a homologous chimeric karyotype, which has only one X chromosome, and the other is a normal female karyotype. Chromosome abnormalities in patients with this type of disease have not been reported in the relevant literature, which is the first discovery. Studies have found that 48% of patients with pituitary stalk interruption syndrome have extrapituitary malformations, which may be caused by gene mutations. Among them, pituitary-specific transcription factor ancestral protein (PROP1) gene mutation is the most common genetic cause of pituitary dysfunction.13,14 The patient in this study refused to undergo genetic testing, so it could not be excluded that the patient was a disease caused by gene mutation. Many studies have found that fetuses with breech delivery, neonatal asphyxia and dystocia have a higher risk of pituitary stalk interruption syndrome, which may be related to pituitary stalk and pituitary injury caused by perinatal injury.15,16 The patients in this study belonged to full-term spontaneous delivery, and the fetal position was anterior occiput. The patients in this study denied perinatal injury, so perinatal events were less likely to cause the disease. The parents of the patient in this study are close relatives, and the patient has congenital absence of the uterus and ovaries. Because the patient ‘s parents were married to a close relative, and the patient ‘s chromosome examination confirmed the presence of an X chromosome deletion, so the patient may be due to genetic factors caused by congenital absence of the uterus and ovaries.
Because the patient ‘s age and the degree of damage to the pituitary stalk are different, the clinical manifestations of pituitary stalk interruption syndrome are complex and diverse. In the neonatal period, hypoglycemia, prolonged jaundice, micropenis malformation and cryptorchidism were more common. In older children and adults, it is mainly manifested as growth retardation, short stature and most of them have abnormal sexual development.17–19 Studies have found that there are 100% growth hormone deficiency, 97.2% gonadotropin deficiency, 88.2% adrenocorticotropic hormone deficiency and 70.3% thyroid stimulating hormone deficiency in patients with this disease.20 The growth hormone, gonadotropin, adrenocorticotropic hormone and thyroid stimulating hormone in the patients of this study are obviously insufficient, which is consistent with the literature report. In this study, the patient had a lack of growth hormone but no short stature. On the contrary, the patient ‘s epiphysis was not closed and its height showed a continuous abnormal growth trend, which was a rare feature of pituitary stalk interruption syndrome. A patient with pituitary stalk interruption syndrome was previously reported in the literature. The patient had recurrent hypoglycemia since birth and had abnormal height growth at the age of 13, which was similar to the patient in this study.21 Although growth hormone plays an important role in the growth and development of the human body, some studies have found that in patients with growth hormone deficiency, the levels of insulin and prolactin increase and play an antagonistic role in insulin-like growth factor binding protein-3, which relatively increases the concentration of free insulin-like growth factor-1 in the blood, and ultimately makes insulin-like growth factor-1 play a sufficient biological effect to promote human growth.22–24 The concentration of insulin-like growth factor binding protein-3 in the blood of the patient in this study was 2.63ug / mL (3.5–7.8ug/mL), which was significantly lower than the concentration range of normal people, but it was also found that the concentration of insulin-like growth factor in the blood of the patient was less than 15ng/mL (71–234ng/mL), and its value was also much lower than the normal reference range, which was inconsistent with the literature report. There are also studies that leptin can be used as a bone growth factor in patients with growth hormone deficiency, involved in promoting the growth and development of the body.25 The patients in this study did not carry out the relevant test of leptin index, whether it is related to the abnormal growth of height in patients with growth hormone deficiency remains to be discussed. In short, the mechanism of the continuous growth of height in patients with growth hormone deficiency remains to be further studied and explored.
