")
Back to Journals » Journal of Inflammation Research » Volume 17
NAD-Dependent Protein Deacetylase Sirtuin-1 Mediated Mitophagy Regulates Early Brain Injury After Subarachnoid Hemorrhage
Received 27 November 2023
Accepted for publication 14 March 2024
Published 28 March 2024 Volume 2024:17 Pages 1971—1981
DOI https://doi.org/10.2147/JIR.S451922
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Subashchandrabose Chinnathambi
Gen Wang, Ning Lin
Department of Neurosurgery, The Affiliated Chuzhou Hospital of Anhui Medical University (The First People’s Hospital of Chuzhou), Chuzhou, Anhui Province, People’s Republic of China
Correspondence: Ning Lin, Email [email protected]
Background: This study focuses on the role of SIRT1 in neuroinflammation caused by early brain injury (EBI) after subarachnoid hemorrhage (SAH), and explores its mechanism in mitophagy after SAH.
Methods: C57BL/6J mice and primary microglia SAH in vivo and in vitro models were constructed to explore the expression level of SIRT1 in neuroinflammation after SAH. Subsequently, the brain edema content, blood–brain barrier (BBB) damage and neurological function scores of the mice were observed after using the SIRT1 inhibitor EX-527. q-PCR and Western blot were used to detect relevant genes and proteins, and enzyme-linked immunosorbent assay (ELISA) was used to detect the levels of IL-6, IL-1β, and TNF-α inflammatory factors. Immunofluorescence staining was used to observe the positive level of SIRT1 and the degree of mitochondria-lysosome fusion, and transmission electron microscopy was used to observe mitochondrial damage and autophagosome levels.
Results: In in vivo and in vitro experiments, we found that SIRT1 expression increased after SAH, and neurological deficits, brain edema, and blood–brain barrier damage after SAH were aggravated. Inhibiting SIRT1 further aggravates the aforementioned damage. In addition, EX-527 can also inhibit the level of mitophagy and aggravate neuroinflammation after SAH.
Conclusion: Our results indicated that SIRT1 promotes mitophagy and alleviates neuroinflammation after SAH.
Keywords: subarachnoid hemorrhage, SIRT1, mitophagy, early brain injury
Introduction
Subarachnoid hemorrhage (SAH) is a severe hemorrhagic cerebrovascular disease, which mainly caused by the rupture of aneurysm in clinically and is one of the most devastating cerebrovascular diseases. One-third of the patients usually die at the time, and the other one-third develop permanent disability, which seriously affects the patient’s quality of life.1–3 Studies4,5 have found that early brain injury (EBI) is one of the pivotal factors causing this outcome after SAH. EBI involves a variety of physiological disorders, including neuroinflammatory injury, blood–brain barrier disruption, nerve cell apoptosis, etc. and neuroinflammation plays a vital role in EBI.6,7 Therefore, a deeper understanding of the neuroinflammation in EBI may provide new preventive strategies for SAH patients. Evidences have shown that microglia participate in the immune response of the central nervous system and participate in the regulation of inflammatory factor levels through cell phenotype conversion after SAH. However, the mechanisms that lead to microglial inflammation after SAH have not yet been clearly understood. Therefore, targeting the microglial inflammatory response may help search for therapeutic targets for SAH.
NAD-dependent protein deacetylase sirtuin-1 (SIRT1) is one of the most well-studied members of the Sirtuin family. Which mainly distributes in the nucleus and cytoplasm, and participates in the regulation of DNA damage repair, such as oxidative stress, immune response, mitochondrial biogenesis and apoptosis/autophagy by exercising its NAD+ dependent deacetylation activity.8–10 Previous studies have shown that SIRT1 can not only be expressed in microglia, but also participate in neuroprotective effects by regulating microglia function in SAH. Chen et al11 used 2,3,5,6-tetramethylpyrazine to activate SIRT1 in BV2 cell, which alleviated neuroinflammation after SAH by inhibiting M1 microglial polarization; Arc could regulate brain injury and neuroinflammation after SAH through SIRT1 in a SAH mouse;12 in addition, SIRT1 could promote M2 microglia polarization by reducing ROS-mediated NLRP3 inflammasome signaling and alleviate neuroinflammatory damage after SAH.13 What’s more, SIRT1 can also play a role in the central nervous system by participating in regulating mitophagy. Yao et al14 induced autophagic cell death/mitophagy through activators of SIRT1 as a potential therapeutic strategy for glioblastoma. TUG1 knockdown promotes SIRT1-induced mitophagy by inhibiting FBXW7-mediated degradation of SIRT1, thereby alleviating neuronal apoptosis induced by cerebral ischemia-reperfusion injury.15 Of course, there are also studies reported the role of mitophagy in SAH. T3 promotes mitophagy through the PINK1-parkin pathway and alleviates EBI after SAH.16 Zhang et al17 reported that NL-1 promotes PINK1-Parkin-mediated mitophagy through MitoNEET inhibition in SAH and reduces EBI after SAH in rats. Liu et al18 demonstrated that inhibiting USP30 promotes mitophagy by regulating ubiquitination of MFN2 by Parkin to attenuate EBI after SAH. However, whether SIRT1 plays a role through mitophagy in SAH has not been reported. Therefore, this study aims to explore the role of SIRT1 in mitophagy after SAH, and provide a reliable target for the treatment of EBI.
