")
Back to Journals » Research and Reports in Urology » Volume 15
Mechanistic Insights on Localized to Metastatic Prostate Cancer Transition and Therapeutic Opportunities
Authors Yu EM, Hwang MW, Aragon-Ching JB
Received 27 September 2023
Accepted for publication 15 November 2023
Published 29 November 2023 Volume 2023:15 Pages 519—529
DOI https://doi.org/10.2147/RRU.S386517
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Panagiotis J Vlachostergios
Eun-mi Yu,1 Min Woo Hwang,2 Jeanny B Aragon-Ching1
1GU Medical Oncology, Inova Schar Cancer Institute, Fairfax, VA, USA; 2Department of Internal Medicine, Inova Fairfax Hospital, Fairfax, VA, USA
Correspondence: Jeanny B Aragon-Ching, Clinical Program Director of Genitourinary Cancers, Inova Schar Cancer Institute, Associate Professor of Medical Education, University of Virginia School of Medicine, 8081 Innovation Park Drive, Fairfax, VA, USA, Tel +1-571-472-4724, Fax +1-571-472-1180, Email [email protected]
Abstract: Prostate cancer is the most common non-cutaneous cancer among American men. Multiple mechanisms are involved in tumorigenesis and progression to metastases. While androgen deprivation therapy remains the cornerstone of treatment, progression to castration-resistant disease becomes inevitable. Aberrant pathway activations of PI3K/AKT due to PTEN loss, epithelial–mesenchymal transition pathways, homologous recombination repair, and DNA repair pathway mechanisms of resistance and cross-talk lead to opportunities for therapeutic targeting in metastatic castration-resistant prostate cancer. This review focuses on mechanisms of progression and key trials that evaluate the drugs and combinations that exploit these pathways.
Keywords: prostate cancer, metastasis, castration-resistant prostate cancer, epithelial mesenchymal transition, androgen receptor pathway inhibitors, poly-ADP ribose pathway inhibitors
Introduction
An estimated 288,300 new prostate cancer cases will be diagnosed in 2023, and about 34,700 patients will die due to prostate cancer. Although the rate of death due to prostate cancer has declined by half since 1993, in more recent years, an increase in advanced-stage diagnosis has been observed.1 Localized prostate cancer is typically treated and cured with local therapy, including prostatectomy or radiotherapy. The use of androgen deprivation therapy (ADT) in the localized setting is generally limited to certain prostate cancer patients receiving radiotherapy who are at higher risk of disease recurrence and ADT is received for a finite period. However, 20–50% of prostate cancer patients develop disease recurrence after local therapy which often implies lifelong systemic therapy.2 Patients presenting with de novo metastatic prostate cancer are also primarily treated with systemic therapy, though there is an increasing role for radiotherapy in low-volume metastatic castration-sensitive prostate cancer based on data from the STAMPEDE and PEACE-1 trials.3,4 In the metastatic castration-resistant setting, there has been an increasing role for PARP inhibition and radioligand therapy in recent years.
Prostate cancer metastasis entails a complex interplay of cells escaping the primary tumor, spreading and settling down at another site that is primed for growth and further metastasis.5 Numerous mechanisms of tumorigenesis have been proposed for certain prostate cancers. There are historical risk factors that predict for higher risk of metastasis. However, Stephen Paget’s “seed and soil” hypothesis laid the foundation of the metastatic phenomenon and there is now an extensive body of data that supports his theory.6 Though the development of metastases is extraordinarily complex, put simply, metastases are the result of tumor cells (the “seed”) thriving in their ideal microenvironment (the “soil”). In prostate cancer, mechanisms of cancer progression can be divided into the epithelial-to-mesenchymal transition (EMT), which defines epigenetic drivers for cancer cell migration and metastasis, and stromal interactions within the tumor microenvironment (TME) that allow for the survival of tumor cells in distant organs, such as the bone.7 Although ADT is initially universally effective in untreated prostate cancer, the development of castration-resistance is inevitable and this ultimately limits the life expectancy of advanced prostate cancer patients. Though much progress has been made in the treatment landscape of advanced metastatic castration-resistant prostate cancer (mCRPC) within the last 15 years with the initial approval of abiraterone acetate in this setting, responses to oral secondary antiandrogens are not durable. It goes without saying that the ongoing pursuit of highly effective and well-tolerated treatment strategies in advanced prostate cancer remains a worthwhile and important cause and patients will be better served as we deepen our knowledge of prostate cancer tumorigenesis and progression.
Epithelial–Mesenchymal Transition (EMT)
The EMT transition has long been considered a paradigm of metastatic cancer progression and it refers to the transition of tumor cells with epithelial characteristics to cells with mesenchymal properties which facilitates mobility and metastases of the cancer cells within distant tissues and organs.8 It describes the process by which circulating tumor cells undergo remodeling as they enter the bloodstream and this process is, in part, regulated via epigenetic modifications.9,10 In the setting of prostate cancer progression, EMT involves the upregulation of growth factors including transforming growth factor-beta (TGF-β) and insulin-like growth factor (IGF-1), but also signaling pathways such as mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) which are intertwined with androgen receptor (AR) signaling.8 Upregulation of these signaling pathways ultimately induces the expression of key transcription factors such as SNAI1/2, ZEB1, TWIST1, ETS, as well as tumor suppressor genes such as RB1, PTEN, and TP53 which all have a role in EMT regulation.11 Tumor progression occurs as cancer cells lose expression of epithelial markers, E-cadherin and β-catenin, and acquire mesenchymal markers such as vimentin and N-cadherin, which leads to loss of cell–cell adhesion and increase in cell migration.12
Many patients with high-risk localized and advanced prostate cancer ultimately develop progression to mCRPC because of the eventual development of resistance to standard drugs that suppress testosterone production and target the androgen receptor signaling pathway. Given the potential role that EMT has in prostate cancer metastasis, the pathways involved in EMT induction, potentially, can be harnessed for drug targeting.
