")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 16
In silico Study of Antiviral Activity of Polyphenol Compounds from Ocimum basilicum by Molecular Docking, ADMET, and Drug-Likeness Analysis
Authors Kurnia D , Putri SA , Tumilaar SG, Zainuddin A, Dharsono HDA , Amin MF
Received 11 January 2023
Accepted for publication 15 April 2023
Published 26 April 2023 Volume 2023:16 Pages 37—47
DOI https://doi.org/10.2147/AABC.S403175
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Dikdik Kurnia,1 Salsabila Aqila Putri,1 Sefren Geiner Tumilaar,1 Achmad Zainuddin,1 Hendra Dian Adhita Dharsono,2 Meiny Faudah Amin3
1Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Sumedang, West Java, Indonesia; 2Department of Conservative Dentistry, Faculty of Dentistry, Universitas Padjadjaran, Sumedang, West Java, Indonesia; 3Dental Conservation, Faculty of Dentistry, Trisakti University, Jakarta, Indonesia
Correspondence: Dikdik Kurnia, Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Sumedang, West Java, 45363, Indonesia, Tel/Fax +62-22-7794391, Email [email protected]
Aim: The SARS-CoV-2 virus is a disease that has mild to severe effects on patients, which can even lead to death. One of the enzymes that act as DNA replication is the main protease, which becomes the main target in the inhibition of the SARS-CoV-2 virus. In finding effective drugs against this virus, Ocimum basilicum is a potential herbal plant because it has been tested to have high phytochemical content and bioactivity. Apigenin-7-glucuronide, dihydrokaempferol-3-glucoside, and aesculetin are polyphenolic compounds found in Ocimum basilicum.
Purpose: The purpose of this study was to analyze the mechanism of inhibition of the three polyphenolic compounds in Ocimum basilicum against the main protease and to predict pharmacokinetic activity and the drug-likeness of a compound using the Lipinski Rule of Five.
Patients and Methods: The method used is to predict the molecular docking inhibition mechanism using Autodock 4.0 tools and use pkcsm and protox online web server to analyze ADMET and Drug-likeness.
Results: The binding affinity for apigenin-7-glucuronide was − 8.77 Kcal/mol, dihydrokaempferol-3-glucoside was − 8.96 Kcal/mol, and aesculetin was − 5.79 Kcal/mol. Then, the inhibition constant values were 375.81 nM, 270.09 nM, and 57.11 μM, respectively. Apigenin-7-glucuronide and dihydrokaempferol-3-glucoside bind to the main protease enzymes on the active sites of CYS145 and HIS41, while aesculetin only binds to the active sites of CYS145. On ADMET analysis, these three compounds met the predicted pharmacokinetic parameters, although there are some specific parameters that must be considered especially for aesculetin compounds. Meanwhile, on drug-likeness analysis, apigenin-7-glucuronide and dihydrokaempferol-3-glucoside compounds have one violation and aesculetin have no violation.
Conclusion: Based on the data obtained, Apigenin-7-glucuronide and dihydrokaempferol-3-glucoside are compounds that have more potential to have an antiviral effect on the main protease enzyme than aesculetin. Based on pharmacokinetic parameters and drug-likeness, three compounds can be used as lead compounds for further research.
Keywords: Apigenin-7-glucuronide, aesculetin, dihydrokaempferol-3-glucoside, main protease, Ocimum basilicum
Introduction
COVID-19 is a global disease that began at the end of 2019. In January 2022, the World Health Organization confirmed that the cumulative cases of COVID-19 were 323,610,370 with a cumulative death of 5,529,693.1 SARS-CoV-2 is one of seven types of coronaviruses that cause severe lower respiratory tract dysfunction.2 This virus will bind to ACE2 as the main receptor and spread through the innate immune response in humans. This is a disease that attacks the respiratory tract, which is a mild disease in about 80% of patients. As for about 20% of patients, it can become a severe disease. In a study of 292 COVID-19 patients in Wuhan, there were elderly patients who had an increased risk of 15.15%. Then, congenital diseases such as hypertension, malignant tumors, chronic obstructive pulmonary disease, coronary heart disease, and chronic kidney disease will also be dangerous. This can lead to death if the patient is elderly and has comorbidities. Fifty-one patients out of 145 cases with comorbidities were reported to have died and 90.2% of them were 60 years old.3–5
Viruses have structural and non-structural proteins, which are classified according to their function. Structural proteins serve as protection against host enzymes so that DNA is not degraded as in the nuclear capsid. While a non-structural protein, the main protease (Mpro), is a cysteine protease enzyme that functions for replication such as chymotrypsin.6,7 The main protease (Mpro) is an important enzyme that functions as the replication of the corona virus (CoV). It has been identified that Mpro homologues are absent in humans.8,9 Therefore, research on Mpro inhibitors is very potential and effective because it does not cause side effects on human proteases.10 Currently, vaccines are the main solution to prevent COVID-19. However, a good immune system is important to fight the SARS-CoV-2 virus. Researchers and health professionals are still looking for therapeutic treatments and vaccines that have been promised to date.11 One of the complementary and alternative medicines that has been widely developed is herbal products as immunomodulators for the prevention and treatment of Covid-19 disease.12
Ocimum basilicum is one of the potential herbal plants. Its essential oils have been used in dental and oral products, foodstuffs, and fragrances for many years.13 O. basilicum contains phytochemical compounds such as alkaloids, tannins, flavonoids, and saponins. O. basilicum is known to have anti-inflammatory, antioxidant, and antimicrobial activity.14 A study of antiherpatic activity, aqueous extract of O. basilicum showed EC50 of 90.9 mg/L, ethanol extract of 108.3 mg/L, linalool of more than 200 mg/L, apigenin of 6.7 mg/L, and ursolic acid of 6,6 mg/L. They have also been tested against antiadenovirals with EC50 values of 174.1, over 1000, 24.4, 11.1, and more than 200 mg/L, respectively.13 Polyphenol compounds are known to reduce low-level inflammation thereby boosting the immune system. This makes polyphenols a good antiseptic to prevent the SARS-CoV-2 virus.15 O. basilicum contains several polyphenols such as apigenin-7-glucuronide, dihydrokaempferol-3-glucoside, and aesculetin. To investigate the antiviral activity of the three polyphenolic compounds, a molecular docking approach can be used. This method is one of the in silico studies that uses Computer Aided Drug Design (CADD) so that it can predict the interaction of natural compounds that act as ligands to target receptors.16,17 This study aims to test the inhibition of the main protease enzyme in the SARS-CoV-2 virus using these three compounds in silico.