Materials and Methods
Animal
The hospital’s Animal Ethical Review Committee in the affiliated Chuzhou hospital of Anhui Medical University approved all procedures. This study was conducted in accordance with the National Institutes of Health Guide for the Care of Laboratory Animals. Health adult male C57BL/6J mice (8–10 weeks, 18‒22 g) were purchased from the Animal Center of Zhejiang University. Mice were placed in a laboratory environment with a temperature of 22‒24°C and a humidity of 55‒60%, with free access to water and sleep.
SAH Model
The intravascular filament perforation SAH model was adopted in mice.19 Briefly, mice were anesthetized with 3% inhaled isoflurane, and then, the left internal carotid artery (ICA) was dissected from adjacent tissues in the neck. Use monofilament nylon (diameter: 0.18 ± 0.01 mm, Beijing, China) to puncture the bifurcation of the left middle cerebral artery and anterior cerebral artery through the ICA, then withdraw the nylon guidewire and suture the tissue and skin layer by layer. The sham group underwent the same surgical procedure except that the sutures did not penetrate the artery. During operation, use a thermostatic pad to maintain body temperature at 37°C ± 0.5°C. They were transferred to the animal room and continued to be raised after recovering from anesthesia. During this period, jelly was given to provide energy.
Experiment Study
A total of 155 mice were used for the following study, except for 25 mice that died and were excluded from the 180 mice. Twenty-five mice in experiment 1 were randomly divided into five groups (sham, 6 h, 12 h, 24 h, 72 h, n = 5). The level of SIRT1 was detected by q-PCR and Western blot analysis, and the positive of SIRT1 was detected by immunofluorescence. Another 30 mice in experiment 2 were randomly divided into six groups (sham, SAH, SAH + 2.5 μM, SAH + 5 μM, SAH + 10 μM, and SAH + 20 μM, n = 5) was used to explore the optimal concentration of EX-527. Forty mice in experiment 3 were randomly divided into four groups (sham, SAH, SAH + vehicle, and SAH + EX-527, n = 10); each group of mice was used to evaluate brain edema, BBB injury, and neurological deficit. Another 60 mice in experiment 4 were randomly divided into four groups (sham, SAH, SAH + vehicle, and SAH + EX-527, n = 15), and the Western blot analysis was used to detect mitophagy relative proteins.
Intraventricular Injection
Intracerebroventricular administration was performed as previously described.20 Briefly, mice were anesthetized with 3% inhaled isoflurane and then placed in a stereotaxic frame. A 10 μL microinjector (Shanghai, China) was inserted into the right cerebral ventricle at the following coordinates: 0.4 mm posterior and 1.0 mm lateral to bregma, and 3.0 mm below the dural layer. Ten μg of EX-527 (Selleck, USA) was injected into the ventricle through the lateral ventricle, and its dosage concentration was based on the previous dosage.21,22
Brain Edema Measurement
Brain edema was measured 24 hours after SAH according to previous methods.23 Mouse brains were harvested and weighed immediately to obtain wet weight and dried in a 105°C oven for 24 h before weighing again to obtain dry weight. The percentage of moisture content is calculated according to the following formula: ([wet weight – dry weight]/wet weight) × 100%.
Behavior Analysis
Neurological deficits were assessed using Behavior scores.24 Three behavioral activity checks are used in the scoring method, including appetite (0–2), activity (0–2), and neurological deficits (0–2), where higher scores indicate worse functioning.
Elisa
The mice were anesthetized and brain tissue was obtained at 24 h after SAH. The brain tissue was homogenized in 0.9% saline and centrifuged at 12,000 × g for 10 min at 4°C. Protein concentrations of IL-1β, IL-6 and TNF-α in brain tissue homogenates or cell culture supernatants were measured using ELISA kits (ab255730, ab22503 and ab208348) according to the manufacturer’s instructions.
Immunofluorescence Staining and Laser Confocal Imaging
C57/B6J mice were euthanized at appropriate times according to the experimental design. The brain tissue was obtained after intraventricular perfusion of phosphate-buffered saline (PBS) and 4% paraformaldehyde (PFA). The tissues were fixed in 4% PFA and dehydrated in a gradient of 15% and 30% sucrose, and then embedded and frozen in Tissue-Tek O.C.T. A Leica CM1900 cryostat was used to cut 20 μm coronal brain sections 1.0 mm posterior to bregma. The sections were permeabilized, blocked, and incubated with primary antibodies overnight at 4°C. The following antibodies were used: rabbit anti-SIRT1 (1:200, MCE, USA) and goat anti-Iba1 (1: 200, Abcam, USA). After washing three times in PBS, tissue samples were incubated for 2 h with the following secondary antibodies: Alexa Fluor 488 mouse anti-rabbit IgG or Alexa Fluor 594 goat anti-mouse IgG (Invitrogen, USA), followed by antifade reagent containing DAPI (P0131, Beyotime, China) were covered on the brain slices and fixed with coverslips for fluorescence imaging. Mito Tracker@Red FM (M22425, Invitrogen, USA) was used to identify mitochondria, and Lyso-Tracker Red DND-99 (MX4317, Invitrogen, USA) was used to identify lysosomes. All images were taken using an Olympus microscope (APX100).