Epigenetic Mechanisms in Prostate Cancer
Epigenetics, a concept developed by Conrad Waddington, refers to the chemical modifications of genes or gene products that alter cell phenotypes without changes in the DNA sequence.13 Epigenetic modifications, including DNA methylation, play a major role in regulating EMT and prostate cancer cell remodeling in the context of their microenvironment.9,10,14 On a broad level, epigenetic modifications can be subdivided into those that exert their effect at the chromosome level (DNA methylation, histone modification, nucleosome positioning, and higher-order chromatin organization to name a few) and those that are RNA-mediated (RNA methylation or modifications, gene regulation by non-coding RNA such as microRNAs).15
DNA methylation refers to the addition of a methyl group (CH3), commonly at the 5-carbon position of cytosine which is mediated by DNA methyltransferases.16,17 Methylation of 5’ carbocytidine (5mC) on CpG islands associated with gene promoter regions maintains gene silencing.18 Dysregulation of DNA methylation, an example of which is the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) by the TET family enzymes, results in the reversal of DNA methylation which is thought to play a key role in tumorigenesis. In one study of 5hmC sequencing in prostate cancer tumor specimen, 5hmC modification was increased in some pathways, such as TGF-β, in mCRPC tumors but not in localized prostate cancer samples. 5hmC levels at the binding sites of key prostate cancer oncogenes including AR, FOXA1, and HOXB13 were assessed and found to be enriched at these loci in mCRPC tumor samples.19 This implies a potential role for 5hmC modification in gene reprogramming and metastatic progression of prostate cancer.
Histones are proteins that form structural units with DNA called nucleosomes and they have an important role in gene regulation and DNA replication. The N-terminal ends of histones are where post-transcriptional modifications, including acetylation, methylation, phosphorylation, and ubiquitination, occur. Several enzymes involved in histone modification have been implicated in progressive prostate cancer. The polycomb group protein enhancer of zeste homolog 2 (EZH2) is a histone-lysine N-methyltransferase enzyme and its activity leads to the transcriptional repression of specific genes and modulation of cell growth pathways via its SET domain. EZH2 expression was found to be increased in metastatic versus localized prostate cancer and benign prostatic tissue in a study utilizing the significance analysis of microarrays (SAM) technique.20 Expression of EZH2 was higher in castration resistant, relative to, castration sensitive metastatic prostate cancer specimen. This study also revealed a correlation between increased EZH2 expression in localized prostate cancer tissue and disease recurrence after treatment, suggesting that dysregulated EZH2 expression has a role in prostate cancer progression.20 Lysine-specific demethylase 1 (LSD1) and histone methyltransferase SET9 are other histone-modification-related enzymes believed to be associated with the progression of prostate cancer, as well.21,22
Multiple Myeloma SET domain (MMSET) is a histone methyltransferase that is overexpressed in several metastatic cancers, including metastatic prostate cancer. One study found that overexpression of MMSET in immortalized, non-transformed prostate cells led to increased cell migration, invasiveness, and EMT. TWIST1, an EMT-promoting gene, was found to be strongly activated in response to MMSET. Chromatin immunoprecipitation analysis revealed that MMSET binds to the TWIST1 locus and leads to increased demethylation of lysine 36 on histone H3 (H3K36me2), which highlights the potential role of MMSET as an epigenetic regulator of metastasis-promoting genes.23
These are just a few examples of epigenetic mechanisms in the context of prostate cancer progression and there are numerous others, the discussion of which is beyond the scope of this review. Suffice to say, however, the metastatic progression of prostate cancer is a highly complex and dynamic process and there remain many opportunities for clinical applications that have yet to be discovered.
Factors That Promote Bone Metastases
As cancers metastasize from their primary site of origin, the tumor microenvironment (TME) must evolve in parallel to the cancer cell clones for the successful colonization and spread of cancer cells at distant sites. Changes in the extracellular matrix, angiogenesis, recruitment of tumor supporting cells and inflammatory cytokines are among some of the changes that occur in the TME that promote cancer metastases. Prostatic adenocarcinoma has a high propensity for metastasizing to the bones and over 80% of patients with advanced prostate cancer will develop and die with bone metastases.24 The molecular mechanisms by which bone metastases occur in prostate cancer are not fully known, but there is evidence that certain prostate tumor-derived factors promote changes in the bone microenvironment that allow for the colonization of circulating prostate cancer cells. Specifically, primary prostatic tumors may stimulate the establishment of premetastatic niches and bone remodeling via the secretion of exosomes and microRNAs as a form of intercellular communication.25 Also, the role of genetic alterations in the progression of prostate cancer is well-established. In prostate cancer, the primary oncogenic drivers are chromosomal rearrangements and copy number alterations.26,27 Certain genomic alterations are specific to metastatic tumors in prostate cancer, including those involving the AR, CDK12, ZFHX3, RB1, GNAS, and OR5L1 genes. There appears to be a pattern of genomic changes that occur more often in mCRPC tumors, including mutations in SPOP, FOXA1, TP53, BRCA2, PTEN, and CHD1, as well.27–29 Mutations at this stage of prostate cancer appear to confer resistance to androgen deprivation and other systemic therapies such as AR-directed drugs.27,30
Certain genetic alterations in metastatic prostate cancers appear to occur in concert with changes in the TME that clinically manifest as an increasingly more aggressive malignancy. In one study, 260 prostate biopsy samples were assessed for loss of PTEN, a tumor suppressor gene, by immunohistochemistry. Eighty-eight (34%) of the samples were confirmed to have PTEN loss. Logistic regression analysis revealed that PTEN loss, compared to PTEN intact cases, was significantly associated with certain tumor morphologic features including large gland pattern, poorly formed glands, cribriform Gleason pattern 4, solid architecture, intraductal carcinoma, and stromogenic prostate cancers. In this study, tumor-associated stroma, otherwise known as the TME, was graded based on the presence of reactive stroma (disorganized, pale, loose, and rich in extracellular matrix with infiltrating angulated and distorted glands) and the epithelial-to-stroma ratio. Stromogenic prostate cancer was defined by a stroma grade of 3 (>50% reactive stroma).31 Another study found an association between PTEN loss and increased CXCL8 expression, tumor-associated macrophage infiltration, and increased tumor sensitivity to inhibitor of apoptosis protein (IAP) antagonist therapy.32
In the case of bone metastases in prostate cancer, mutations in the E26 transformation specific (ETS) family of transcription factors, including ERG, ETV1, ETV4, and ETV5 are common. Specifically, TMPRSS2-ERG gene fusions occur most often, and these appear to promote the development of bone metastases. Transmembrane Protease Serine 2 (TMPRSS2) is a prostate-specific androgen-regulated gene and rearrangement of this gene leads to increased expression of the ETS transcription factors. In one mouse model study, cell lines expressing TMPRSS2-ERG fusion were found to increase cell migration, subcutaneous tumor size, and the number of bone metastases. There also appeared to be a correlation between TMPRSS2-ERG and an increased propensity for bone metastases involving the hind limbs and spine.33 Though other frequently-occurring genomic changes in metastatic prostate cancers have not yet been proven to have a direct role in bone metastasis development, these mutations have been identified in bone metastasis specimen. Thus, ongoing genomics-based analyses are crucial to gaining a better understanding of tumor biology and clinical behavior in advanced prostate cancer.