Materials and Methods
Materials
Materials which used for in silico assay were three dimensions structure of Main protease (Mpro) enzyme with PDB ID 6LU7 obtained from RCSB protein data bank (https://www.rcsb.org/). The chemical structure of O. basilicum was obtained from PubChem compound database (https://pubchem.ncbi.nlm.nih.gov/) with the compound IDs 1 for A, 2 for B, and 3 for C. The structure of ritonavir as a positive control was also obtained from PubChem compound database with ID of 4.
In silico Assay Against SARS-CoV-2 Enzyme
Compounds apigenin-7-glucuronide, dihydrokaempferol-3-glucoside, and aesculetin were obtained from the PubChem compound database, and then the three-dimensional structures were converted using ChemDraw 3D format and saved as PDB format. The Mpro enzyme was extracted from the RCSB protein database with PDB ID 6LU7 and saved as PDB format. The active site determination on the receptor was obtained from the Computed Atlas of Surface Topography of proteins (CASTp) web server. In silico test was analyzed by molecular docking method. The tools used are Autodock 4.0. The Mpro enzyme was isolated from its native ligand by the BIOVIA discovery studio program and saved in PDB format. Mpro enzymes free from native ligands were molecularly anchored to apigenin-7-glucuronide, dihydrokaempferol-3-glucoside, aesculetin, and ritonavir using Autodock 4.0. Receptors and ligands stored in pdbqt format were opened to set grid box and docking area. Then, it is saved in gpf format. Binding of receptor and ligand using genetic algorithm parameters saved in dpf format. Both files that have been saved and docked on the command prompt using Autogrid4 and Autodock4 formulas. The best conformation was selected by the lowest binding affinity. Then, the interaction of Mpro enzyme with the three compounds was visualized using BIOVIA discovery studio program.18
ADMET and Drug-Likeness Analysis
Absorption, distribution, metabolism, and excretion (ADME) can be predicted using the online web server pkCSM (http://biosig.unimelb.edu.au/pkcsm/) while toxicity predictions and Lipinski Rule of Five can be predicted using the online web server (https://tox-new.charite.de/protox_II/).19,20
Results
Prediction mechanism of antiviral compounds to Mpro enzyme was carried out using Autodock 4.0 tools. Table 1 shows binding affinity of apigenin-7-glucuronide is −8.77 Kcal/mol, dihydrokaempferol-3-glucoside is −8.96 Kcal/mol, and aesculetin is −5.79 Kcal/mol, while the positive control, ritonavir is −9.77 Kcal/mol. The value of binding affinity showed the correlation to inhibition constant (Ki), where the more negative the binding affinity, the better the Ki value. Apigenin-7-glucuronide, dihydrokaempferol-3-glucoside, and aesculetin showed the Ki value of 375.81 nM, 270.09 nM, and 57.11 µM, respectively, while ritonavir was 68.40 nM.
 |
Table 1 Prediction of Antiviral Activity Against Mpro Enzyme |
Furthermore, to identify the interaction between residues of Mpro enzyme and inhibitor compounds, it was visualized using BIOVIA discovery studio program. Based on Table 2, apigenin-7-glucuronide had several interactions to Mpro enzyme, which are conventional and carbon hydrogen bonds, Pi-alkyl, Pi–Pi T-shaped, Pi-lone pair, unfavorable donor–donor, and Van der Waals. It had conventional hydrogen bonds to glutamic acid (GLU A:166), histidine (HIS A:163), serine (SER A:144), glycine (GLY A:143), and threonine (THR A:190). Then, it had carbon hydrogen bonds to histidine (HIS A:172).
 |
Table 2 Molecular Interaction Between Apigenin-7-Glucuronide and Mpro Enzyme |
Table 3 shows the molecular interaction between dihydrokaempferol-3-glucoside and residues of Mpro enzyme. It interacts by conventional and carbon hydrogen bonds, Pi-alkyl, Pi-cation, unfavorable donor–donor, and Van der Waals. It interacts by conventional hydrogen bonds with amino acids aspartic acid (ASP A:187), leucine (LEU A:141), glutamic acid (GLU A:166), glutamine (GLN A:192), threonine (THR A:190), and arginine (ATG A:188). Then, it had carbon hydrogen bonds to glutamic acid (GLU A:166).