Cell Counting Kit-8 Assay
The CCK-8 experiment was detected with a CCK-8 kit (C0038, Beyotime, China) and performed according to the instructions. Usually, 100 μL of 5000 cells was added to each well for cytotoxicity experiments and 2 μL of drug stimulation was given. Add 10 μL of CCK-8 solution to each well. The wells in which corresponding amounts of cell culture medium and CCK-8 solution without cells were added were used as blank controls. Continue incubation in the cell culture incubator at 37° for 1 h and measure the absorbance at 450 nm with a microplate reader.
Nissl Staining
After the coronal sections were dehydrated with 4% PFA and xylene, the sections were placed in Nissl staining solution and dip-stained in an incubator at 56°C for 1 h, then differentiate with Nissl differentiation medium for 1~3 min and observe under a microscope until the background is close to colorless. Dehydrate quickly with absolute ethanol, become transparent with conventional xylene or dewaxed transparent liquid, and seal with neutral gum. Then the staining was observed with a fluorescence microscope and the normal neurons with round and pale stained nuclei were counted.
Cell Culture and in vitro Models
Primary microglia were cultured according to our previous methods.25 Briefly, cortical brain tissue from D1 old mice was collected and incubated with 0.25% trypsin for 10 min at 37°C. After terminating digestion with 10% FBS/DMEM, filter the supernatant of the tissue mixture using a 75 μm cell strainer. Then the cells were resuspended in 10% FBS/DMEM and evenly seeded in T75 cell culture dishes (Corning, USA). The medium was changed on days 3 and 7, respectively, and the mature microglia were harvested on day 10. Oxyhemoglobin (OxyHb) was produced using fetal bovine hemoglobin (Sigma-Aldrich, USA) according to the manufacturer’s instructions. To simulate SAH in vitro, primary microglia were incubated with OxyHb at a concentration of 25 μM according to previous report.26 For in vitro SIRT1 inhibition experiments, microglia were treated with EX-527 (10 μM) at the same time as OxyHb incubation.
Q-Rt-Pcr
Total RNA was extracted from primary microglia and ipsilateral cortical tissue using TRIzol Universal reagent (Tiangen, DP424, China). 500ng of total RNA was reverse transcribed into cDNA using the Quantscript RT Kit (Tiangen, KR103, China). We then performed real-time PCR using FastReal qPCR PreMix (Tiangen, FP217, China) under the following conditions: pre-denaturation at 95°C for 2 min, then pre-denaturation at 95°C for 5 sec, 60°C 15 sec, 40 cycles in total. The primers used in this study are listed in Table 1. GAPDH was used as an internal control to normalize the mRNA. GAPDH (2- ΔΔCt) was utilized as the reference gene for determining relative gene expressions. Results are based on at least three independent experiments.
 |
Table 1 Primer Sequences for RT-qPCR |
Western Blotting
Total protein was prepared using RIPA lysis buffer. Proteins were loaded and separated on a 7.5–12.5% SDS-PAGE, followed by electrophoresis and transfer to polyvinylidene difluoride membrane PVDF (Millipore, USA). The membrane was blocked with 5% skim milk for 1 h and washed three times with 1 x TBST, then incubated with the following primary antibodies for 4°C overnight. SIRT1 (Rabbit, 1:1000, 13,161-1-AP, Proteintech), P62 (Rabbit, 1:5000, HA721171, HuaAn), Pink1 (Rabbit, 1:1000, 23,274-1-AP, Proteintech), Parkin (Rabbit, 1:2000, 14,060-1-AP, Proteintech), Tom20 (Rabbit, 1:5000, 11,802-1-AP, Proteintech), LC3 (Rabbit,1:1000,12,741, CST), GAPDH (Mouse, 1:50,000, 60,004-1-Ig, Proteintech), Acetyl-FOXO1A (Rabbit, 1:1000, AF2305, Affinity Biosciences). After washing three times with TBST, the corresponding secondary antibody was added and incubated for 1 h. Proteins were detected by Tanon 4800 Multi (Tanon, Shanghai), and quantitative analysis was performed using Image J software (NIH, USA).
Transmission Electron Microscope (TEM)
For microglia, we removed the upper culture medium and add pre-chilled fixative. After fixation for 15 min, gently scrape off the cells, then centrifuge at 2000 × g for 10 min to collect the cell pellet. Then, the cells were spread flat in 1% osmium tetroxide and dehydrated in increasing concentrations of ethanol. They were treated with propylene oxide and soaked in resin overnight at room temperature. After embedding, cure at 60°C for 48 h. we re-embedded the region of interest into the tip of the resin block and cut it to 70 nm using an ultramicrotome. Ultrathin sections were collected and examined under a Hitachi TEM at 80 kV.