The diagnosis of pituitary stalk interruption syndrome is mainly based on its clinical manifestations, laboratory tests and imaging examinations. In this study, the patient ‘s height continued to increase abnormally, and now the height is 180 cm. In clinical diagnosis, it is necessary to pay attention to the identification of pituitary giant, familial high body, Marfan syndrome and other diseases. The laboratory examination of pituitary stalk interruption syndrome is mainly based on the hypothalamic-pituitary-gonadal axis, hypothalamic-pituitary-growth hormone axis, hypothalamic-pituitary-thyroid axis and hypothalamic-pituitary-adrenal axis to reasonably evaluate the function of the anterior pituitary lobe. For patients with urinary specific gravity less than 1.005, water deprivation pressure test was performed to evaluate the function of the posterior pituitary lobe.26 The patients in this study were tested for pituitary and target gland function, and the laboratory test results were consistent with the diagnosis of pituitary stalk interruption syndrome. Pituitary stalk interruption syndrome has characteristic imaging findings. The characteristic MRI triads are absence or shortness or thinning of the pituitary stalk, dysplasia or thinning of the anterior pituitary lobe, and ectopic T1 high signal in the posterior pituitary lobe.10,27 In this study, the pituitary volume of the patient was reduced, the pituitary stalk was absent, and there was no T1 high signal shadow in the normal neurohypophysis, which seemed to move upward. The enhanced scan of pituitary showed uniform enhancement and shallow sella, which was consistent with the literature report. It is worth noting that when MRI examination finds pituitary stalk lesions, we should distinguish them from pituitary inflammatory changes, pituitary or sellar tumors, vacuolar sella and optic septal dysplasia.28 In conclusion, MRI has unique advantages and value in the classification and prediction of the severity of pituitary stalk interruption syndrome, which is of great significance for the diagnosis and treatment of this kind of disease.
Pituitary stalk interruption syndrome is currently unable to be cured by drugs and surgery. Its treatment is mainly the application of physiological doses of hormone replacement therapy, so it is very important to detect the lack of hormones as early as possible.29 The principle of hormone supplementation is to give priority to the supplement of cortisol hormones, and then supplement the remaining hormones that are lacking. Especially for patients with hypothyroidism, thyroid hormone should be supplemented after cortisol hormone supplementation to avoid adrenocortical crisis. In this study, the patients were found to have obvious deficiencies in growth hormone, gonadotropin, adrenocorticotropic hormone and thyroid stimulating hormone. Therefore, oral physiological dose of glucocorticoid, estradiol valerate tablets and levothyroxine sodium tablets were given for hormone replacement therapy. Studies have found that long-term use of hormone replacement can increase bone mineral density to normal levels in patients with pituitary stalk interruption syndrome.30,31 The bone mineral density of the patients in this study showed low bone mass. Considering that due to the lack of hormones, calcium treatment was not supplemented during hospitalization. In short, patients with pituitary stalk interruption syndrome should adhere to the use of hormone replacement therapy, regular follow-up, to avoid disease progression.
Conclusion
Patients with X chromosome abnormalities with excessive height growth and congenital absence of the uterus and ovaries are extremely rare, and there is no similar case reported in the relevant literature. Pituitary stalk interruption syndrome is a very rare congenital disease. Its etiology and pathogenesis are still unclear and need further study. We focus on this special case and review the relevant literature, hoping to provide a reference for the diagnosis and treatment of the disease, so as to achieve early detection and timely medication intervention to avoid affecting the growth and development of patients.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article. Further inquiries can be directed to the corresponding author.
