")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 6
Pharmacokinetics and pharmacodynamics of acetylsalicylic acid after intravenous and oral administration to healthy volunteers
Authors Nagelschmitz J, Blunck M, Kraetzschmar J, Ludwig M, Wensing G, Hohlfeld
Received 10 May 2013
Accepted for publication 16 July 2013
Published 19 March 2014 Volume 2014:6 Pages 51—59
DOI https://doi.org/10.2147/CPAA.S47895
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
J Nagelschmitz,1 M Blunck,1 J Kraetzschmar,1 M Ludwig,1 G Wensing,1 T Hohlfeld2
1Bayer HealthCare AG, Clinical Pharmacology, Wuppertal, Germany; 2Institut für Pharmakologie und Klinische Pharmakologie, Heinrich-Heine Universität Düsseldorf, Düsseldorf, Germany
Background: The pharmacology of single doses of acetylsalicylic acid (ASA) administered intravenously (250 or 500 mg) or orally (100, 300, or 500 mg) was evaluated in a randomized, placebo-controlled, crossover study.
Methods: Blood and urine samples were collected before and up to 24 hours after administration of ASA in 22 healthy volunteers. Pharmacokinetic parameters and measurements of platelet aggregation were determined using validated techniques.
Results: A comparison between administration routes showed that the geometric mean dose-corrected peak concentrations (Cmax/D) and the geometric mean dose-corrected area under the curve (AUC0–∞/D) were higher following intravenous administration of ASA 500 mg compared with oral administration (estimated ratios were 11.23 and 2.03, respectively). Complete inhibition of platelet aggregation was achieved within 5 minutes with both intravenous ASA doses, reflecting a rapid onset of inhibition that was not observed with oral dosing. At 5 minutes after administration, the mean reduction in arachidonic acid-induced thromboxane B2 synthesis ex vivo was 99.3% with ASA 250 mg intravenously and 99.7% with ASA 500 mg intravenously. In exploratory analyses, thromboxane B2 synthesis was significantly lower after intravenous versus oral ASA 500 mg (P<0.0001) at each observed time point up to the first hour after administration. Concentrations of 6-keto-prostaglandin1α at 5 and 20 minutes after dosing were also significantly lower with ASA 500 mg intravenously than with ASA 500 mg orally.
Conclusion: This study demonstrates that intravenous ASA provides more rapid and consistent platelet inhibition than oral ASA within the first hour after dosing.
Keywords: intravenous acetylsalicylic acid, oral acetylsalicylic acid, pharmacodynamics, pharmacokinetics, platelet aggregation, cyclooxygenase-1, thromboxane formation
Introduction
Low doses of orally administered acetylsalicylic acid (ASA) irreversibly inhibit platelet cyclooxygenase (COX)-1, thereby preventing the enzymatic formation of thromboxane A2, a potent activator of both platelet aggregation and vasoconstriction. Inhibition of COX-2-dependent prostaglandins occurs at much higher oral ASA doses than those used in cardiovascular prophylaxis. The ratio of IC50 values (the concentration producing 50% enzyme inhibition) for inhibition of COX-2/COX-1 is 166.1 As a result, the antiplatelet and vasodilatory effects of the COX-2-mediated metabolite, prostaglandin I2 (PGI2), should be less affected at low doses of ASA.
The pharmacokinetics of oral ASA have been assessed using high-performance liquid chromatography assays,2 and the pharmacodynamics have been measured using validated radioimmunoassays for markers of platelet aggregation such as serum thromboxane B2 (formed by nonenzymatic hydrolysis of thromboxane A2) and 6-keto-prostaglandin F1α (6-keto-PGF1α), which is a metabolite of PGI2.3–5 It has been shown that different formulations of oral ASA can have varying pharmacokinetic properties, depending on the clinical indication for which they were developed.6
In one study in healthy volunteers, the peak plasma concentration (Cmax) following administration of soluble ASA 600 mg/day was significantly higher than that of other oral formulations (13.82 μg/mL versus 5.51 μg/mL with plain ASA 650 mg/day orally) and the half-life (t1/2) was significantly shorter (16 minutes versus 32 minutes, respectively).7 However, this study did not include intravenous ASA, which has been shown to have a t1/2 of 2.8 minutes and 15 minutes in the initial distribution phase and the elimination phase, respectively.8 This indicates that intravenous ASA is rapidly converted to salicylic acid by hydrolysis and presystemic COX acetylation. Thus intravenous administration likely offers a means of achieving rapid inhibition of platelet function in an emergency setting. However, there are only a few studies that have compared intravenous ASA and oral ASA, and these were performed in animal models.5,9 Human data are even more scarce and have used doses (ASA 1 g/day intravenously or orally) that are not recommended as atherothrombotic prophylaxis.10
The aim of this study was to compare the pharmacokinetics and pharmacodynamics of intravenously administered ASA (D,L-lysine acetylsalicylate · glycine, 250 and 500 mg intravenously) with orally administered ASA (100, 300, and 500 mg orally) in healthy volunteers. The intravenous doses of ASA were chosen because previous studies have shown that ASA 250 mg intravenously reduces the rate of thromboembolic events without increasing intraoperative bleeding.11 This dose is also close to guideline recommendations for patients with acute coronary syndrome (ie, ASA 162–325 mg initially, reduced to 75–162 mg daily for indefinite use),11–13 while ASA 500 mg intravenously is widely used as a loading dose in patients with acute coronary syndrome. The 100 mg dose of ASA administered orally was also studied because this is close to the minimum oral dose approved for the treatment of acute coronary syndrome.