Statistical Analysis
Statistical analysis was performed using SPSS 22.0 software (IBM, USA). All data were first tested for normal distribution, those conform to the normal distribution were expressed as mean and standard deviation (mean ± SD), and t test was used for two group comparisons. P < 0.05 was considered statistically significant.
Results
Expression of SIRT1 in Cortex After SAH
To determine whether SIRT1 is activated after SAH, we detected the expression of SIRT1 by q-PCR and Western blot respectively. As shown in Figure 1A, SIRT1 expression increased at 6 h and reached a peak at 24 h after SAH, and then decreased at 72 h. Western blot showed that SIRT1 protein expression in cortical neurons increased significantly after SAH, which consistent with the expression of mRNA level change (Figure 1B–C). Double immunofluorescence labeling of SIRT1 and Iba1 showed that the number of SIRT1-positive cells was increased in SAH mice (24 h) compared with Sham group (Figure 1D).
 |
Figure 1 Temporal expression of SIRT1 in the cortex after SAH. (A–C), q-PCR and Western blot showed SIRT1 expression in sham at 6h, 12h, 24h, and 72h after SAH. (D), Co-staining of SIRT1 (green) and Iba1 (red) demonstrated that SIRT1 was upregulated in microglia 24 h after SAH. **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bar: 50 μm. |
EX-527 Treatment Aggravates Neurological Deficits and BBB Damage After SAH
To evaluate the role of SIRT1 in EBI after SAH, the SIRT1 inhibitor EX-527 was injected intracerebroventricularly. We set drug concentrations with different concentration gradients (2.5μM, 5.0μM, 10μM, 20μM) to treat mice for 24 h according to the previous studies and the instruction. We found that the cell viability decreased significantly at 10 μM (P < 0.01), while the concentration increased to 20 μM, the cell viability decreased insignificantly (P > 0.05), as shown in Figure 2A. Therefore, we selected EX-527 at a dose of 10 μM for the follow-up experiment.
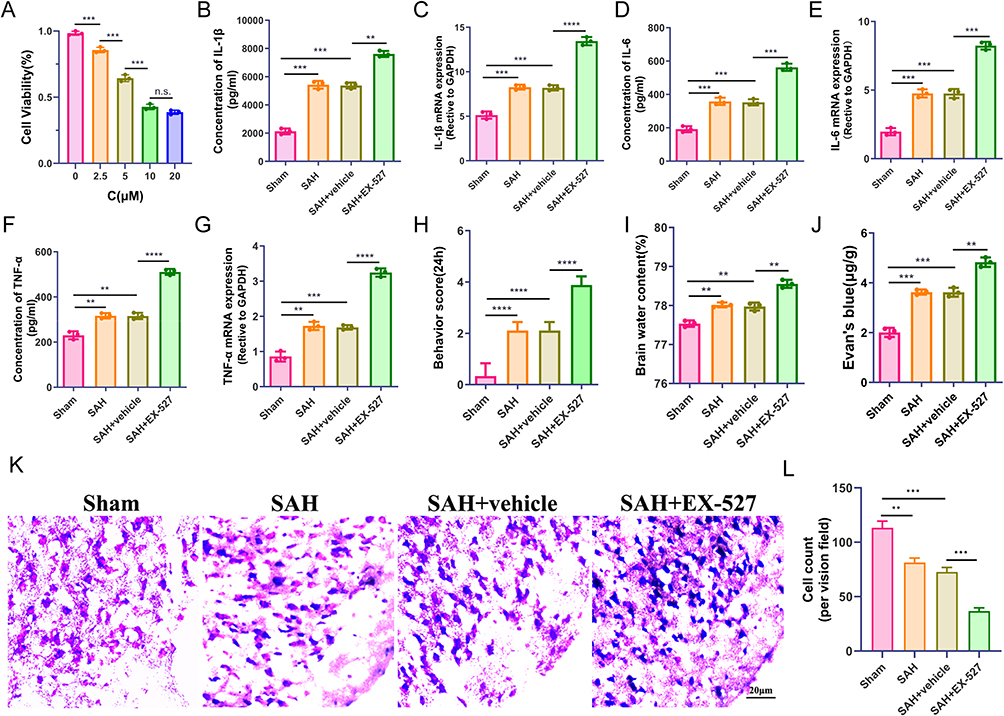 |
Figure 2 SIRT1 inhibition aggravates inflammation damage and neurological functions and attenuates brain edema after SAH. (A) CCK-8 assay to detect the optimal concentration of the SIRT1 inhibitor of EX-527. (B–G) the levels of IL-1β, IL-6 and TNF-α in the cortex and primary microglia were analyzed by q-PCR and ELISA. (H) Neurological deficits, (I) brain water content, and (J) Evan’s blue in Sham, SAH, SAH + vehicle, and SAH + EX-527 groups at 24h after SAH. (K–L) Nissl staining in the cortex with quantification. **P < 0.01, ***P < 0.001, ****P < 0.0001, Scale bar: 20 μm. Abbreviations: n.s, no significance. |
In order to explore the effect of EX-527 on neuroinflammation after SAH, we used q-PCR and ELISA to detect the levels of inflammatory factors, such as IL-1β, IL-6, and TNF-α, respectively. Compared with the Sham group, the levels of inflammatory factors in the SAH group and the vehicle group were moderate higher, when EX-527 was administered, their levels were significantly increased, which aggravated the neuroinflammatory damage after SAH. The difference was statistically significant (Figure 2B–G, P < 0.001). Subsequently, we evaluated the functional status of the mice at 24h after SAH. Compared with the sham group, Significant neurological dysfunction and Evan’s blue were observed in the SAH group and the vehicle group, neurological dysfunction and Evan’s blue were further exacerbated when EX-527 was administered (Figure 2H and J, P < 0.01). On the contrary, the brain water content in the ipsilateral hemisphere of mice in SAH group and vehicle group increased slightly, while the brain water content was further increased after treatment with EX-527 (Figure 2I, P < 0.01). Then, we tested whether EX-527 could rescue neuronal degeneration by Nissl staining. The results showed that the number of Nissl-stained neurons was significantly reduced after SAH due to SIRT1 inhibition (Figure 2K–L, P < 0.01).