The TME is comprised of fibroblasts, immune cells, endothelial cells, as well as extracellular matrix (ECM) proteins including laminin, fibronectin, collagen, and hyaluronate. These components of the TME interact with malignant cells via multiple complex pathways comprised of cytokines, chemokines, growth factors, and matrix remodeling enzymes.34 The process by which tumor cells promote the development of a permissive secondary microenvironment, or premetastatic niche, has generally been well demonstrated in many types of cancers, including prostate cancer.35 In the primary prostate cancer TME, cancer-associated fibroblasts (CAFs) appear to promote the progression of prostate cancer by inducing ECM remodeling and the release of ECM-bound growth factors which sustain cancer cell proliferation and invasion.36,37 Human prostate fibroblasts also appear to have a role in inducing castration resistance and metastases formation.38 Prostate cancer cells and TME components may be involved in priming the bone microenvironment for metastases. Prostate cancer cells in the blood circulation interact with surface proteins on endothelial cells and home in on the bone microenvironment. The prostate cancer cells, which express cell surface αβ-integrins, bind to bone ECM proteins. Through the secretion of PTHrP and cytokines, prostate cancer cells influence the RANK-RANKL interaction between osteoblasts and osteoclasts and alter the activity of the latter, leading to bone matrix resorption and the release of ECM-bound growth factors, such as TGF-β, FGFs, and HGF, which promote tumor cell growth at the metastatic site.39
ADT appears to influence the prostate stroma and contribute to the development of castration-resistance via CAF-mediated pathways.40 There is also evidence that androgen ablation is associated with increased bone resorption, loss of bone volume, and growth of disseminated prostate cancer cells in bone tissue via osteoclast-mediated mechanisms in mouse models. In the same study, the administration of zoledronic acid appeared to prevent the development of bone metastases.41 There has been an increasing body of evidence, in general, of stroma or TME-mediated treatment resistance.42 For the purpose of developing effective drug targets, it is imperative to further define the relationship between genetic changes in prostate cancer cells and the TME to the development of skeletal metastases, but this has proven to be challenging. While many mouse models mimicking the earlier stages of prostatic adenocarcinoma genesis have been developed, relatively fewer genetically engineered mouse models to date include those that reflect the natural formation of bone metastases.43
A few studies of prostate cancer mouse models have demonstrated co-activation of Myc and Ras pathways.44,45 Loss of p53 and phosphatase and tensin homologue (PTEN) have been associated with loss of hormone sensitivity and the development of metastatic CRPC.46 However, lymph node, lung, and liver are the most frequent sites of metastases in mouse models for reasons that are not fully elucidated aside from the differences in overall physiology and bone microenvironment in mice and humans.47 To date, microenvironment-directed treatments, such as anti-CXCR4 agents, have yet to show clinical activity in metastatic prostate cancer.48 Novel approaches of developing humanized models including in vivo tissue-engineered xenografts, in vitro microtissue-engineered models, and ex vivo manipulation and serial re-transplantation are promising as the intricacies of prostate cancer progression are further studied for the purpose of developing new drug targets.49–51
Therapeutic Opportunities
Targeting the PI3K Pathway
There are multiple mechanisms involved in activating the PI3K-AKT-mTOR (phosphoinositide 3-kinase-RAC-alpha serine/threonine-protein kinase-mammalian target of rapamycin) pathway which is implicated in the progression of a wide range of cancers. This is especially prevalent in metastatic prostate cancer, where PI3K pathway alterations can occur in up to 50% of patients,52 and contributes to the emergence of resistance to androgen deprivation therapy.52,53 In addition, genomic aberration of PTEN with loss of function of this tumor suppressor gene leads to activation of the PI3K-AKT pathway. PTEN loss in prostate cancer has been shown to be associated with poor prognosis,54 which makes therapeutic targeting of PTEN particularly attractive especially for advanced metastatic CRPC where the inactivation via deletion or mutation is seen in 50% of cases.55 PTEN is also a known negative regulator of the PI3K/AKT pathway,56 hence, inactivation or loss of PTEN often leads to activation of this pathway that leads to cellular growth and signaling driving further tumorigenesis.57 Several studies revealed crucial reciprocal signaling and cross-talk between AR and the PI3K/AKT/mTOR pathway. As such, PI3K, AKT, and mTOR inhibitors have been studied in prostate cancer, though to date, none have demonstrated considerable efficacy with an acceptable level of toxicity at therapeutic doses.58 While manipulating PI3K-AKT and mTOR in metastatic prostate cancer has been attempted, utility has been limited in several Phase I and II trials, whether alone or in combination,59 hence no resultant FDA approval as of yet using these drugs in mCRPC. However, there are several drugs that have advanced to Phase III trials that are worthy of discussion (see Table 1).