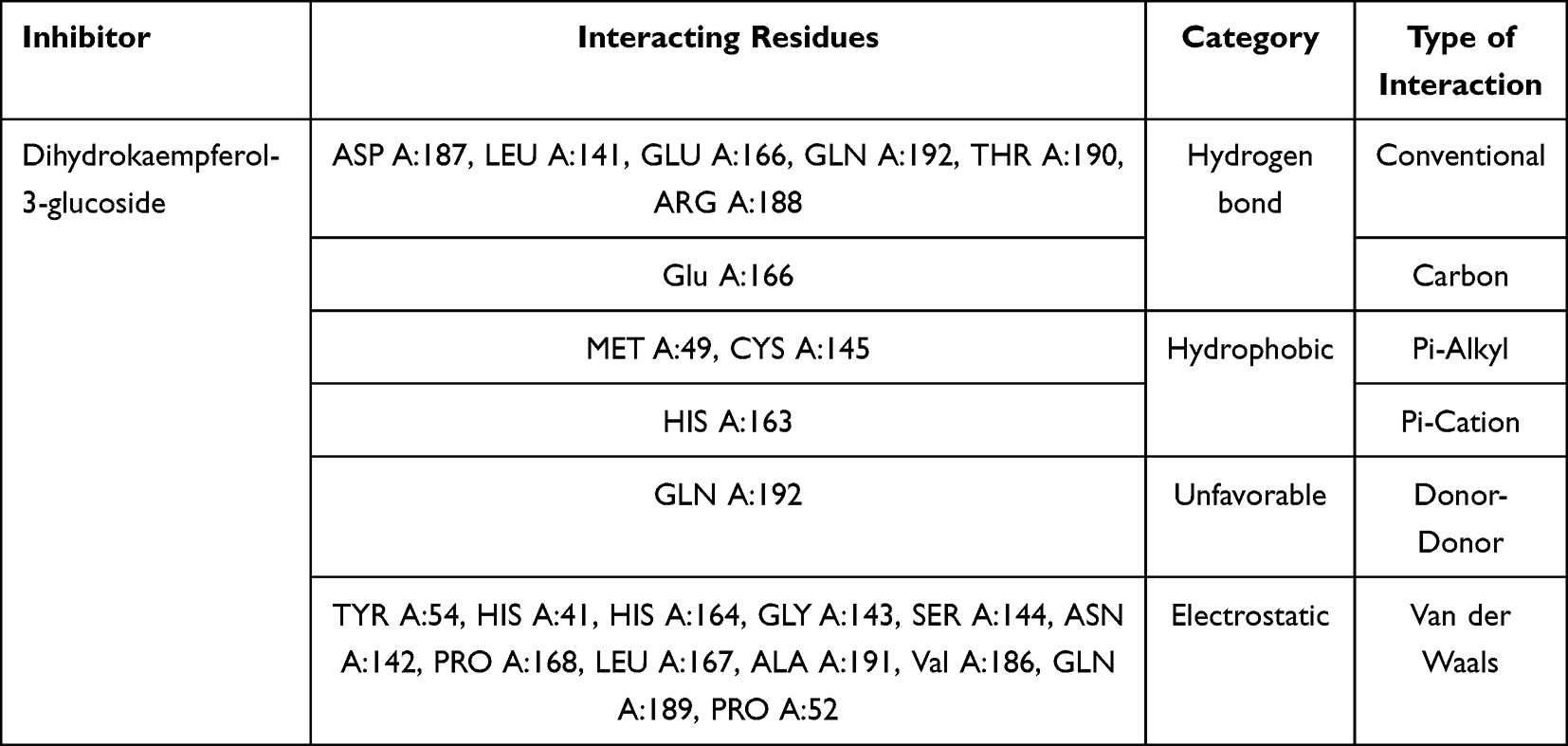 |
Table 3 Molecular Interaction Between Dihydrokaempferol- 3-Glucoside and Mpro Enzyme |
Interaction of aesculetin to Mpro enzyme residues is shown in Table 4. It had the interaction by conventional hydrogen bonds, Pi-alkyl, amide-Pi-stacked, and Van der Waals. The conventional hydrogen bonds were with histidine (HIS A:172 and HIS A:163), cystine (CYS A:145). Moreover, ritonavir as positive control had interaction of conventional and carbon hydrogen bonds, Pi-sulfur, sulfur-x, amide-Pi stacked, Pi-alkyl, and Van der Waals. Ritonavir interacts with glutamic acid (GLU A:166) and glutamine (GLN A:189) by conventional hydrogen bonds, to leucine (LEU A:167) by carbon hydrogen bonds as shown in Table 5. Visualization of inhibitor compounds to Mpro enzyme can be seen in Figures 1 and 2.