Consent for Publication
Written informed consent for publication of their details was obtained from the patient. Based on the hospital there is no need for ethical clearance for the case report.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation of Jiangxi Province (20224ACB206010), Clinical Medical Research Center Programs of JiangXi Province (grant number 20221ZDG02011 and 2020BCG74001).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fujisawa I, Kikuchi K, Nishimura K, et al. Transection of the pituitary stalk: development of an ectopic posterior lobe assessed with MR imaging. Radiology. 1987;165(2):487–489. doi:10.1148/radiology.165.2.3659371
2. Vergier J, Castinetti F, Saveanu A, Girard N, Brue T, Reynaud R. Diagnosis of endocrine disease: pituitary stalk interruption syndrome: etiology and clinical manifestations. Eur J Endocrinol. 2019;181(5):R199–R209. doi:10.1530/EJE-19-0168
3. Wang CZ, Guo LL, Han BY, Su X, Guo QH, Mu YM. Pituitary stalk interruption syndrome: from clinical findings to pathogenesis. J Neuroendocrinol. 2017;29(1). doi:10.1111/jne.12451
4. Turton JP, Mehta A, Raza J, et al. Mutations within the transcription factor PROP1 are rare in a cohort of patients with sporadic combined pituitary hormone deficiency (CPHD). Clin Endocrinol. 2005;63(1):10–18. doi:10.1111/j.1365-2265.2005.02291.x
5. Wang Q, Hu Y, Li G, Sun X. Pituitary stalk interruption syndrome in 59 children: the value of MRI in assessment of pituitary functions. Eur J Pediatr. 2014;173(5):589–595. doi:10.1007/s00431-013-2214-1
6. Wu ZY, Li YL, Chang B. Pituitary stalk interruption syndrome and liver changes: from clinical features to mechanisms. World J Gastroenterol. 2020;26(44):6909–6922. doi:10.3748/wjg.v26.i44.6909
7. Yang A, Kim J, Cho SY, Lee JE, Kim HJ, Jin DK. A case of de novo 18p deletion syndrome with panhypopituitarism. Ann Pediatr Endocrinol Metab. 2019;24(1):60–63. doi:10.6065/apem.2019.24.1.60
8. Guo Q, Yang Y, Mu Y, et al. Pituitary stalk interruption syndrome in Chinese people: clinical characteristic analysis of 55 cases. PLoS One. 2013;8:1.
9. Karaca E, Buyukkaya R, Pehlivan D, et al. Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. 2015;100(1):E140–E147. doi:10.1210/jc.2014-1984
10. Reynaud R, Jayakody SA, Monnier C, et al. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab. 2012;97(6):E1068–E1073. doi:10.1210/jc.2011-3056
11. El Chehadeh-Djebbar S, Callier P, Masurel-Paulet A, et al. 17q21.31 microdeletion in a patient with pituitary stalk interruption syndrome. Eur J Med Genet. 2011;54(3):369–373. doi:10.1016/j.ejmg.2011.03.001
12. Vetro A, Pagani S, Silengo M, et al. Severe growth hormone deficiency and pituitary malformation in a patient with chromosome 2p25 duplication and 2q37 deletion. Mol Cytogenet. 2014;7(1):41. doi:10.1186/1755-8166-7-41
13. Correa FA, Nakaguma M, Madeira JLO, et al. Combined pituitary hormone deficiency caused by PROP1 mutations: update 20 years post-discovery. Arch Endocrinol Metab. 2019;63(2):167–174. doi:10.20945/2359-3997000000139
14. Correa-Silva SR, Kunii I, Mitne-Neto M, Moreira CM, Dias-da-Silva MR, Abucham J. Copy number variation in pituitary stalk interruption syndrome: a large case series of sporadic non-syndromic patients and literature review. J Neuroendocrinol. 2023;35(1):e13221. doi:10.1111/jne.13221
15. Sridhar S, Raja BR, Priyanka R, Natarajan S, Soundararajan S, Natarajan V. Clinico-radiological correlation of pituitary stalk interruption syndrome in children with growth hormone deficiency. Pituitary. 2023;26(5):622–628. doi:10.1007/s11102-023-01351-2