Materials and methods
Subjects
A total of 22 healthy volunteers (14 males and eight postmenopausal females) aged >40 years with a body mass index range of 20–37 kg/m2 were studied. The subjects abstained from ASA or any other medication that could interfere with platelet function for 2 weeks prior to the study. Informed consent was obtained from all subjects prior to inclusion.
Protocol
Each subject was randomized to receive a single dose of NaCl (placebo) solution (5 mL intravenously), ASA (D,L-lysine acetylsalicylate · glycine) 250 mg or 500 mg intravenously, or ASA 100, 300, or 500 mg plain tablets orally (supplied by Bayer HealthCare AG, Wuppertal, Germany) in the fasted state, according to the randomization list (Global Biometry, Bayer HealthCare AG). Randomization was carried out after the prestudy examination, with subjects being assigned to random numbers starting with 01 in ascending order as they were listed, and the individual sequence of the treatments was then determined by the randomization list; female subjects were randomized into a subgroup of eight random numbers.
For oral dosing, subjects were asked to chew the tablets and swallow them with approximately 240 mL of water. Intravenous ASA was dissolved in 5 mL of sterile water and injected into a forearm vein within one minute by the study physician. All medication was administered in the morning. Treatments were given in a crossover design, with washout periods of >3 weeks between each dose. The study protocol was approved by the ethics committee of the North-Rhine Medical Council, Düsseldorf, Germany, and the German Bundesinstitut für Arzneimittel und Medizinprodukte.
Venous blood samples (5 mL) were collected using an indwelling cannula (Sarstedt Group, Sarstedt, Germany) prior to ASA administration (baseline, 0) and 5, 30, 45, and 60 minutes and 1.5, 2, 3, 6, 8, and 24 hours after administration of oral ASA, and one, 3, 5, 10, 15, 20, 30, 40, and 60 minutes and 1.5, 2, 3, 6, 8, and 24 hours after administration of intravenous ASA. Blood for pharmacokinetic analysis was collected into lithium heparin monovettes containing approximately 50 mg of solid sodium fluoride. The plasma was separated immediately and stored frozen at −70°C or below. Assays were performed within 3 months of sampling. Blood samples for measurements of platelet aggregation were collected into citrated tubes and centrifuged for 10 minutes at 100× g and room temperature to obtain plasma rich in platelets. After removal of the plasma, the remaining blood was centrifuged for 10 minutes at 2,000× g to obtain plasma poor in platelets. Platelet aggregation was measured as described below within 2 hours of sample collection. Blood samples for measurement of serum thromboxane B2 and 6-keto-PGF1α were collected into Sarstedt tubes and shaken for 30 minutes at 37°C to allow for coagulation. Serum was separated by centrifugation for 10 minutes at 2,000× g and stored in 0.5 mL fractions at −20°C or −70°C.
Urine samples were collected over 0–8 and 8–24 hours after drug administration and 9 mL samples were stored frozen for analysis of ASA and salicylic acid.
Pharmacokinetics
Plasma concentrations of ASA and salicylic acid were measured by means of a validated and internally standardized high-performance liquid chromatography assay. The method employed liquid/liquid extraction using diethylether under acidic conditions, followed by reverse-phase chromatography on an Aqua C18® column (Phenomenex, Niederlassung, Germany). The analytes and the internal standard (2-acetylbenzoic acid) were measured by ultraviolet detection at 230 nm. The linear regression curves were calculated from peak height ratios versus spiked concentrations, with 1/x2 weighing. For each analyte, two independently prepared stock solutions in lithium heparin human plasma stabilized with sodium fluoride (3 mg/mL) were used for preparation of calibration standards and quality control samples. The lower limits of quantification for ASA and salicylic acid in plasma were 0.29 mg/L and 0.66 mg/L, respectively, and the precision ranged from 2.1% to 4.7% and 3.8% to 9.7%, respectively.
Urinary concentrations of salicylic acid were measured using a validated high-performance liquid chromatography/ultraviolet assay. The method consists of incubation of urine with concentrated hydrochloric acid, followed by reverse phase chromatographic separation of extracted analytes, and detection by ultraviolet at a wavelength of 310 nm. The internal standard was 4-acetylbenzoic acid. Calibration curves for analytes were constructed using linear least square regression of the ratio of peak heights analyte/internal standard, with a weighting of 1/x. Calibration standards and quality control samples were prepared in human urine. The lower limit of quantification was 2.03 mg/L and the precision ranged from 4.0% to 7.6%.