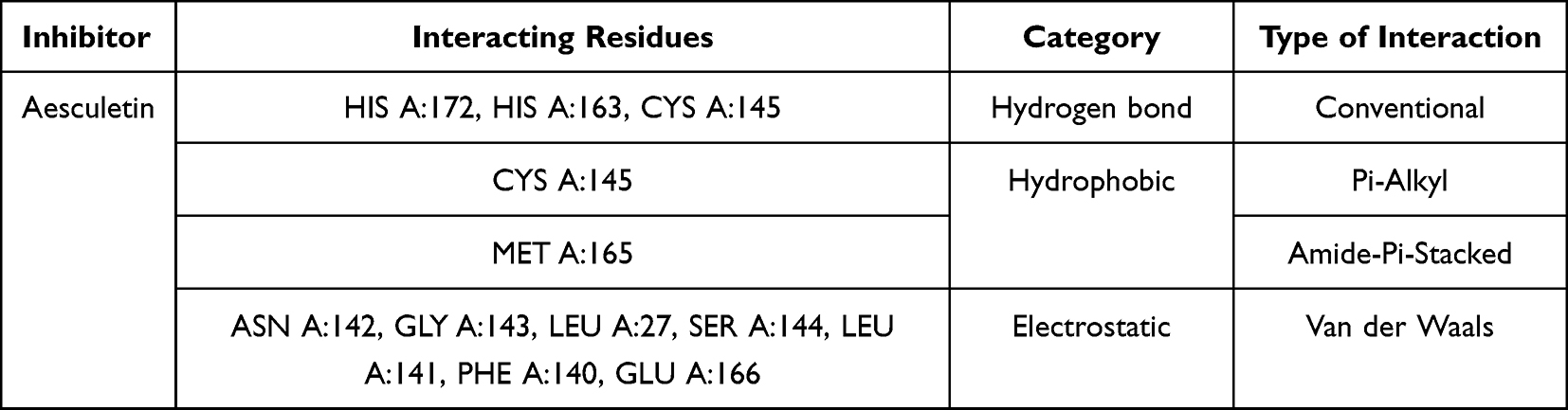 |
Table 4 Molecular Interaction Between Aesculetin and Mpro Enzyme |
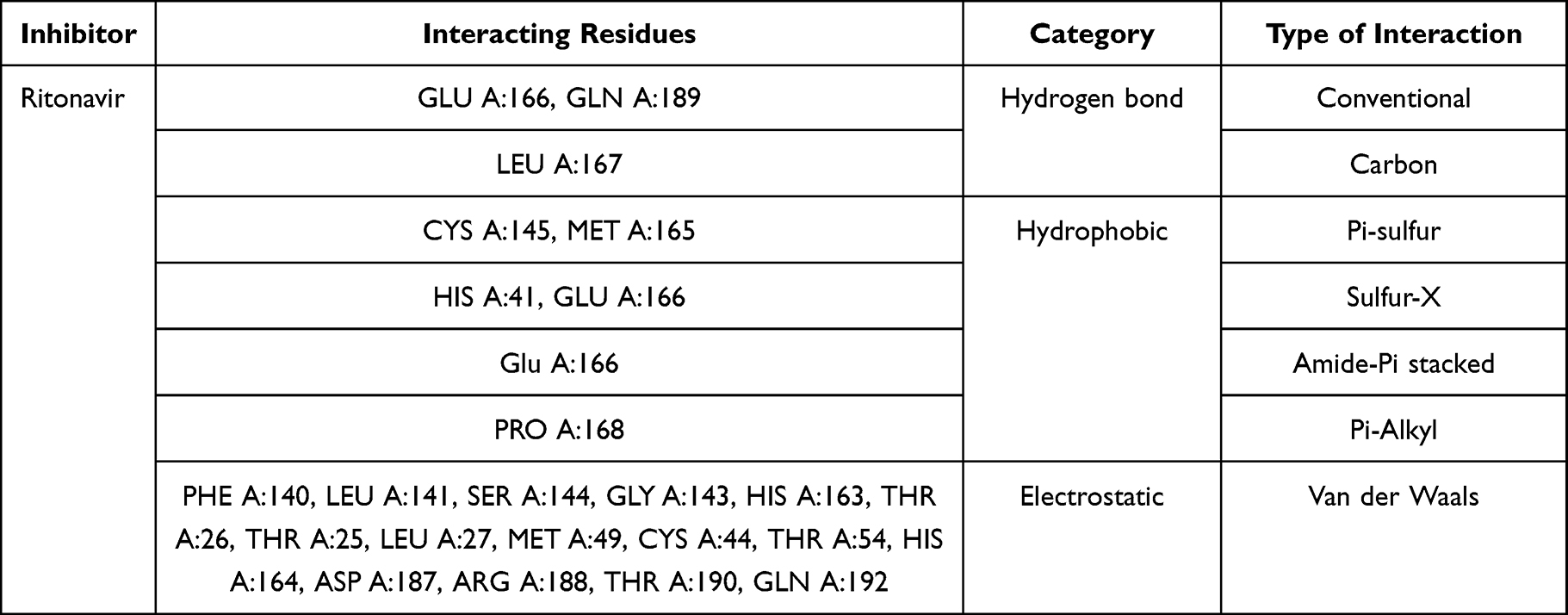 |
Table 5 Molecular Interaction Between Ritonavir and Mpro Enzyme |
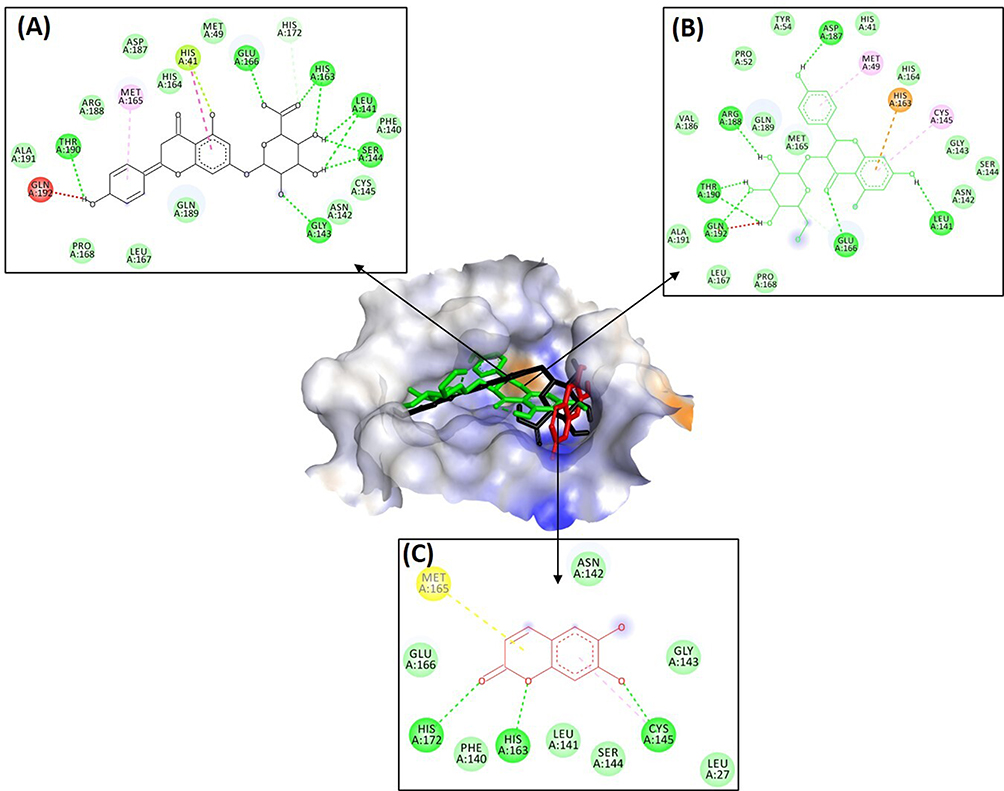 |
Figure 1 Molecular Interaction of (A) Apigenin-7-glucuronide, (B) Dihydrokaempferol-3-glucoside, and (C) Aesculetin to Mpro Enzyme. |
 |
Figure 2 Molecular Interaction of Ritonavir to Mpro Enzyme. |
For the ADMET analysis, Table 6 shows the pharmacokinetic data on the absorption, distribution, metabolism, excretion, and toxicity of apigenin-7-glucuronide, dihydrokaempferol-3-glucoside, and aesculetin. The absorption parameter consists of the category of water solubility, intestinal absorption, and distribution parameters are divided into three categories: volume distribution (VDss), BBB permeability, and CNS permeability. Metabolism parameters determined the inhibition of CYP enzymes, while the excretion parameter is seen from the total clearance value. Then, a drug-likeness analysis is shown in Table 7 which is divided into five parameters, namely molecular mass, hydrogen bond donors, hydrogen bond acceptors, LogP, and molar refractivity.
 |
Table 6 ADMET Prediction of Apigenin-7-Glucuronide, Dihydrokaempferol-3-Glucoside, Esculetin |
 |
Table 7 Analysis Results of Lipinski’s Rule of Five |
Discussion
Molecular Docking Analysis
O. basilicum is rich in polyphenols, and the total phenolic content of ethanolic extract has been tested with a value of 29.60 mg GAE/g.21 Some polyphenol compounds present in O. basilicum are apigenin-7-glucuronide, dihydrokaempferol-3-glucoside, and aesculetin.22,23 The previous study reported that apigenin-7-glucuronide has antimicrobial activity against Staphylococcus epidermidis, Escherichia coli, Klebsiella pneumoniae, Proteus vulgaris, and Pseudomonas aeruginosa.24 Dihydrokaempferol-3-glucoside was reported to have antioxidant activity of 1.0 mmol Trolox/mmol by Trolox equivalent assay.25 Then, aesculetin has been tested to have antimicrobial activity against Ralstonia solanacearum with MIC and MBC values of 192 mg/L.26 Therefore, these three compounds were selected to evaluate the mechanism of their inhibition against the Mpro enzyme. The structure of three compounds can be seen in Figure 3.