SIRT1 Activation Triggers Mitophagy After SAH
Sirt1 plays an important role in promoting mitophagy.27 Next, we investigated whether mitophagy is affected by SIRT1 after SAH. First, we detected the expression levels of mitophagy-related proteins by Western blot. The results showed that compared with the Sham group, Parkin, Pink1 and LC3 protein levels were significantly increased in SAH mice and vehicle groups. However, the use of EX-527 reduced the levels of Parkin, Pink1 and LC3. On the contrary, the level of P62 and Tom 20 were decreased in SAH mice and vehicle groups, when EX-527 was administered, the levels of P62 and Tom20 were reduced, and the difference was statistically significant (Figure 3A–F, P < 0.05). Meanwhile, similar results in vitro experiments were consistent with in vivo experiments. The levels of Parkin, Pink1 and LC3 were significantly increased in the OxyHb group and DMSO group, and the levels were reduced while EX-527 was used (Figure 3G–L, P < 0.01). We speculated that SIRT1 may play a role by promoting mitophagy, while EX-527 inhibited the role of SIRT1.
 |
Figure 3 SIRT1 promotes mitophagy after SAH. (A–F) Western blot and quantitative analysis of Parkin, P62, Pink1, Tom20 and LC3 in the cortex at 24h after SAH in mice. (G–L) Western blot and quantitative analysis of Parkin, P62, Pink1, Tom20 and LC3 in primary microglia in four groups. (M–N) The co-localization of Mito-Green and Lyso-Red with laser confocal and quantitative analysis in different groups (Con, OxyHb, OxyHb + DMSO, OxyHb + EX-527). Scale bar: 10 μm. (O–P) Representative TEM images of mitophagosomes after SAH cell model (black triangle refers to the mitophagosomes and the * refers to mitochondria). Scale bar: 5 μm (upper panels), 1 μm (lower panels). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
In order to confirm this conjecture, we used Mito-Tracker and Lyso-Tracker to perform immunofluorescence staining of mitochondria and lysosomes respectively. The results showed that compared with Con group, the number of mitolysosomes in OxyHb group and DMSO group increased significantly; however, the number of mitolysosomes decreased after using EX-527, and it was still more than those in Con group (Figure 3M–N). In order to more intuitively observe the effect of SIRT1 on mitophagy, we conducted transmission electron microscope observations. The results were consistent with immunofluorescence staining. Mitophagosomes in SAH group and vehicle group were significantly increased compared with Sham group; while those in the EX-527 group were decreased (Figure 3O–P). The above results showed that SIRT1 promotes mitophagy and exerts anti-inflammatory effects.
Inhibition of SIRT1 Enhances Foxo1 Acetylation After SAH
As the main target of SIRT1, foxo1 plays an important role in neuroinflammation after SAH.28 As a deacetylase, it is not clear whether sirt1 participates in regulating mitophagy through acetylation modification of foxo1 after SAH. Therefore, we explored the effect of SIRT1 on foxo1 signaling after SAH. The results showed that the expression level of foxo1 was decreased in SAH mice and OxyHb-treated microglia, and its expression level was even lower after treatment with EX-527. In contrast, when detecting acetylated foxo1 (ac-foxo1), increased expression levels of ac-foxo1 were detected in SAH mice and OxyHb-treated microglia, with higher expression levels after EX-527 treatment (Figure 4, P < 0.01). These results suggested that sirt1 can participate in the regulation of mitophagy through foxo1, which is achieved by inhibiting the acetylation of foxo1, and this effect increases the acetylation level of foxo1 due to the inhibition of sirt1 by EX-527.
 |
Figure 4 Inhibition of SIRT1 enhances Foxo1 acetylation after SAH. Western blot and quantitative analysis of foxo1, ac-foxo1 in mice (A–B) and primary microglia (C–D). **P < 0.01 vs Sham/Con group; ##P < 0.01, ###P < 0.001 vs Sham/Con group; &&P < 0.01, &&&P < 0.001 vs (SAH + vehicle) / (OxyHb + DMSO) group. |
Discussion
Neuroinflammatory damage caused by EBI seriously affects the prognosis of patients after SAH, and SIRT1 played a neuroprotective role after being activated.29–32 What’s more, SIRT1 can also play a role in the central nervous system through mitophagy.33 Mitophagy plays an important role in regulating inflammation, promoting mitophagy, and alleviating neuroinflammatory damage after EBI in SAH.16,17 However, the role of SIRT1 about mitophagy in SAH has been rarely reported. This study aims to study SIRT1-mediated mitophagy in SAH.