Pharmacokinetic parameters were calculated by model-independent (compartment-free) methods using WinNonlin® software, version 4.1.a. (Pharsight Corporation, Mountain View, CA, USA). The primary pharmacokinetic parameters were the area under the plasma concentration versus time curve from zero to infinity after study drug dosing divided by the dose (AUC0–∞/D), Cmax divided by dose (Cmax/D), and absolute bioavailability (F). Secondary plasma pharmacokinetic parameters were AUC0–∞, Cmax, time to reach maximum drug concentration in plasma (Tmax), and the half-life associated with the terminal slope (t½). Other parameters included clearance (CL) or apparent clearance (CL/f) for intravenous and oral application respectively, and amount excreted in urine (Ae).
Platelet aggregation and thromboxane B2 production
Measurements of platelet aggregation were carried out by light transmission aggregometry. Platelet-poor reference plasma was used to calibrate a 100% aggregation level, and the transmission of the platelet-rich sample to be analyzed was converted to percentage values of aggregation accordingly. Platelet aggregation was triggered by addition of 20 μL of 10 mM arachidonic acid to 180 μL of the platelet-rich plasma sample.
Serum thromboxane B2 and 6-keto-PGF1α concentrations were determined in serum by radioimmunoassay following in vitro conversion of thromboxane A2 and PGI2, respectively. The radioimmunoassays used polyclonal rabbit antisera previously generated and validated in the analytical laboratory. The lower limits of quantification were 100 pg/mL and 250 pg/mL for thromboxane B2 and 6-keto-PGF1α, respectively.
Adverse events
Adverse events were identified by: questioning at baseline, 20 minutes, 3 hours, and 24 hours after intravenous dosing; by questioning at baseline and at 1, 3, and 24 hours after oral dosing; and by spontaneous reporting throughout the study. Adverse events were recorded by the investigator and classified according to the Medical Dictionary for Regulatory Activities (MedDRA, version 9.1). The severity of adverse events was rated as mild (usually transient and generally not interfering with normal activities), moderate (sufficiently discomforting to interfere with normal activities), or severe (preventing normal activities). Treatment-emergent adverse events were defined according to the International Conference on Harmonization E9 guidelines as events that emerged during treatment, having been absent pretreatment, or that worsened relative to the pretreatment state.14
Statistical analysis
The statistical analysis was performed using SAS® software (version 8.2, Cary, NC, USA). The concentration-time courses of ASA and salicylic acid were calculated as geometric means with geometric coefficients of variation, except for Tmax, for which medians were reported. Log-transformed primary pharmacokinetic parameters were analyzed using analysis of variance including sequence, subject (sequence), period, and treatment effects. Point estimates (least squares means) and exploratory 90% confidence intervals for drug ratios were calculated by retransformation of the logarithmic data using the intraindividual standard deviation of the analysis of variance.
Descriptive statistics were used for analyzing platelet aggregation, lag time of aggregation, and thromboxane B2 and 6-keto-PGF1α concentrations. Because of the “all-or-none” nature of platelet aggregation, aggregation values below 10% (the threshold for discrimination of noise from true aggregation) were replaced by zero in the statistical analysis. Dose-effect relationships for intravenous and oral treatments were evaluated by correlation analysis (Spearman’s correlation coefficient, rho). Comparisons of the pharmacodynamic parameters between treatments were performed by nonparametric Wilcoxon tests.
All statistical analyses were performed without adjustment for multiple comparisons, and with unadjusted significance levels of α =0.05. No formal sample size calculation was performed due to the exploratory nature of the study. Equivalence was determined using the standard ranges (0.8–1.25 90% confidence interval ranges).
Results
Subjects
The study was carried out in 14 healthy male and eight healthy female volunteers with a mean age of 49 (range 40–58) years) and a mean body mass index of 26 ± 3.9 kg/m2. All 22 subjects received at least one dose of trial medication and were included in the safety analysis. A total of 21 subjects completed all six study periods and were included in the pharmacodynamic and pharmacokinetic analyses.
Pharmacokinetics of ASA according to route of administration
The derived pharmacokinetic parameters for single doses of intravenous and oral ASA are summarized in Table 1 and the estimates for salicylic acid are summarized in Table 2. There were differences in the pharmacokinetic variables between the formulations, as expected due to the differing modes of administration. The median Tmax was much shorter with intravenous ASA than with oral ASA (0.017 versus 0.500 hours, respectively), and both Cmax and AUC0–∞ were higher (Cmax, geometric mean 54.25 mg/L with ASA 500 mg intravenously versus 4.84 mg/L with ASA 500 mg orally; and AUC0–∞, geometric mean 10.31 mg · hour/L with ASA 500 mg intravenously versus 5.12 mg · hour/L with ASA 500 mg orally). In most cases, dose proportionality between intravenous and oral dosing was demonstrated for AUC/D and Cmax/D. The only exception was the comparison between ASA 100 mg orally and ASA 500 mg orally, where the upper limit of the 90% confidence interval for the ratio of AUC0–∞/D was higher than the prespecified limit of 1.25. The variability of the AUC0–∞ of ASA was considerably higher after oral ASA than after intravenous ASA.