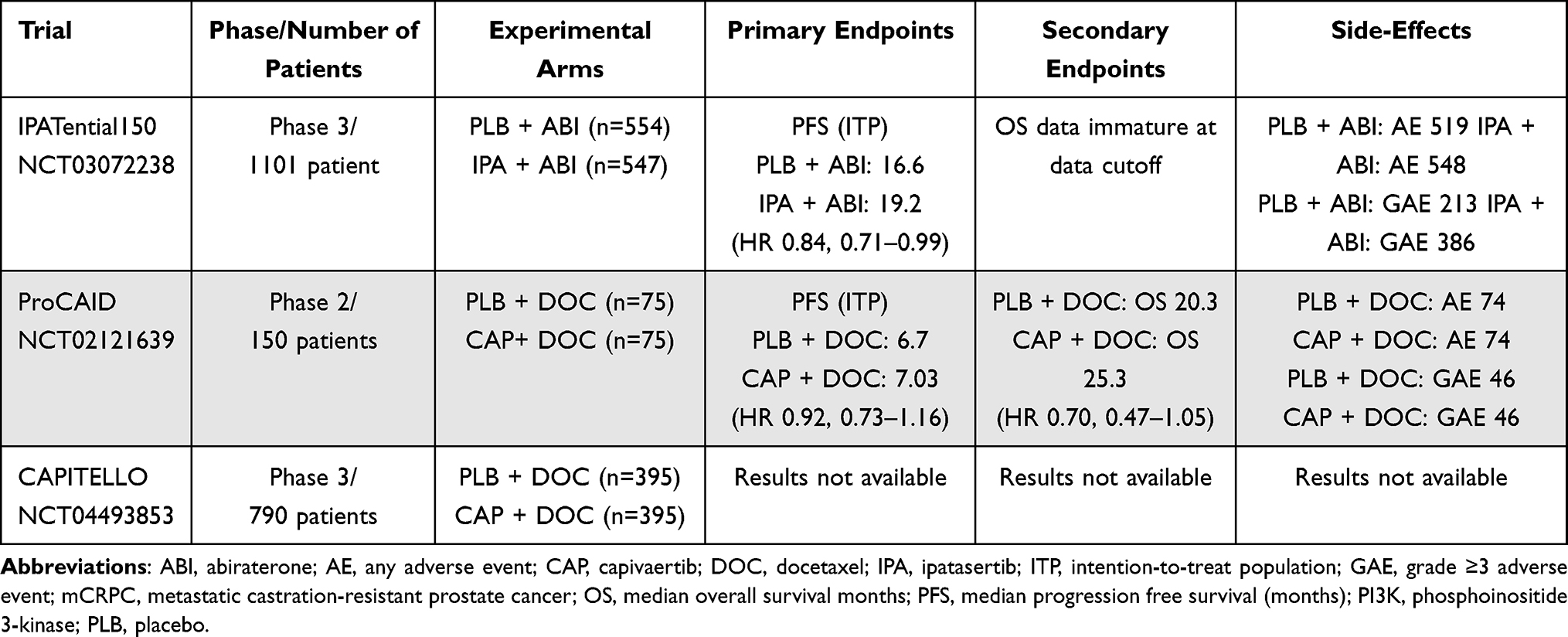 |
Table 1 Selected PI3K Trials in Prostate Cancer |
Ipatasertib is an oral small-molecule inhibitor of all isoforms of AKT and was studied early on in phase I and II trials in prostate cancer. Given promising Phase II radiographic progression-free survival (rPFS) outcomes with ipatasertib, particularly in those with prostate cancers with PTEN loss,60 a phase III trial IPATential150 was launched.61 The trial enrolled 1101 mCRPC patients of whom 47% were found to have PTEN loss by immunohistochemistry (IHC). Randomization occurred in a 1:1 fashion to either ipatasertib with abiraterone/prednisone versus abiraterone/prednisone with placebo. The trial had co-primary endpoints with investigator-assessed rPFS assessed in the intention-to-treat (ITT) population and investigator-assessed rPFS in mCRPC patients whose cancers harbored PTEN loss using the VENTANA assay by IHC. While the results showed that one of the co-primary endpoint of rPFS in the PTEN loss group showed better rPFS outcomes in the ipatasertib with abiraterone group at 18.5 months compared to the placebo with abiraterone group at 16.5 months (95% CI 13.9–17.0) with an HR of 0.77 [95% CI 0.61–0.98]; p = 0.034; significant at α = 0.04), the ITT group did not meet statistical significance. The median PFS in the ITT population was 16.6 months in the placebo-abiraterone cohort compared to 19.2 months in the ipatasertib-abiraterone cohort (HR 0.84 [95% CI 0.71–0.99]; p = 0.043; not significant at α = 0.01).
Capivasertib is another oral AKT inhibitor that demonstrated preclinical activity in both hormone-sensitive and CRPC models and was studied in a phase II trial called ProCAID that included patients with mCRPC. While the trial did not show adding capivasertib to docetaxel improved PFS, it resulted in improvement in overall survival (OS) with median OS of 25.3 months for capivasertib with docetaxel compared to 20.3 months for the placebo plus docetaxel (hazard ratio [HR] 0.70, 95% confidence interval [CI] 0.47–1.05; nominal p = 0.09), in the 110 patients included in the ITT population.62 Exploratory analyses also suggested benefit in a cohort of patients treated with an androgen receptor pathway inhibitor (ARPI). Therefore, a phase III trial CAPItello-281 was launched.63 This randomized-controlled trial has opened to enrollment and aims to determine the efficacy of capivasertib in combination with abiraterone acetate and prednisone in de novo mCSPC patients looking at rPFS as a primary endpoint.
Co-Targeting of AR and DDR Mechanism
Poly(ADP-ribose) polymerase (PARP) belongs to a family of proteins responsible for key regulatory cellular processes involved in DNA repair, chromatin functions, and genomic stability.64 Early clinical trials in mCRPC suggest increased PARP-1 activity as a pro-tumorigenesis pathway that supports AR-mediated growth, prostate cancer proliferation, and cellular survival in BRCA-deficient cells.65,66 Trials studying sequential PARP inhibition after failure from prior ARPI use therefore led to multiple studies showing PFS benefit in the setting of DNA damage response and repair (DDR) mutations, with drugs like olaparib in the PROfound trial,67 niraparib in the GALAHAD trial,68 and rucaparib in the TRITON269 and TIRITON3 trials.70 While sequential PARPi monotherapy trials showed improvement in PFS outcomes, early preclinical studies evaluating the downregulation of DNA repair genes with the use of ARPIs formed the rationale for synergistic responses with ionizing radiation as a way to increase DNA damage and decrease the clonogenic survival of prostate cancer cells.71 In addition, early preclinical studies suggested that the loss of BRCA2 in prostate cancer cell lines led to the emergence of a more aggressive and castration-resistant phenotype.72 Dual loss of BRCA2 and RB1 in human prostate cancer cell lines may further induce EMT, resulting in an aggressive phenotype which, in part, partly justifies co-inhibition of AR and poly-ADP ribose.73 Early preclinical models suggest that enhanced recruitment of PARP-1 in AR-positive cancer cells promotes AR occupancy, AR transcription, and the emergence and maintenance of castration resistance (see Figure 1 for proposed mechanism).74 Conversely, AR inhibition was shown to upregulate PARP activity and downregulate homologous recombination repair (HRR) gene expression,75 and this resultant upregulation of PARP activity leads to prostate cancer cell survival, hence, demonstrating synthetic lethality between ADT and PARP inhibition in preclinical models,76 leading to clinical trials investigating co-targeting AR and PARP.