16. Diwaker C, Thadani P, Memon SS, et al. Pituitary stalk interruption syndrome: phenotype, predictors, and pathophysiology of perinatal events. Pituitary. 2022;25(4):645–652. doi:10.1007/s11102-022-01243-x
17. Bar C, Zadro C, Diene G, et al. Pituitary stalk interruption syndrome from infancy to adulthood: clinical, hormonal, and radiological assessment according to the initial presentation. PLoS One. 2015;10(11):e0142354. doi:10.1371/journal.pone.0142354
18. Pham LL, Lemaire P, Harroche A, Souberbielle JC, Brauner R. Pituitary stalk interruption syndrome in 53 postpubertal patients: factors influencing the heterogeneity of its presentation. PLoS One. 2013;8(1):e53189. doi:10.1371/journal.pone.0053189
19. Wang Q, Meng X, Sun Y, et al. Hypoglycemia and jaundice in newborns with pituitary stalk interruption syndrome. Medicine. 2021;100(19):e25843. doi:10.1097/MD.0000000000025843
20. Wang W, Wang S, Jiang Y, et al. Relationship between pituitary stalk (PS) visibility and the severity of hormone deficiencies: PS interruption syndrome revisited. Clin Endocrinol. 2015;83(3):369–376. doi:10.1111/cen.12788
21. Lee SS, Han AL, Ahn MB, et al. Growth without growth hormone in combined pituitary hormone deficiency caused by pituitary stalk interruption syndrome. Ann Pediatr Endocrinol Metab. 2017;22(1):55–59. doi:10.6065/apem.2017.22.1.55
22. Bucher H, Zapf J, Torresani T, Prader A, Froesch ER, Illig R. Insulin-like growth factors I and II, prolactin, and insulin in 19 growth hormone-deficient children with excessive, normal, or decreased longitudinal growth after operation for craniopharyngioma. N Engl J Med. 1983;309(19):1142–1146. doi:10.1056/NEJM198311103091902
23. Hathout EH, Baylink DJ, Mohan S. Normal growth despite GH, IGF-I and IGF-II deficiency. Growth Horm IGF Res. 1999;9(4):272–277. doi:10.1054/ghir.1999.0106
24. Hackney AC, Davis HC, Lane AR. Growth hormone-insulin-like growth factor axis, thyroid axis, prolactin, and exercise. Front Horm Res. 2016;47:1–11.
25. Holloway WR, Collier FM, Aitken CJ, et al. Leptin inhibits osteoclast generation. J Bone Miner Res. 2002;17(2):200–209. doi:10.1359/jbmr.2002.17.2.200
26. Liu W, Hou J, Liu X, Wang L, Li G. Causes and follow-up of central diabetes insipidus in children. Int J Endocrinol. 2019;2019:5303765. doi:10.1155/2019/5303765
27. Bozzola M, Mengarda F, Sartirana P, Tato L, Chaussain JL. Long-term follow-up evaluation of magnetic resonance imaging in the prognosis of permanent GH deficiency. Eur J Endocrinol. 2000;143(4):493–496. doi:10.1530/eje.0.1430493
28. Xu C, Zhang X, Dong L, Zhu B, Xin T. MRI features of growth hormone deficiency in children with short stature caused by pituitary lesions. Exp Ther Med. 2017;13(6):3474–3478. doi:10.3892/etm.2017.4377
29. Wang F, Han J, Wang Z, Shang X, Li G. Growth and adult height during human growth hormone treatment in Chinese children with multiple pituitary hormone deficiency caused by pituitary stalk interruption syndrome: a single centre study. J Clin Res Pediatr Endocrinol. 2020;12(1):71–78. doi:10.4274/jcrpe.galenos.2019.2019.0086
30. Zhu Y, Nie M, Wang X, et al. Bone mineral density in pituitary stalk interruption syndrome: the role of insulin-like growth factor-1 and testosterone at different skeletal sites. Endocr Pract. 2022;28(11):1118–1124. doi:10.1016/j.eprac.2022.07.011
31. Laitinen EM, Hero M, Vaaralahti K, Tommiska J, Raivio T. Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism. Int J Androl. 2012;35(4):534–540. doi:10.1111/j.1365-2605.2011.01237.x
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.