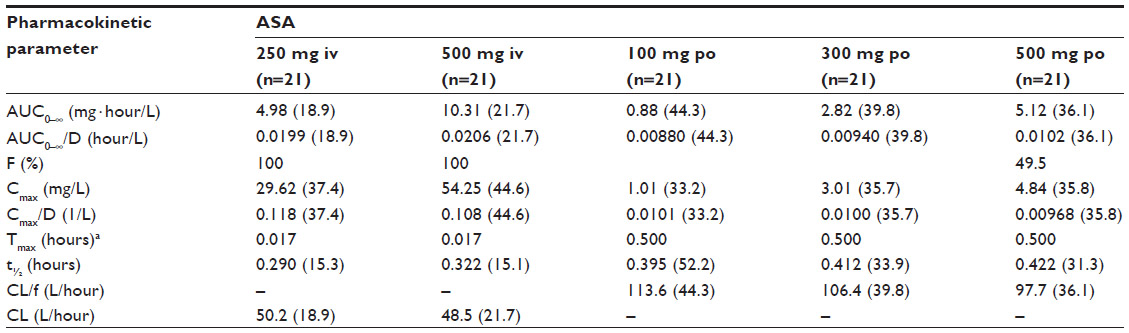 | Table 1 Geometric means (geometric coefficients of variation, %) and ranges for pharmacokinetic parameters of ASA following administration of ASA 250 mg and 500 mg intravenously and ASA 100 mg, 300 mg, and 500 mg orally |
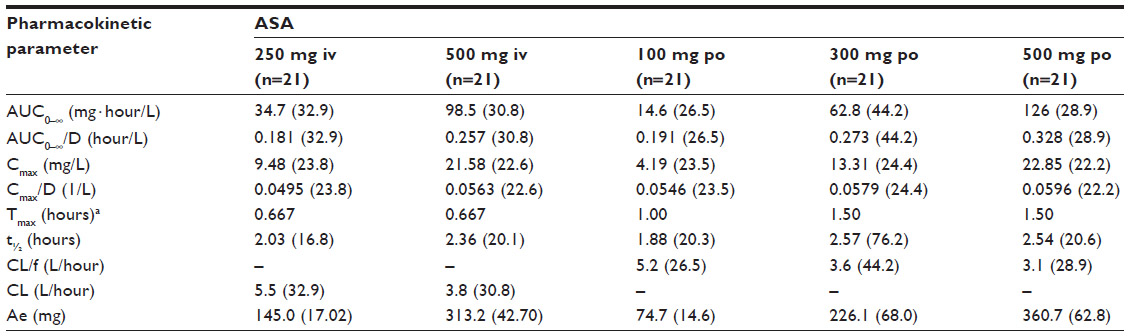 | Table 2 Geometric means (geometric coefficients of variation, %) and ranges for pharmacokinetic parameters of salicylic acid following administration of acetylsalicylic acid 250 mg and 500 mg intravenously and acetylsalicylic acid 100 mg, 300 mg, and 500 mg orally |
Platelet aggregation and thromboxane B2 production
The effect of intravenous and oral ASA on arachidonic acid-induced platelet aggregation is shown in Figure 1. Complete inhibition of platelet aggregation was achieved within 5 minutes with both intravenous ASA doses, whereas this was not found following any of the oral doses. The mean (standard deviation) platelet aggregation values at 5 minutes were 87.4% ± 10.7%, 75.6% ± 36.2%, and 66.1% ± 39.7% with ASA doses of 100, 300, and 500 mg orally, respectively. Complete inhibition was achieved by 20 minutes with ASA 300 mg and 500 mg orally, while the remaining mean platelet aggregation value was 31.6% ± 46.0% for ASA 100 mg orally at this time point. Complete inhibition with ASA 100 mg orally was seen after 40 minutes. The mean platelet aggregation with intravenous placebo was 92.0% ± 10.7% at 5 minutes and 89.0% ± 9.9% at 3 hours, reflecting normal platelet function in the study participants in the absence of any aspirin treatment.