In this study, we extracted primary microglia from mouse brain tissue and established SAH model in vivo and vitro to detect the expression levels of SIRT1 at different time points and found that its expression level was highest at 24 h. In order to inhibit the expression of SIRT1, we selected EX-527, a specific inhibitor of SIRT1, to detect cell viability, neurological function, and Evan’s blue and brain edema content in microglia and SAH mice respectively. Compared with the Sham group, we found that the Evan’s blue and brain edema levels in SAH group and vehicle group were increased. However, when the inhibitor EX-527 was given, the levels were significantly higher than before. At the same time, Nissl staining experiments showed that the number of neuronal cells in the SAH group was reduced compared with the Sham group, and the number of neuronal cells was even further reduced after EX-527 treatment. Not only that, the expression levels of inflammatory factors such as IL-6, IL-1β and TNF-α were higher in the SAH group than in the Sham group. After EX-527 was administered, their expression levels were actually higher. It further illustrates that inhibiting the expression of SIRT1 can aggravate brain damage after SAH. In other words, SIRT1 plays a neuroprotective role after SAH.
In fact, many studies have also reported similar effects of SIRT1 after SAH. Activation of SIRT1 promotes the survival rate of CA1 neurons and promotes neurological functional recovery after SAH.31 Clarke et al34 reported that SIRT1 mediated hypoxic postconditioning-induced protection of neurological function after SAH. Han et al35 found that Oleanolic acid reduced the acetylation of HMGB1 by activating SIRT1 and exerted anti-inflammatory effects to alleviate EBI after SAH. Berberine could relieve injury by inducing sirt1 and inhibit the HMGB1/NF-κB pathway after SAH.36 In our study, we found that by inhibiting the expression of SIRT1, the levels of Parkin, Pink1, LC3 decreased, and the levels of P62 and Tom20 increased, indicating that the level of mitophagy was inhibited. At the same time, ELISA and q-PCR showed that the levels of inflammatory factors such as IL-6, IL-1β, and TNF-α increased. We concluded that inhibiting the expression of SIRT1 can reduce the clearance of damaged mitochondria and promote inflammation-related factors by mitophagy, which aggravate the inflammatory damage after SAH.
Foxo1, as the target of sirt1, plays an important role in mitophagy.37 In order to explore its possible mechanism in SAH, we detected the level of foxo1 and ac-foxo1. The results shown that inhibiting the expression of sirt1 increased the level of ac-foxo1, thereby reducing the level of mitophagy. In other words, SIRT1 alleviates the inflammatory damage of SAH by reducing foxo1 acetylation levels and promoting mitophagy. In addition, studies have found38 that in the endotoxin tolerance process of sepsis, SIRT1 accumulates in the IL-1β, TNF-α and NAD+ levels increase, which enhances H3K16 deacetylation. Zhang et al39 demonstrated that SIRT1 can reduce histone H3K9 acetylation in the promoters of IL-6 and TNF-α and block their expression. In addition, Chen et al40 reported that SIRT1 targets the TNF-α promoter, reduces H3K16 acetylation and inhibits TNF-α transcription during sepsis-induced inflammation. Therefore, it remains unclear whether SIRT1 regulates inflammatory damage after SAH directly through foxo1 deacetylation or directly through acetylation modification of inflammatory factors, which need to further explore the underlying mechanism in future studies.
Conclusion
In this study, we found that the expression of SIRT1 in microglia was increased after SAH, and the levels of inflammatory factors were increased after specifically blocking SIRT1 activation; however, the levels of mitophagy were decreased. We then clarified that SIRT1 exerts a protective effect after SAH through mitophagy, which is achieved by regulating foxo1 acetylation levels. Our study deepens the understanding of microglia function after SAH and provides evidence for further regulation.