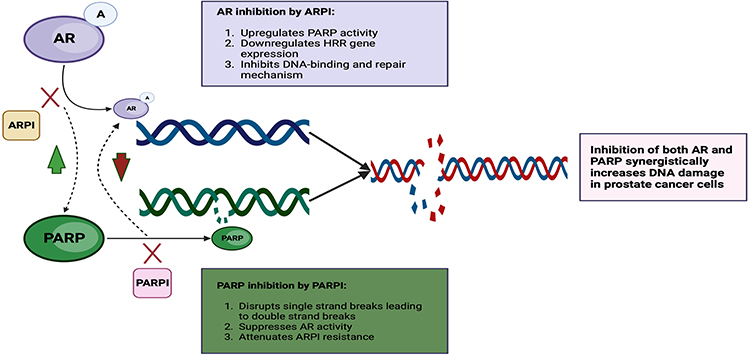 |
Figure 1 AR and PARP inhibition interactions. Diagram shows AR inhibition upregulates PARP activity and downregulates HRR gene expression and converse PARP inhibition suppresses AR activity and attenuates resistance to ARPI apart from disrupting single-stranded breaks. Created with BioRender.com. Abbreviations: AR, androgen receptor; ARPI, androgen receptor pathway inhibitors; DNA, double-stranded nucleic acid; PARP, poly-adenosine diphosphate ribose polymerase; PARPi, poly-adenosine diphosphate ribose polymerase inhibitor. |
One of the early clinical trials that explored co-targeting of AR and PARP was the NCI 9012 trial using the combination of the CYP17 inhibitor abiraterone with veliparib.77 Co-targeting AR and PARP had a solid scientific rationale, especially since canonical ETS gene fusions which are commonly overexpressed in prostate cancer, are known to induce DNA damage in a manner similar to BRCA1/2 that is potentiated by PARP inhibition.78 The primary endpoints of NCI 9012 were confirmed prostate-specific antigen (PSA) response rate (RR) and whether ETS fusions predicted response, although the study was deemed a negative trial since PSA response rates were no different in the two arms (abiraterone at 63.9% versus abiraterone and veliparib at 72.4%; P = 0.27). This may be explained by weaker PARP-trapping activity with veliparib use.79 However, three subsequent randomized controlled trials (see Table 2) showed that the combination of an ARPI and a PARPi with abiraterone and olaparib in the PROpel trial,80 abiraterone and niraparib in the MAGNITUDE trial,81 and enzalutamide with talazoparib in the TALAPRO-2 trial,82 all showed improvement in rPFS when compared to an ARPI alone in the ARPI-naïve, mCRPC setting. PROpel was a phase III trial that randomly assigned 796 patients with mCRPC in a 1:1 fashion to either abiraterone with olaparib compared to abiraterone alone with the primary endpoint of imaging-based progression-free survival (ibPFS) by investigator assessment and overall survival as a secondary endpoint. HRR mutation status was not mandated but pre-defined upon study enrollment and both tumor tissue and circulating tumor DNA (ctDNA) test were allowed after study enrollment and prior to primary analyses. The primary endpoint was met with the combination arm of abiraterone and olaparib with longer ibPFS of 24.8 months compared to 16.6 months in the abiraterone only arm; hazard ratio, 0.66; 95% confidence interval [CI], 0.54 to 0.81; P<0.001) and improvement in the blinded independent central review (BICR) at 27.6 months in the combination arm compared to 16.4 months in the abiraterone and placebo arms.80 Further exploratory analyses revealed benefits across subgroups in both homologous recombination repair mutation (HRRm) versus non-HRRm groups, though hazard ratios approaching 1 in the non-HRRm group, with ibPFS by investigator assessment: hazard ratio, 0.50; 95% CI, 0.34 to 0.73 in the HRRm and non-HRRm: hazard ratio, 0.76; 95% CI, 0.60 to 0.97. Therefore, the US FDA granted approval for the combination restricting the use of combination olaparib and abiraterone to adult patients with deleterious or suspected deleterious BRCA-mutated metastatic castration-resistant prostate cancer (see Table 2).83 MAGNITUDE was a phase III trial that randomized patients with mCRPC to niraparib and abiraterone with prednisone compared to abiraterone and prednisone for a pre-defined HRRm patient population (n = 423) and an HRRm negative population (n=247). One of the differences in the two trials was the HRR biomarker status was determined prior to random assignment. HRRm status was also further stratified into BRCA 1/2 status versus other HRRm alterations. The primary results showed significantly longer rPFS in the combination of niraparib with abiraterone/prednisone at 16.5 months compared to abiraterone/prednisone at 13.7 months; HR, 0.73; 95% CI, 0.56 to 0.96; P = 0.022). Significant differences were seen for the BRCA 1/2 subgroup with mPFS of 16.6 months in the niraparib with abiraterone/prednisone subgroup compared to abiraterone/prednisone at only 10.9 months; HR, 0.53; 95% CI, 0.36 to 0.79; P = 0.001), suggesting a greater magnitude of benefit in the BRCA 1/2 subset, leading to US FDA approval that is similar to the olaparib with abiraterone approval in adult patients with mCRPC who harbor deleterious or suspected deleterious BRCA mutations,84 though formulation of two drugs in one pill might lead to easier adherence and access to the drug. It is noteworthy that the HRRm negative cohort of 233 patients was halted early by the IDMC after meeting a pre-specified futility analysis, a composite endpoint with a hazard ratio of 1.09, 95% CI [0.75, 1.59] suggesting lack of benefit in the HRRm-negative population. On the other hand, another phase III trial TALAPRO-2 has the broadest indication of all HRRm patients with mCRPC, since it randomized 805 patients in a 1:1 fashion based on HRR gene alterations in tumor tissue to either talazoparib with enzalutamide versus enzalutamide with placebo. HRRm included the following 12 genes: BRCA1, BRCA2, PALB2, ATM, ATR, CHEK2, FANCA, RAD51C, NBN, MLH1, MRE11A, and CDK12. The primary endpoint of rPFS as determined by BICR in the ITT population was met, with a median rPFS of not reached after a median follow-up of 25 months (95% CI 27.5 months–not reached) for talazoparib plus enzalutamide compared to 21.9 months (16.6–25.1) for the enzalutamide arm (HR 0.63; 95% CI 0.51–0.78; p < 0.0001), which led to the US FDA approval for this combination in the HRRm population.85 Combination ARPI and PARPi regimens appear to show PFS benefit particularly in the BRCA 1/2 positive prostate cancer patients. The combination of talazoparib with enzalutamide, specifically, appears to be active for all HRRm patients, although for the BRCA1/2 subgroup population, the HR was 0.23 (0.10–0.53; p = 0.0002) suggesting further benefit.82