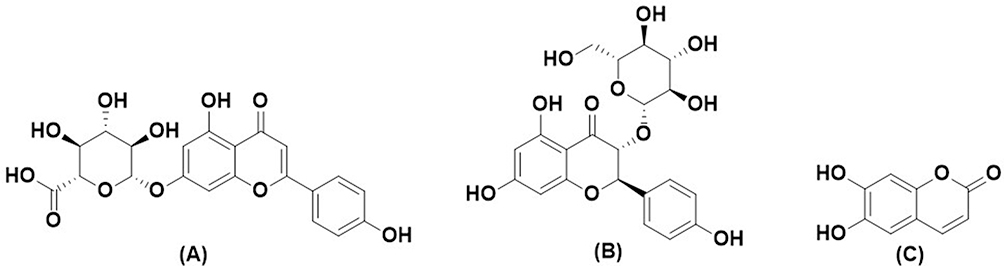 |
Figure 3 Chemical Structure of (A) Apigenin-7-glucuronide, (B) Dihydrokaempferol-3-glucoside, and (C) Aesculetin. |
In this study, ritonavir was used as positive control. It is a protease inhibitor, which is used for the treatment of HIV and has been tested to have antiviral activity against SARS-CoV viruses.27 It provides good inhibitory effect with strong binding pattern to aromatic groups.28 The binding affinity of ritonavir was used as a comparison for the three inhibitor compounds. Based on the results, apigenin-7-glucuronide and dihydrokaempferol-3-glucoside showed a better value than aesculetin. They also have binding affinity values that are not significantly different from ritonavir. Furthermore, this value was used to understand the correlation with Ki. Based on Mulu et al,29 the greater binding affinity makes the smaller Ki value, so it only requires a small amount of drug to provide antiviral activity. This value is important to show the ability of the compound to inhibit the virus, because the results of the inhibition constant correlated with the IC50 value regardless of competitive or uncompetitive kinetics.30 Based on Table 1, the inhibitor compound that has the smallest Ki value is dihydrokaempferol-3-glucoside, then apigenin-7-glucuronide, then aesculetin with the highest value. Therefore, dihydrokaempferol-3-glucoside and apigenin-7-glucuronide were more potential to be Mpro enzyme inhibitors.
Furthermore, the catalytic site of the Mpro enzyme is important to evaluate. Enzyme Mpro was obtained from RCSB Protein Data Bank with enzyme code 6LU& which has an RMSD value of 2.16 Å. The grid box area of Mpro enzyme is located at positions −10.712 (X), 12.411 (Y), and 68.831 (Z). In molecular docking using Autodock 4.0 with running 100 times showed the number of clusters apigenin-7-glucuronide (−8.77 Kcal/mol) 63 times, dihydrokaempferol-3-glucoside (−8.96 Kcal/mol) 6 times, and aesculetin (−5.79 Kcal/mol) 52 times. The Mpro enzyme has catalytic sites on CYS145 and HIS41.9,31 Apigenin-7-glucuronide is known to interact with CYS145 via Van der Waals interaction and with HIS41 through hydrophobic Pi–Pi T-shaped and Pi-lone pair interactions. Then, dihydrokaempferol-3-glucoside has the interaction with CYS145 through hydrophobic Pi–alkyl interaction and with HIS41 by Van der Waals interaction. While aesculetin just interacts to CYS145 through conventional hydrogen bond and Pi–alkyl interactions. This could be one of the reasons why aesculetin provides the lowest binding affinity. The positive control, ritonavir, also interacts to CYS145 and HIS41 through hydrophobic Pi–sulfur and sulfur–X interactions, respectively.