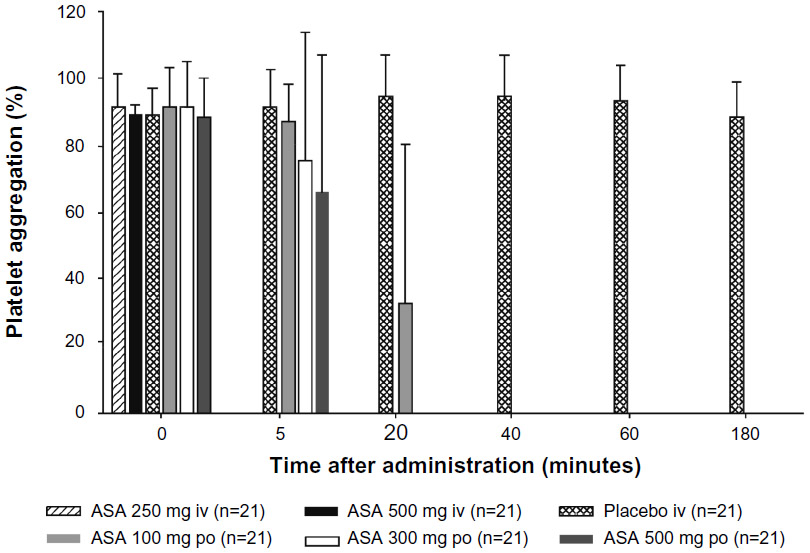 | Figure 1 The mean (standard deviation) inhibition of arachidonic acid-induced platelet aggregation measured after administration of a single dose of ASA administered intravenously (250 mg or 500 mg) or orally (100, 300, or 500 mg), or saline (placebo intravenously). |
The mean serum thromboxane B2 concentrations at 5 minutes after intravenous drug administration were 0.82 ± 0.81 ng/mL for ASA 250 mg intravenously and 0.69 ± 2.21 ng/mL for ASA 500 mg intravenously, which translated into a mean reduction of 99.3% ± 1.5% and 99.7% ± 0.9%, respectively (Figure 2). There was a significant difference between intravenous doses at this time point (P<0.0001, Wilcoxon test), and a negative correlation between intravenous doses and thromboxane B2 concentrations (Spearman’s rho =−0.57). The mean serum thromboxane B2 concentrations at 5 minutes after oral drug administration were 157.01 ± 92.44 ng/mL with ASA 100 mg orally, 108.33 ± 58.43 ng/mL with ASA 300 mg orally, and 83.74 ± 55.49 ng/mL with ASA 500 mg orally. There was a significant difference at 5 minutes after administration between ASA 100 mg orally and ASA 300 mg orally (P=0.0091, Wilcoxon test), and between ASA 100 mg orally and ASA 500 mg orally (P=0.0009, Wilcoxon test). At this time point, there was a negative correlation between oral doses and thromboxane B2 concentrations (Spearman’s rho =−0.37). ASA doses of 300 mg orally and 500 mg orally did not produce clinically relevant inhibition of platelet function (>95% reduction in thromboxane B2) within 20 minutes, and the 100 mg oral dose did not reach this threshold at any time point; the maximum inhibition of thromboxane B2 with ASA 100 mg orally was 89.4%. There were significant differences in thromboxane B2 concentrations between ASA 500 mg intravenously and ASA 500 mg orally at 5, 20, 40, and 60 minutes (all P<0.0001, Wilcoxon test).
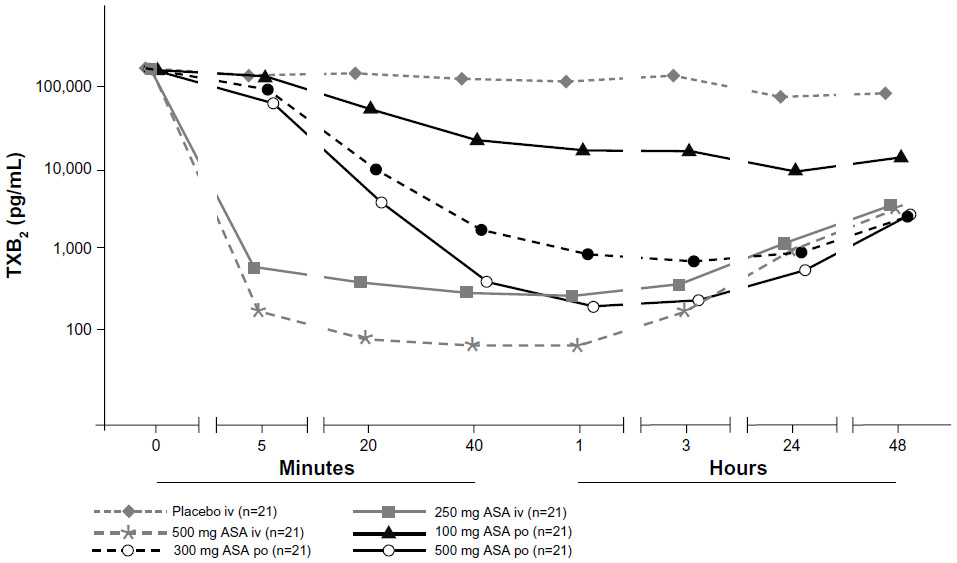 | Figure 2 Mean serum TXB2 concentrations after administration of a single dose of ASA administered either intravenously (250 mg or 500 mg) or orally (100, 300, or 500 mg), or saline (placebo intravenously). Data are presented using a semilogarithmic scale. |
The time course of the reductions in serum 6-keto-PGF1α concentrations following administration of ASA is shown in Figure 3. Concentrations declined rapidly within 5 minutes with both intravenous ASA doses compared with oral ASA doses. There was evidence of dose-related reductions in serum 6-keto-PGF1α with oral ASA. The serum 6-keto-PGF1α concentrations following administration of ASA 500 mg intravenously and ASA 500 mg orally differed significantly at 5 minutes (mean difference −0.061 ng/mL, P=0.0002) and 20 minutes (mean difference −0.015 ng/mL, P=0.0086), but not at 40 minutes (mean difference −0.004 ng/mL, P=0.7216).