Data Sharing Statement
Data will be made available on request.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Tholance Y, Aboudhiaf S, Balanca B, et al. Early brain metabolic disturbances associated with delayed cerebral ischemia in patients with severe subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2023;43(11):1967–1982. doi:10.1177/0271678X231193661
2. Tokareva B, Meyer L, Heitkamp C, et al. Early and recurrent cerebral vasospasms after aneurysmal subarachnoid hemorrhage: the impact of age. Eur Stroke J. 2023;23969873231209819. doi:10.1177/23969873231209819
3. Dodd WS, Dayton O, Lucke-Wold B, Reitano C, Sorrentino Z, Busl KM. Decrease in cortical vein opacification predicts outcome after aneurysmal subarachnoid hemorrhage. J Neurointerv Surg. 2023;15(11):1105–1110. doi:10.1136/jnis-2022-019578
4. Savarraj JPJ, McBride DW, Park E, et al. Leucine-rich alpha-2-glycoprotein 1 is a systemic biomarker of early brain injury and delayed cerebral ischemia after subarachnoid hemorrhage. Neurocrit Care. 2023;38(3):771–780. doi:10.1007/s12028-022-01652-7
5. Caylor MM, Macdonald RL. Pharmacological prevention of delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2023. doi:10.1007/s12028-023-01847-6
6. Sun XG, Chu XH, Godje Godje IS, et al. Aerobic glycolysis induced by mtor/hif-1alpha promotes early brain injury after subarachnoid hemorrhage via activating m1 microglia. Transl Stroke Res. 2022;15(1):1–15. doi:10.1007/s12975-022-01105-5
7. Tao Q, Qiu X, Li C, et al. S100a8 regulates autophagy-dependent ferroptosis in microglia after experimental subarachnoid hemorrhage. Exp Neurol. 2022;357:114171. doi:10.1016/j.expneurol.2022.114171
8. Qiu G, Li X, Che X, et al. Sirt1 is a regulator of autophagy: implications in gastric cancer progression and treatment. FEBS Lett. 2015;589(16):2034–2042. doi:10.1016/j.febslet.2015.05.042
9. Yi W, Chen F, Yuan M, et al. High-fat diet induces cognitive impairment through repression of sirt1/AMPK-mediated autophagy. Exp Neurol. 2023;371:114591. doi:10.1016/j.expneurol.2023.114591
10. Chen F, Zhang X, Chen S, et al. 5-(3’,4’-dihydroxyphenyl)-gamma-valerolactone, a microbiota metabolite of flavan-3-ols, activates sirt1-mediated autophagy to attenuate h(2)o(2)-induced inhibition of osteoblast differentiation in mc3t3-e1 cells. Free Radic Biol Med. 2023;208:309–318. doi:10.1016/j.freeradbiomed.2023.08.018
11. Chen Y, Peng F, Yang C, et al. Sirt1 activation by 2,3,5,6-tetramethylpyrazine alleviates neuroinflammation via inhibiting m1 microglia polarization. Front Immunol. 2023;14:1206513. doi:10.3389/fimmu.2023.1206513
12. Chen T, Xu YP, Chen Y, Sun S, Yan ZZ, Wang YH. Arc regulates brain damage and neuroinflammation via sirt1 signaling following subarachnoid hemorrhage. Brain Res Bull. 2023;203:110780. doi:10.1016/j.brainresbull.2023.110780
13. Xia DY, Yuan JL, Jiang XC, et al. Sirt1 promotes m2 microglia polarization via reducing ros-mediated nlrp3 inflammasome signaling after subarachnoid hemorrhage. Front Immunol. 2021;12:770744. doi:10.3389/fimmu.2021.770744
14. Yao ZQ, Zhang X, Zhen Y, et al. A novel small-molecule activator of sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis. 2018;9(7):767. doi:10.1038/s41419-018-0799-z
15. Xue LX, Chen SF, Xue SX, Liu PD, Liu HB. Lncrna tug1 compromised neuronal mitophagy in cerebral ischemia/reperfusion injury by targeting sirtuin 1. Cell Biol Toxicol. 2022;38(6):1121–1136. doi:10.1007/s10565-022-09700-w
16. Chang H, Lin C, Li Z, et al. T3 alleviates neuroinflammation and reduces early brain injury after subarachnoid haemorrhage by promoting mitophagy via pink 1-parkin pathway. Exp Neurol. 2022;357:114175. doi:10.1016/j.expneurol.2022.114175
17. Zhang T, Zhang M. Nl-1 promotes pink1-parkin-mediated mitophagy through mitoneet inhibition in subarachnoid hemorrhage. Neurochem Res. 2023. doi:10.1007/s11064-023-04024-5
18. Liu Y, Yao C, Sheng B, et al. Inhibition of usp30 promotes mitophagy by regulating ubiquitination of mfn2 by parkin to attenuate early brain injury after sah. Transl Stroke Res. 2023. doi:10.1007/s12975-023-01228-3
19. Demura M, Ishii H, Takarada-Iemata M, et al. Sympathetic nervous hyperactivity impairs microcirculation leading to early brain injury after subarachnoid hemorrhage. Stroke. 2023;54(6):1645–1655. doi:10.1161/STROKEAHA.123.042799
20. Nishikawa H, Liu L, Nakano F, et al. Modified citrus pectin prevents blood-brain barrier disruption in mouse subarachnoid hemorrhage by inhibiting galectin-3. Stroke. 2018;49(11):2743–2751. doi:10.1161/STROKEAHA.118.021757