The strength of the interaction is also affected by the hydrogen bond to the Mpro enzyme. In drug design, hydrogen bonding is influenced by the dipole moment where the higher the dipole moment, the greater the ligand bond. In drug design, hydrogen bonding is affected by the dipole moment where the higher the dipole moment, the greater the ligand bond. This polarizability can increase the formation of covalent bonds. The size of the compound is one of the factors that increase the polarization. The larger the size of the molecule, the more polarized the compound is.32 The results in Tables 2–4 show that apigenin-7-glucuronide and dihydrokaempferol-3-glucoside interact more with amino acids in Mpro enzyme by hydrogen bonding than aesculetin. This is influenced by the presence of a hydroxyl group in the molecule. Apigenin-7-glucuronide and dihydrokaempferol-3-glucoside have more hydroxyl groups than aesculetin, thus providing more potent antiviral activity. It was reported that the antiviral mechanism is provided by the number of hydroxyl groups present in the benzene ring.33–35
ADMET and Drug-Likeness Analysis
Currently, computational studies are in the modeling stage of studying the drug’s pharmacokinetics, including absorption, distribution, metabolism, excretion, and toxicity (ADMET).36 Pharmacokinetics prediction of a compound needs to be seen using computational studies because this method is more cost-effective and more efficient. Bioactive compounds that have been isolated from plants can be said to be lead compounds if they have a good ADMET profile.37
In the absorption parameter, there are two things that need to be considered, namely water solubility and intestinal absorption. Water solubility is a key factor in the process of drug bioavailability.38 Solubility in water is categorized as the best if it is in the range of values less than 0 and greater than −0.5.39 In this study, the water solubility of these three compounds is within the specified range. Based on research from Khan et al,40 the best average intestinal absorption of a compound is above 80%. Aesculetin has an intestinal absorption value of 86.29%, which indicates that the compound is well absorbed in the intestines, while apigenin-7-glucuronide and dihydrokaempferol-3-glucoside have a poor intestinal absorption value of 15.25% and 36.32%, respectively. Prediction of drug or lead compound distribution in the body is divided into three parameters including volume distribution, BBB permeability, and CNS permeability.41,42 Generally, the volume of distribution of a drug or compound ranges from 0.5 to 3 L/Kg.43 Dihydrokaempferol-3-glucoside and aesculetin have a fairly good drug delivery system in the blood, while apigenin-7-glucuronide is in the category of poor drug distribution in the blood. A drug can be categorized as good if it is difficult to penetrate the Central Nervous System (CNS) or even up to the blood-brain barrier (BBB).44 Determination of the value of drug absorption into the CNS and BBB is divided into three categories: high absorption if it has a value of more than 2.0, moderate absorption if it has a value between 0.1 and 2.0, and low absorption if it has a value of less than 0.1.45 Three test compounds have an absorption value of less than 0.1 so they are classified as low absorption. This means that these three compounds are difficult to enter in the CNS and BBB. Prediction of the metabolism of a drug can be seen whether the drug can inhibit CYP enzymes or not. CYP or Cytochrome 450 is an enzyme that plays a role in the digestive system and Phase 1 metabolic processes.46 These three test compounds had no inhibition of these various types of CYP enzymes. The last pharmacokinetic parameter to consider is the excretory system. The faster the process of excretion of a molecule, the higher the total clearance value. This has a positive effect on the body.47 When compared with apigenin-7-glucuronide, the total clearance value is greater than that of the other two compounds. This means that the compound has a fairly good excretion process.
Toxicology prediction science uses computational techniques as an effort to reduce toxicity testing in experimental animals because the results obtained in computational predictions are similar to in vivo tests.48,49 The ratio of the level of toxicity is seen from the Lethal Dose value. The lethal dose of 50 (LD50) is a parameter that determines whether the compound is toxic or not.50 LD50<5 mg/kg is fatal if swallowed, 50 mg/kg≤LD50>5 mg/kg is fatal if swallowed, 300 mg/kg≤LD50>50 mg/kg is classified as toxic if swallowed, 2000 mg/kg≤LD50>300 mg/kg is classified as harmful if swallowed, 5000 mg/kg≤LD50>2000 mg/kg is classified as possibly harmful if swallowed, LD50>5000 mg/kg is classified as non-toxic.20,51 Apigenin-7-glucuronide and dihydrokaempferol-3-glucoside are categorized as potentially possibly harmful if swallowed, while aesculetin is classified as a harmful if swallowed compound.
Drug-likeness analysis of a compound can follow the Lipinski Rule of Five (RO5). A compound can be evaluated for chemical and physical properties to be used as an active drug.52 Lipinski RO5 complies with the following rules: molar refractivity value of 40–130, Log P less than 5, number hydrogen bond acceptors less than 10, number hydrogen bond donors less than 10, and molecular mass less than 500 Dalton.53,54 Table 7, shows that apigenin-7-glucuronide and dihydrokaempferol-3-glucoside have one violation from the RO5 rule, while aesculetin has no violation. This rule also states that a compound can be used as a drug orally if it does not have more than one violation.55 In addition, if the compounds exhibit two or more RO5 violations, the solubility and permeability of the compound is very low.56
Conclusion
The conclusion of this study is that the polyphenolic compound O. basilicum has the potential as an antiviral against the Mpro enzyme for SARS-CoV-2 disease, which is predicted in silico. Based on the molecular docking data, apigenin-7-glucuronide (−9.77 Kcal/mol) and dihydrokaempferol-3-glucoside (−8.96 Kcal/mol) have greater activity than aesculetin (−5.79 Kcal/mol). The binding accuracy between the polyphenolic compound and the receptor is at the appropriate active site characterized by the amino acids involved. Furthermore, this study also analyzed the pharmacokinetic parameters and drug-likeness, where the three compounds can be used as lead compounds for further research.
Acknowledgments
The authors are grateful to Universitas Padjadjaran for Grant of Riset Daftar 305 Pustaka dan Daring (RDPD) 2022.
Funding
This study was funded by Grant of Riset Daftar Pustaka dan Daring (RDPD) Prof. Dikdik Kurnia, M.Sc., Ph.D, Indonesia (1318/UN6.3.1/PT.00/2022, May 12, 2022).
Disclosure
The authors declare no conflicts of interest, financial or otherwise.