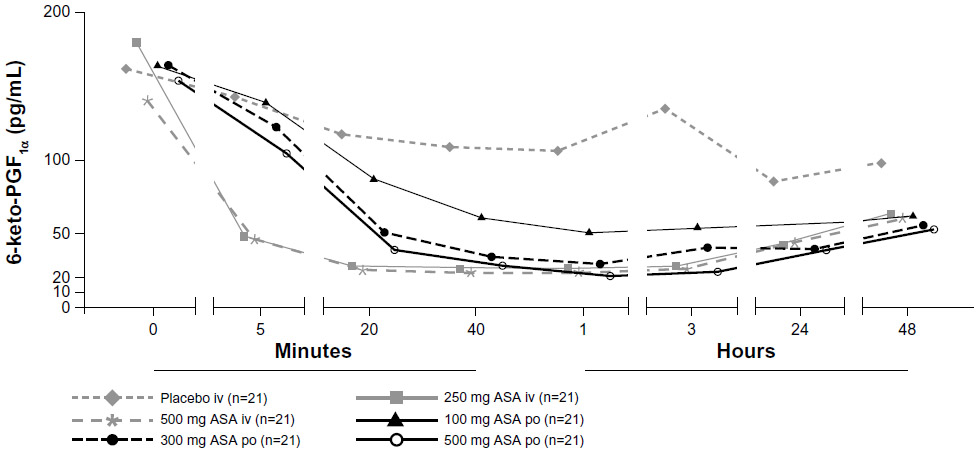 | Figure 3 Mean serum 6-keto-PGF1α concentrations after administration of a single dose of ASA administered either intravenously (250 mg or 500 mg) or orally (100, 300, or 500 mg) or saline (placebo). |
Adverse events
There were no serious adverse events in any subject following administration of either intravenous or oral ASA at any of the doses studied. In total, 15 subjects reported at least one treatment-emergent adverse event, all but two of which were mild in intensity; headache and migraine, both of moderate intensity, were each reported by one subject after administration of intravenous placebo. There were no differences between the five active treatments in the incidence and spectrum of treatment-emergent adverse events.
Discussion
Differences in onset of action have implications for determining the minimally effective dose that should be used in an intravenous preparation and also for the adverse event profile. The current study showed that although intravenous ASA had a faster onset of action than oral ASA, with a Tmax of 0.02 hours and 0.5 hours, respectively, the geometric mean Cmax/D was 11 times higher with intravenous than oral ASA 500 mg (estimated ratio 11.23) and the geometric mean AUC0–∞/D was twice as high (estimated ratio 2.03). For salicylic acid, there was a slight increase in AUC0–∞/D with dose (estimated ratio 1.41 for ASA 500 mg intravenously versus ASA 250 mg intravenously), which was expected. The current study also shows that there were no clinically relevant differences in treatment-emergent adverse events, but using a lower intravenous dose might have implications in this regard, particularly in relation to renal adverse events. However, this was beyond the scope of our study.
Clinically relevant inhibition of platelet function, defined as >95% reduction in serum thromboxane B2 concentrations from baseline, was achieved with intravenous ASA within 5 minutes of dosing. The mean inhibition of serum thromboxane B2 was 99.3% and 99.7% with intravenous ASA 250 mg and 500 mg, respectively. Complete inhibition of platelet aggregation was achieved within 5 minutes with both intravenous ASA doses. By contrast, complete inhibition was only achieved by 20 minutes with ASA 300 mg and 500 mg administered orally, and incomplete inhibition of platelet aggregation with ASA 100 mg orally was observed at this time (the mean residual aggregation value was only 31.6% ± 46.0%). The difference in serum thromboxane B2 reductions between ASA 500 mg intravenously and ASA 500 mg orally was statistically significant at each time point up to one hour after administration (P<0.0001).