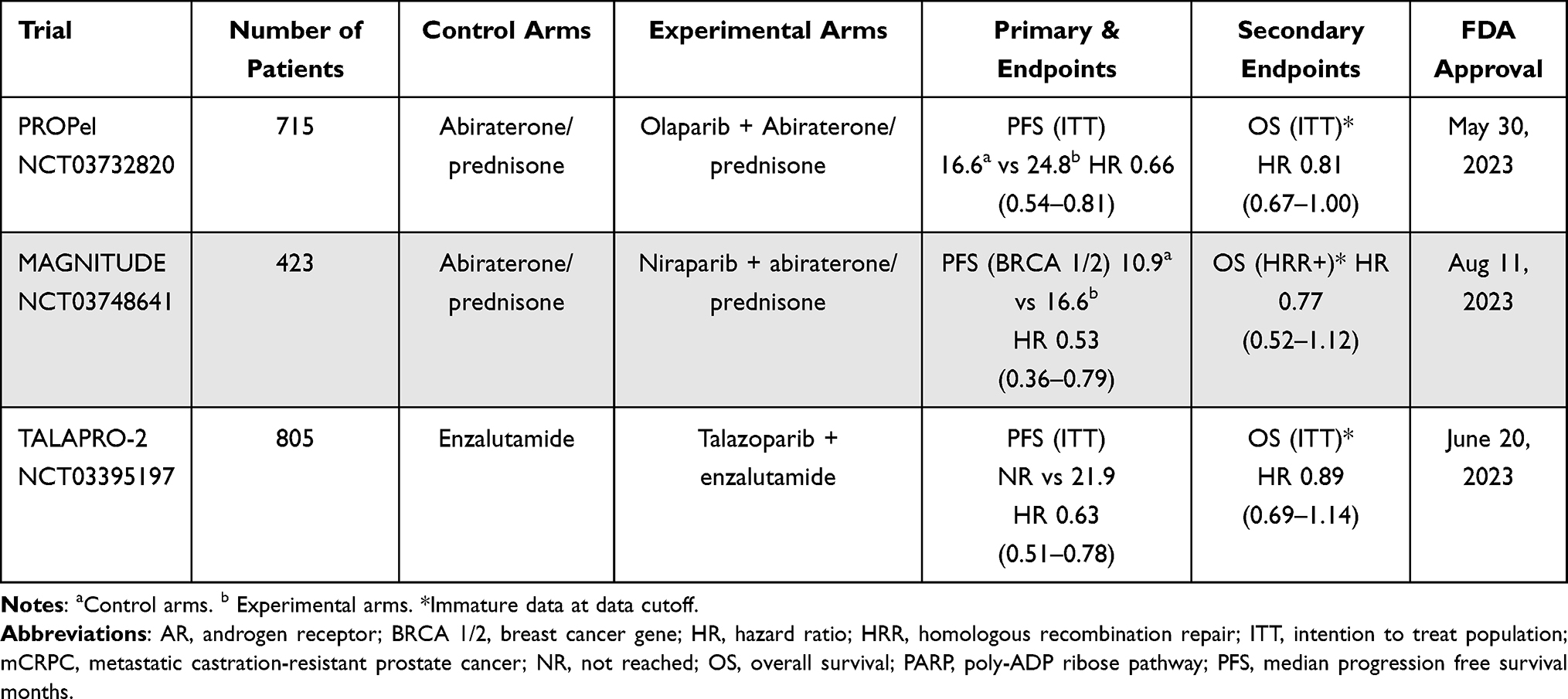 |
Table 2 Phase III mCRPC Trials Co-Targeting AR and PARP |
While the combination of ARPI and PARPi yields promising results, side effects are worthy of mention, including the higher incidence of anemia and pulmonary embolism in the PROpel trial, and 39% of patients required transfusion of blood products in the TALAPRO-2 trial. In addition, there was an incidence of myelodysplasia (<1%) and acute myelogenous leukemia (<1%) in the TALAPRO-2 trial with 4% incidence of venous thromboembolism. Therefore, as with most cancer therapies, comorbid conditions and baseline patient factors need to be carefully considered with these treatment regimens.
Conclusions
Understanding the pathways involved in the natural progression of prostate cancer provides the impetus for discovering unique mechanisms of hormone-resistance and metastases and developing targeted therapies that are highly efficacious and safe. We have taken advantage of the pathways and mechanisms we know and understand so far, which has led to the development and approval of targeted therapies in metastatic prostate cancer, including PARP inhibitors. One significant limitation of the aforementioned trials evaluating PARP inhibitor therapy in mCRPC is that the studied patient populations were mostly ARPI-naïve. So, the question of whether a doublet regimen with a PARP inhibitor plus an ARPI post-progression on an ARPI in the mCSPC setting is beneficial remains unanswered. Furthermore, targeting other pathways, such as PI3K, has not yet been proven to be successful to date, though early phase data on capivasertib are promising and phase III trials are underway. However, that there are an infinite number of opportunities for further progress in prostate cancer therapeutics ahead of us, yet to be discovered. Continued efforts to better define specific prostate cancer patient populations and minimize drug toxicities remain important considerations as we try to advance and individualize the care of our prostate cancer patients.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Acknowledgement using the David Wohlscheid Fund.
Disclosure
Dr Jeanny Aragon-Ching has served as a member of Advisory Board/speaker’s Bureau for Pfizer, EMD Serono, Astellas/Seagen, BMS, Exelixis, AZD, and AVEO, during the conduct of the study. The other authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48. doi:10.3322/caac.21763
2. Freedland SJ, Humphreys EB, Mangold LA, et al. Risk of prostate cancer–specific mortality following biochemical recurrence after radical prostatectomy. JAMA. 2005;294(4):433. doi:10.1001/jama.294.4.433
3. Parker CC, James ND, Brawley CD, et al. Radiotherapy to the prostate for men with metastatic prostate cancer in the UK and Switzerland: long-term results from the STAMPEDE randomised controlled trial. PLoS Med. 2022;19(6):e1003998. doi:10.1371/journal.pmed.1003998
4. Bossi A, Foulon S, Maldonado X, et al. Prostate irradiation in men with de novo, low-volume, metastatic, castration-sensitive prostate cancer (mCSPC): results of PEACE-1, a phase 3 randomized trial with a 2x2 design. JCO. 2023;41(17_suppl):LBA5000–LBA5000. doi:10.1200/JCO.2023.41.17_suppl.LBA5000
5. Liu M, Yang J, Xu B, Zhang X. Tumor metastasis: mechanistic insights and therapeutic interventions. MedComm (2020). 2021;2(4):587–617. doi:10.1002/mco2.100
6. Paget S. The distribution of secondary growths in cancer of the breast. The Lancet. 1889;133(3421):571–573. doi:10.1016/S0140-6736(00)49915-0
7. Chu GCY, Chung LWK, Gururajan M, et al. Regulatory signaling network in the tumor microenvironment of prostate cancer bone and visceral organ metastases and the development of novel therapeutics. Asian J Urol. 2019;6(1):65–81. doi:10.1016/j.ajur.2018.11.003
8. Odero-Marah V, Hawsawi O, Henderson V, Sweeney J. Epithelial-Mesenchymal Transition (EMT) and prostate cancer. Adv Exp Med Biol. 2018;1095:101–110. doi:10.1007/978-3-319-95693-0_6
9. Goel S, Bhatia V, Biswas T, Ateeq B. Epigenetic reprogramming during prostate cancer progression: a perspective from development. Semin Cancer Biol. 2022;83:136–151. doi:10.1016/j.semcancer.2021.01.009
10. López J, Añazco-Guenkova AM, Monteagudo-García Ó, Blanco S. Epigenetic and epitranscriptomic control in prostate cancer. Genes (Basel). 2022;13(2):378. doi:10.3390/genes13020378
11. Chaves LP, Melo CM, Saggioro FP, Dos Reis RB, Squire JA. Epithelial–mesenchymal transition signaling and prostate cancer stem cells: emerging biomarkers and opportunities for precision therapeutics. Genes (Basel). 2021;12(12):1900. doi:10.3390/genes12121900
12. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15(2):117–134. doi:10.1007/s10911-010-9178-9
13. Feinberg AP, Levchenko A. Epigenetics as a mediator of plasticity in cancer. Science. 2023;379(6632):eaaw3835. doi:10.1126/science.aaw3835
14. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–1449. doi:10.1038/nm.3336
15. Zhang S, Shen T, Zeng Y. Epigenetic modifications in prostate cancer metastasis and microenvironment. Cancers. 2023;15(8):2243. doi:10.3390/cancers15082243
16. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. doi:10.1038/nbt.1685
17. Skvortsova K, Stirzaker C, Taberlay P. The DNA methylation landscape in cancer. Blewitt M, ed. Essays Biochem. 2019;63(6):797–811. doi:10.1042/EBC20190037
18. Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286(5439):481–486. doi:10.1126/science.286.5439.481
19. Sjöström M, Zhao SG, Levy S, et al. The 5-hydroxymethylcytosine landscape of prostate cancer. Cancer Res. 2022;82(21):3888–3902. doi:10.1158/0008-5472.CAN-22-1123
20. Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi:10.1038/nature01075
21. Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. doi:10.1038/nature04020
22. Gaughan L, Stockley J, Wang N, et al. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011;39(4):1266–1279. doi:10.1093/nar/gkq861
23. Ezponda T, Popovic R, Shah MY, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial–mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2013;32(23):2882–2890. doi:10.1038/onc.2012.297
24. Whitmore WF. Natural history and staging of prostate cancer. Urol Clin North Am. 1984;11(2):205–220. doi:10.1016/S0094-0143(21)00182-8
25. Akoto T, Saini S. Role of exosomes in prostate cancer metastasis. Int J Mol Sci. 2021;22(7):3528. doi:10.3390/ijms22073528
26. Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clin Oncol. 2011;29(27):3659–3668. doi:10.1200/JCO.2011.35.1916
27. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi:10.1016/j.cell.2015.05.001
28. Hieronymus H, Sawyers CL. Traversing the genomic landscape of prostate cancer from diagnosis to death. Nat Genet. 2012;44(6):613–614. doi:10.1038/ng.2301
29. Mateo J, Seed G, Bertan C, et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J Clin Invest. 2020;130(4):1743–1751. doi:10.1172/JCI132031
30. Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305. doi:10.1038/nm.4045
31. Shah RB, Shore KT, Yoon J, Mendrinos S, McKenney JK, Tian W. PTEN loss in prostatic adenocarcinoma correlates with specific adverse histologic features (intraductal carcinoma, cribriform Gleason pattern 4 and stromogenic carcinoma). Prostate. 2019;79(11):1267–1273. doi:10.1002/pros.23831
32. Armstrong CWD, Maxwell PJ, Ong CW, et al. PTEN deficiency promotes macrophage infiltration and hypersensitivity of prostate cancer to IAP antagonist/radiation combination therapy. Oncotarget. 2016;7(7):7885–7898. doi:10.18632/oncotarget.6955
33. Deplus R, Delliaux C, Marchand N, et al. TMPRSS2-ERG fusion promotes prostate cancer metastases in bone. Oncotarget. 2016;8(7):11827–11840. doi:10.18632/oncotarget.14399
34. Chiarugi P, Paoli P, Cirri P. Tumor microenvironment and metabolism in prostate cancer. Semin Oncol. 2014;41(2):267–280. doi:10.1053/j.seminoncol.2014.03.004
35. Peinado H, Zhang H, Matei IR, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017;17(5):302–317. doi:10.1038/nrc.2017.6
36. Levesque C, Nelson PS. Cellular constituents of the prostate stroma: key contributors to prostate cancer progression and therapy resistance. Cold Spring Harb Perspect Med. 2018;8(8):a030510. doi:10.1101/cshperspect.a030510
37. Bonollo F, Thalmann GN, Kruithof-de Julio M, Karkampouna S. The role of cancer-associated fibroblasts in prostate cancer tumorigenesis. Cancers. 2020;12(7):1887. doi:10.3390/cancers12071887
38. Thalmann GN, Rhee H, Sikes RA, et al. Human prostate fibroblasts induce growth and confer castration resistance and metastatic potential in LNCaP cells. Eur Urol. 2010;58(1):162–172. doi:10.1016/j.eururo.2009.08.026
39. Kang J, La Manna F, Bonollo F, et al. Tumor microenvironment mechanisms and bone metastatic disease progression of prostate cancer. Cancer Lett. 2022;530:156–169. doi:10.1016/j.canlet.2022.01.015
40. Zhang Z, Karthaus WR, Lee YS, et al. Tumor microenvironment-derived NRG1 promotes antiandrogen resistance in prostate cancer. Cancer Cell. 2020;38(2):279–296.e9. doi:10.1016/j.ccell.2020.06.005
41. Ottewell PD, Wang N, Meek J, et al. Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocr Relat Cancer. 2014;21(5):769–781. doi:10.1530/ERC-14-0199
42. Sun Y, Campisi J, Higano C, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18(9):1359–1368. doi:10.1038/nm.2890
43. Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5(1):21–28. doi:10.1038/nrc1528
44. Perez-Stable C, Altman NH, Mehta PP, Deftos LJ, Roos BA. Prostate cancer progression, metastasis, and gene expression in transgenic mice. Cancer Res. 1997;57(5):900–906.
45. Arriaga JM, Panja S, Alshalalfa M, et al. A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance. Nat Cancer. 2020;1(11):1082–1096. doi:10.1038/s43018-020-00125-0
46. Lunardi A, Ala U, Epping MT, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet. 2013;45(7):747–755. doi:10.1038/ng.2650
47. Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–2738. doi:10.4049/jimmunol.172.5.2731
48. Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. A pilot study of mobilization and treatment of disseminated tumor cells in men with metastatic prostate cancer. clinicaltrials.gov; 2019. Available from: https://clinicaltrials.gov/study/NCT02478125.