21. Li HR, Liu Q, Zhu CL, et al. Beta-nicotinamide mononucleotide activates nad+/sirt1 pathway and attenuates inflammatory and oxidative responses in the hippocampus regions of septic mice. Redox Biol. 2023;63:102745. doi:10.1016/j.redox.2023.102745
22. Li N, Bai N, Zhao X, et al. Cooperative effects of SIRT1 and SIRT2 on APP acetylation. Aging Cell. 2023;22(10):e13967. doi:10.1111/acel.13967
23. Xie Y, Guo H, Wang L, et al. Human albumin attenuates excessive innate immunity via inhibition of microglial mincle/syk signaling in subarachnoid hemorrhage. Brain Behav Immun. 2017;60:346–360. doi:10.1016/j.bbi.2016.11.004
24. Zhang XS, Zhang X, Zhou ML, et al. Amelioration of oxidative stress and protection against early brain injury by astaxanthin after experimental subarachnoid hemorrhage. J Neurosurg. 2014;121(1):42–54. doi:10.3171/2014.2.JNS13730
25. Xu P, Liu Q, Xie Y, et al. Breast cancer susceptibility protein 1 (brca1) rescues neurons from cerebral ischemia/reperfusion injury through nrf2-mediated antioxidant pathway. Redox Biol. 2018;18:158–172. doi:10.1016/j.redox.2018.06.012
26. Lu Y, Zhang XS, Zhang ZH, et al. Peroxiredoxin 2 activates microglia by interacting with toll-like receptor 4 after subarachnoid hemorrhage. J Neuroinflammation. 2018;15(1):87. doi:10.1186/s12974-018-1118-4
27. Lin L, Wei J, Zhu C, et al. Sema3A alleviates viral myocarditis by modulating SIRT1 to regulate cardiomyocyte mitophagy. Environ Toxicol. 2023;38(6):1305–1317. doi:10.1002/tox.23765
28. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022. doi:10.1016/j.cell.2014.04.007
29. Azua-Lopez ZR, Pezzotti MR, Gonzalez-Diaz A, et al. HDL anti-inflammatory function is impaired and associated with high saa1 and low apoa4 levels in aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2023;43(11):1919–1930. doi:10.1177/0271678X231184806
30. Zhang XS, Wu Q, Wu LY, et al. Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats. Cell Death Dis. 2016;7(10):e2416. doi:10.1038/cddis.2016.292
31. Chu D, Li X, Qu X, et al. Sirt1 activation promotes long-term functional recovery after subarachnoid hemorrhage in rats. Neurocrit Care. 2023;38(3):622–632. doi:10.1007/s12028-022-01614-z
32. Yuan B, Zhao XD, Shen JD, et al. Activation of sirt1 alleviates ferroptosis in the early brain injury after subarachnoid hemorrhage. Oxid Med Cell Longev. 2022;2022:9069825. doi:10.1155/2022/9069825
33. Shao Z, Dou S, Zhu J, et al. Apelin-36 protects ht22 cells against oxygen-glucose deprivation/reperfusion-induced oxidative stress and mitochondrial dysfunction by promoting sirt1-mediated pink1/parkin-dependent mitophagy. Neurotox Res. 2021;39(3):740–753. doi:10.1007/s12640-021-00338-w
34. Clarke JV, Brier LM, Rahn RM, et al. Sirt1 mediates hypoxic postconditioning- and resveratrol-induced protection against functional connectivity deficits after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2022;42(7):1210–1223. doi:10.1177/0271678X221079902
35. Han Y, Tong Z, Wang C, Li X, Liang G. Oleanolic acid exerts neuroprotective effects in subarachnoid hemorrhage rats through sirt1-mediated hmgb1 deacetylation. Eur J Pharmacol. 2021;893:173811. doi:10.1016/j.ejphar.2020.173811
36. Zhang XH, Peng L, Zhang J, et al. Berberine ameliorates subarachnoid hemorrhage injury via induction of sirtuin 1 and inhibiting hmgb1/nf-kappab pathway. Front Pharmacol. 2020;11:1073. doi:10.3389/fphar.2020.01073
37. Zhou L, Su W, Wang Y, Zhang Y, Xia Z, Lei S. Foxo1 reduces stat3 activation and causes impaired mitochondrial quality control in diabetic cardiomyopathy. Diabetes Obes Metab. 2023. doi:10.1111/dom.15369
38. Yang Y, Zhang S, Fan C, et al. Protective role of silent information regulator 1 against hepatic ischemia: effects on oxidative stress injury, inflammatory response, and mapks. Expert Opin Ther Targets. 2016;20(5):519–531. doi:10.1517/14728222.2016.1153067
39. Zhang Y, Li Y, Li J, et al. Sirt1 alleviates isoniazid-induced hepatocyte injury by reducing histone acetylation in the il-6 promoter region. Int Immunopharmacol. 2019;67:348–355. doi:10.1016/j.intimp.2018.11.054
40. Chen GD, Yu WD, Chen XP. Sirt1 activator represses the transcription of tnf‑alpha in thp‑1 cells of a sepsis model via deacetylation of h4k16. Mol Med Rep. 2016;14(6):5544–5550. doi:10.3892/mmr.2016.5942
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.