The study also determined the effect of ASA on serum 6-keto-PGF1α, which is a surrogate measure of PGI2 formation. It likely reflects both vascular PGI2 synthesis in vivo as well as PGI2 synthesis by leukocytes during in vitro coagulation of the serum samples. While intravenous ASA and the higher doses of oral ASA reduced thromboxane B2 production by at least two orders of magnitude, the maximum inhibition of 6-keto-PGF1α production following oral or intravenous ASA was only ten-fold or less. This lower inhibition may reflect the fact that ASA has some selectivity for the COX-1 isoform,15 and that COX-1 largely predominates in platelets compared with COX-2, which is expressed in vascular and inflammatory cells. It has been shown that long-term suppression of PGI2 formation may augment injury-induced atherogenesis and platelet activation,16 although the clinical relevance of PGI2 inhibition by ASA in cardiovascular disease is unclear. The present results demonstrate that, at doses of 250–500 mg, administration of ASA either orally or intravenously has comparable effects on PGI2 synthesis, because there were few significant differences in 6-keto-PGF2α concentrations following intravenous or oral dosing, except at 5 and 20 minutes after dosing with 500 mg. A hypothetical adverse effect of intravenous ASA on vascular integrity resulting from higher exposure of PGI2-forming vascular cells to ASA is therefore unlikely. It is of note that in this study ASA orally was administered in the fasted state and that the tablets were chewed and swallowed with 240 mL of tap water at room temperature thereafter. These conditions may have facilitated absorption and led to an “optimized” effect on COX inhibition.
Studies in animals have also shown that biochemical selectivity is not apparent with intravenous ASA. An intravenous or oral dose of ASA 5 mg/kg administered to rats resulted in comparable inhibition of portal vein 6-keto-PGF1α and thromboxane B2, but formation of inferior vena cava 6-keto-PGF1α was spared.5 In humans, administration of intravenous or oral ASA 1 g was found to completely suppress serum thromboxane B2, but urinary thromboxane B2 and 6-keto-PGF1α were also suppressed by intravenous ASA.10,17 Oral ASA is a renal-sparing drug compared with other nonsteroidal anti-inflammatory agents; however, in the present study, only one renal/urinary disorder adverse event was reported with ASA 500 mg intravenously.
The present study characterizing the pharmacokinetics and pharmacodynamics of ASA in healthy subjects has some limitations. The need to perform platelet function assays immediately after blood sampling and the yes/no character of the response preclude the possibility of more accurately characterizing the time-dependency and dose-dependency of effects on platelet aggregation after intravenous and oral ASA administration. In addition, disease-related factors may affect the measurement of arachidonic acid-induced platelet activation in patients with acute coronary syndrome. By contrast, serum thromboxane B2 measurements showed more time-related, treatment-related, and dose-related effects of intravenous and oral ASA treatment. As described above, synthesis of thromboxane B2 was almost completely (>95%) inhibited within 5 minutes of treatment with intravenous ASA, and remained low with both intravenous doses. By contrast, with 300 mg and 500 mg of oral ASA, a comparable decrease in thromboxane B2 was not seen until about 40 minutes after administration, and the maximum inhibition of thromboxane synthesis after oral intake of 100 mg ASA was 89.4%. The delayed effect seen after oral treatment was associated with higher interindividual variability in the inhibition of thromboxane synthesis than with intravenous administration. Despite these limitations, the study shows that intravenous ASA provides rapid and consistent inhibition of platelet function in healthy volunteers. Based on its pharmacokinetic/pharmacodynamic profile, ASA 250 mg given intravenously may be useful where rapid inhibition of platelet function is required, such as in the acute management of patients with acute coronary syndrome, and the findings of this study justify comparison of the efficacy of intravenous and oral ASA in an emergency setting in patients according to treatment guidelines.18 Intravenous ASA could also be beneficial in acute-phase Kawasaki disease, where poor absorption of oral ASA is a problem.19
In summary, this study is novel in that it is one of the first to compare intravenous and oral ASA at doses routinely used in the clinical setting and thus generates clear and comparable pharmacokinetic and pharmacodynamic surrogate marker data. For intravenous ASA treatment specifically, it shows the immediate onset of action supported by pharmacokinetic data that describe the action in an acute cardiovascular clinical setting. Extensive research has been performed on the activity of oral ASA, establishing markers of platelet inhibition and aggregation that are widespread as assays today.20–22 The findings of such studies have also established the optimal doses of oral ASA for clinical use, and research groups are currently exploring the intricacies of its mode of action and most effective dosing strategies in more detail.23–25 A study in 41 patients with unstable angina was one of the first to explore the effects of intravenous ASA (60 mg on the first treatment day and 20 mg on successive days).26 The study showed that serum thromboxane B2 decreased from 160 ng/mL to <6 ng/mL (around 96% reduction). Although the reduction was sufficient for effective clinical inhibition, it was still not 100% complete. Overall, this study showed that ASA was not superior to coronary vasodilators (isosorbide dinitrate and oral diltiazem) and patients remained at risk of myocardial ischaemia. Consequently, these authors recommended early aggressive treatment in an acute setting. In the present study, the thromboxane B2 concentration was negligible at 5 minutes with ASA 250 mg intravenously (0.82 ng/mL). Our findings indicate that this dose of ASA could be more beneficial in an acute setting, and these results will be of interest to those devising guidelines for the most appropriate combination strategies in this environment.