49. Martine LC, Holzapfel BM, McGovern JA, et al. Engineering a humanized bone organ model in mice to study bone metastases. Nat Protoc. 2017;12(4):639–663. doi:10.1038/nprot.2017.002
50. Bock N, Kryza T, Shokoohmand A, et al. In vitro engineering of a bone metastases model allows for study of the effects of antiandrogen therapies in advanced prostate cancer. Sci Adv. 2021;7(27):eabg2564. doi:10.1126/sciadv.abg2564
51. McGovern JA, Bock N, Shafiee A, et al. A humanized orthotopic tumor microenvironment alters the bone metastatic tropism of prostate cancer cells. Commun Biol. 2021;4(1):1–14. doi:10.1038/s42003-021-02527-x
52. Pungsrinont T, Kallenbach J, Baniahmad A. Role of PI3K-AKT-mTOR pathway as a pro-survival signaling and resistance-mediating mechanism to therapy of prostate cancer. Int J Mol Sci. 2021;22(20):11088. doi:10.3390/ijms222011088
53. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. 2020;21(12):4507. doi:10.3390/ijms21124507
54. Saal LH, Johansson P, Holm K, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci. 2007;104(18):7564–7569. doi:10.1073/pnas.0702507104
55. Jamaspishvili T, Berman DM, Ross AE, et al. Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol. 2018;15(4):222–234. doi:10.1038/nrurol.2018.9
56. Blanco-Aparicio C, Renner O, Leal JFM, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28(7):1379–1386. doi:10.1093/carcin/bgm052
57. Haddadi N, Lin Y, Travis G, Simpson AM, Nassif NT, McGowan EM. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol Cancer. 2018;17(1):37. doi:10.1186/s12943-018-0803-3
58. Choudhury AD. PTEN-PI3K pathway alterations in advanced prostate cancer and clinical implications. Prostate. 2022;82(Suppl 1):S60–S72. doi:10.1002/pros.24372
59. Cham J, Venkateswaran AR, Bhangoo M. Targeting the PI3K-AKT-mTOR pathway in castration resistant prostate cancer: a review article. Clin Genitourin Cancer. 2021;19(6):563.e1–563.e7. doi:10.1016/j.clgc.2021.07.014
60. de Bono JS, De Giorgi U, Rodrigues DN, et al. Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN Loss. Clin Cancer Res. 2019;25(3):928–936. doi:10.1158/1078-0432.CCR-18-0981
61. Sweeney C, Bracarda S, Sternberg CN, et al. Ipatasertib plus Abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. The Lancet. 2021;398(10295):131–142. doi:10.1016/S0140-6736(21)00580-8
62. Crabb SJ, Griffiths G, Dunkley D, et al. Overall survival update for patients with metastatic castration-resistant prostate cancer treated with capivasertib and docetaxel in the phase 2 ProCAID clinical trial. Eur Urol. 2022;82(5):512–515. doi:10.1016/j.eururo.2022.05.019
63. Fizazi K, George DJ, De Santis M, et al. A phase III trial of capivasertib and Abiraterone versus placebo and Abiraterone in patients with de novo metastatic hormone-sensitive prostate cancer characterized by PTEN deficiency (CAPItello-281). JCO. 2021;39(6_suppl):TPS178–TPS178. doi:10.1200/JCO.2021.39.6_suppl.TPS178
64. Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477(1):97–110. doi:10.1016/S0027-5107(01)00111-7
65. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi:10.1038/nature03443
66. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi:10.1038/nature03445
67. De Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091–2102. doi:10.1056/NEJMoa1911440
68. Smith MR, Scher HI, Sandhu S, et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2022;23(3):362–373. doi:10.1016/S1470-2045(21)00757-9
69. Abida W, Patnaik A, Campbell D, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. 2020;38(32):3763–3772. doi:10.1200/JCO.20.01035
70. Fizazi K, Piulats JM, Reaume MN, et al. Rucaparib or physician’s choice in metastatic prostate cancer. N Engl J Med. 2023;388(8):719–732. doi:10.1056/NEJMoa2214676
71. Polkinghorn WR, Parker JS, Lee MX, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3(11):1245–1253. doi:10.1158/2159-8290.CD-13-0172
72. Castro E, Goh C, Leongamornlert D, et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur Urol. 2015;68(2):186–193. doi:10.1016/j.eururo.2014.10.022
73. Chakraborty G, Armenia J, Mazzu YZ, et al. Significance of BRCA2 and RB1 co-loss in aggressive prostate cancer progression. Clin Cancer Res. 2020;26(8):2047–2064. doi:10.1158/1078-0432.CCR-19-1570
74. Schiewer MJ, Goodwin JF, Han S, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2(12):1134–1149. doi:10.1158/2159-8290.CD-12-0120
75. Li L, Karanika S, Yang G, et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal. 2017;10(480):eaam7479. doi:10.1126/scisignal.aam7479
76. Asim M, Tarish F, Zecchini HI, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8(1):374. doi:10.1038/s41467-017-00393-y
77. Hussain M, Daignault-Newton S, Twardowski PW, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. JCO. 2018;36(10):991–999. doi:10.1200/JCO.2017.75.7310
78. Brenner JC, Ateeq B, Li Y, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19(5):664–678. doi:10.1016/j.ccr.2011.04.010
79. Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362ps17–362ps17. doi:10.1126/scitranslmed.aaf9246
80. Clarke NW, Armstrong AJ, Thiery-Vuillemin A, et al. Abiraterone and olaparib for metastatic castration-resistant prostate cancer. NEJM Evidence. 2022;1(9):EVIDoa2200043. doi:10.1056/EVIDoa2200043
81. Niraparib and abiraterone acetate for metastatic castration-resistant prostate cancer | Journal of Clinical Oncology. Available from: https://ascopubs.org/doi/10.1200/JCO.22.01649?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed.
82. Agarwal N, Azad AA, Carles J, et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): a randomised, placebo-controlled, phase 3 trial. The Lancet. 2023;402(10398):291–303. doi:10.1016/S0140-6736(23)01055-3
83. Research C for DE and. FDA D.I.S.C.O. Burst Edition: FDA approval of Lynparza (olaparib), with Abiraterone and prednisone, for BRCA-mutated metastatic castration-resistant prostate cancer. FDA; July 13, 2023. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-disco-burst-edition-fda-approval-lynparza-olaparib-abiraterone-and-prednisone-brca-mutated.
84. Research C for DE and. FDA approves niraparib and Abiraterone acetate plus prednisone for BRCA-mutated metastatic castration-resistant prostate cancer. FDA; August 11, 2023. Available from: https://cacmap.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-and-abiraterone-acetate-plus-prednisone-brca-mutated-metastatic-castration.
85. Research C for DE and. FDA approves talazoparib with enzalutamide for HRR gene-mutated metastatic castration-resistant prostate cancer. FDA; June 20, 2023. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-talazoparib-enzalutamide-hrr-gene-mutated-metastatic-castration-resistant-prostate.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.