Acknowledgments
The bioanalytical work for ASA and SA was conducted at the laboratory of Dr Ulf Buetehorn. The authors also gratefully acknowledge the technical assistance of Irmhild Rüter.
Disclosure
JN, MB, JK, ML, and GW are employees of Bayer HealthCare AG. TH declares no conflict of interest in this work. The study and analyses were funded by Bayer HealthCare AG.
References
Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A. 1993;90(24):11693–11697. | |
Cham BE, Ross-Lee L, Bochner F, Imhoff DM. Measurement and pharmacokinetics of acetylsalicylic acid by a novel high performance liquid chromatographic assay. Ther Drug Monit. 1980;2(4):365–372. | |
Smith WL. Molecular mechanisms of aspirin action. Drug News Perspect. 1991;4:362–366. | |
Prosdocimi M, Finesso M, Gorio A, et al. Coronary and systemic 6-ketoprostaglandin F1 alpha and thromboxane B2 during myocardial ischemia in dog. Am J Physiol. 1985;248(4 Pt 2):H493–H499. | |
Cerletti C, Gambino MC, Garattini S, de Gaetano G. Biochemical selectivity of oral versus intravenous aspirin in rats. Inhibition by oral aspirin of cyclooxygenase activity in platelets and presystemic but not systemic vessels. J Clin Invest. 1986;78(1):323–326. | |
FitzGerald GA, Lupinetti M, Charman SA, Charman WN. Presystemic acetylation of platelets by aspirin: reduction in rate of drug delivery to improve biochemical selectivity for thromboxane A2. J Pharmacol Exp Ther. 1991;259(3):1043–1049. | |
Muir N, Nichols JD, Clifford JM, Stillings MR, Hoare RC. The influence of dosage form on aspirin kinetics: implications for acute cardiovascular use. Curr Med Res Opin. 1997;13(10):547–553. | |
Rowland M, Rielgelma S. Pharmacokinetics of acetylsalicylic acid and salicylic acid after intravenous administration in man. J Pharm Sci. 1968;57(8):1313–1319. | |
Broome TA, Brown MP, Gronwall RR, Casey MF, Meritt KA. Pharmacokinetics and plasma concentrations of acetylsalicylic acid after intravenous, rectal, and intragastric administration to horses. Can J Vet Res. 2003;67(4):297–302. | |
FitzGerald GA, Pedersen AK, Patrono C. Analysis of prostacyclin and thromboxane biosynthesis in cardiovascular disease. Circulation. 1983;67(6):1174–1177. | |
Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non ST-Elevation Myocardial Infarction): developed in collaboration with the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. Circulation. 2007;116:e148–e304. | |
Gerrard JM. Measurements of 6-keto-prostaglandin F1 alpha and thromboxane B2 in bleeding time blood: relation to bleeding and vascular disorders? Can J Physiol Pharmacol. 1989;67(8):922–928. | |
Zimmermann N, Wenk A, Kim U, et al. Functional and biochemical evaluation of platelet aspirin resistance after coronary artery bypass surgery. Circulation. 2003;108(5):542–547. | |
European Medicines Agency. ICH Topic E9. Statistical principles for clinical trials. Available from: http://www.ema.europa.eu/pdfs/human/ich/036396en.pdf. Accessed June 6, 2011. | |
Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. | |
Cheng Y, Austin SC, Rocca B, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296(5567):539–541. | |
Cerletti C, Gambino MC, Bucchi F, Rajtar G, Riva E, de Goetano G. Comparison of the effects of oral and intravenous aspirin administration on platelet and peripheral vascular cyclo-oxygenase activity: studies in rats and in man. Agents Actions Suppl. 1986;20:239–248. | |
Hamm CW, Bassand J-P, Agewall S, et al. ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2011;32(23):2999–3054. | |
Ito S, Oka R, Tsuchida A, Yoshioka H. Disposition of single-dose intravenous and oral aspirin in children. Dev Pharmacol Ther. 1991; 17(3–4):180–186. | |
Patrono C, García Rodríguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353(22):2373–2383. | |
Patrignani P, Filabozzi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J Clin Invest. 1982;69(6):1366–1372. | |
FitzGerald GA, Oates JA, Hawiger J, et al. Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J Clin Invest. 1983;71(3):676–688. | |
Dillinger JG, Drissa A, Sideris G, et al. Biological efficacy of twice daily aspirin in type 2 diabetic patients with coronary artery disease. Am Heart J. 2012;164(4):600–606. | |
Rocca B, Santilli F, Pitocco D, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost. 2012;10(7):1220–1230. | |
Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewel of the drug target. Blood. 2012;119(15):3595–3603. | |
Vejar M, Hackett D, Brunelli C, et al. Comparison of low-dose aspirin and coronary vasodilators in acute unstable angina. Circulation. 1990;81 Suppl I:I4–I11. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.