References
1. World Health Organization. COVID-19 Weekly Epidemiological Update. Geneva: World Health Organization; 2021.
2. Junaid K, Qasim S, Yasmeen H, et al. Potential inhibitory effect of vitamins against COVID-19. Comput Mater Contin. 2020;66(1):707–714. doi:10.32604/cmc.2020.012976
3. Chowdhury MA, Hossain N, Kashem MA, Shahid MA, Alam A. Immune response in COVID-19: a review. J Infect Public Health. 2020;13:1619–1629. doi:10.1016/j.jiph.2020.07.001
4. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi:10.1016/S0140-6736(20)30211-7
5. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi:10.1016/S0140-6736(20)30183-5
6. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi:10.1126/science.abb3405
7. El-Demerdash A, Metwaly AM, Hassan A, et al. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules. 2021;11:460. doi:10.3390/biom11030460
8. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002;21:3213–3224. doi:10.1093/emboj/cdf327
9. Hu Q, Xiong Y, Zhu GH, et al. The SARS‐CoV‐2 main protease (Mpro): structure, function, and emerging therapies for COVID‐19. MedComm. 2022;3:e151. doi:10.1002/mco2.151
10. Issa SS, Sokornova SV, Zhidkin RR, Matveeva TV. The main protease of SARS-CoV-2 as a target for phytochemicals against coronavirus. Plants. 2022;11:1862. doi:10.3390/plants11141862
11. Tiwari S, Dubey N. Traditional medicinal plants as promising source of immunomodulator against covid-19. J Exper Biol Agric Sci. 2020;8:S126–S138. doi:10.18006/2020.8(Spl-1-SARS-CoV-2).S126.S138
12. Chali BU, Melaku T, Berhanu N, et al. Traditional medicine practice in the context of COVID-19 pandemic: community claim in Jimma zone, Oromia, Ethiopia. Infect Drug Resist. 2021;14:3773. doi:10.2147/IDR.S331434
13. Chiang LC, Ng LT, Cheng PW, Chiang W, Lin CC. Antiviral activities of extracts and selected pure constituents of Ocimum basilicum. Clin Exp Pharmacol Physiol. 2005;32:811–816. doi:10.1111/j.1440-1681.2005.04270.x
14. Shahrajabian MH, Sun W, Cheng Q. Chemical components and pharmacological benefits of Basil (Ocimum basilicum): a review. Int J Food Propert. 2020;23:1961–1970. doi:10.1080/10942912.2020.1828456
15. Brahmi F, Vejux A, Ghzaiel I, et al. Role of diet and nutrients in SARS-CoV-2 infection: incidence on oxidative stress, inflammatory status and viral production. Nutrients. 2022;14. doi:10.3390/nu14112194
16. Chaudhary KK, Mishra N. A review on molecular docking: novel tool for drug discovery. Databases. 2016;3:1029.
17. Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384–13421. doi:10.3390/molecules200713384
18. Ragi K, Kakkassery JT, Raphael VP, Johnson R. In vitro antibacterial and in silico docking studies of two Schiff bases on Staphylococcus aureus and its target proteins. Future J Pharm Sci. 2021;7:1–9.
19. Pires DE, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–4072. doi:10.1021/acs.jmedchem.5b00104
20. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263. doi:10.1093/nar/gky318
21. Nguyen V, Nguyen N, Thi N, Thi C, Truc T, Nghi P, editors. Studies on chemical, polyphenol content, flavonoid content, and antioxidant activity of sweet basil leaves (Ocimum basilicum L.). In: IOP Conference Series: Materials Science and Engineering. IOP Publishing; 2021.
22. Marc RA, Mureșan V, Mureșan AE, et al. Spicy and aromatic plants for meat and meat analogues applications. Plants. 2022;11:960. doi:10.3390/plants11070960
23. Jayasinghe C, Gotoh N, Aoki T, Wada S. Phenolics composition and antioxidant activity of sweet basil (Ocimum basilicum L.). J Agric Food Chem. 2003;51:4442–4449. doi:10.1021/jf034269o
24. Aboulaghras S, Sahib N, Bakrim S, et al. Health benefits and pharmacological aspects of chrysoeriol. Pharmaceuticals. 2022;15:973. doi:10.3390/ph15080973
25. Baderschneider B, Winterhalter P. Isolation and characterization of novel benzoates, cinnamates, flavonoids, and lignans from Riesling wine and screening for antioxidant activity. J Agric Food Chem. 2001;49:2788–2798. doi:10.1021/jf010396d
26. Yang L, Ding W, Xu Y, et al. New insights into the antibacterial activity of hydroxycoumarins against Ralstonia solanacearum. Molecules. 2016;21:468. doi:10.3390/molecules21040468
27. Kang CK, Seong M-W, Choi S-J, et al. In vitro activity of lopinavir/ritonavir and hydroxychloroquine against severe acute respiratory syndrome coronavirus 2 at concentrations achievable by usual doses. Korean J Intern Med. 2020;35:728. doi:10.3904/kjim.2020.157
28. El-Hoshoudy A. Investigating the potential antiviral activity drugs against SARS-CoV-2 by molecular docking simulation. J Mol Liq. 2020;318:113968. doi:10.1016/j.molliq.2020.113968
29. Mulu A, Gajaa M, Woldekidan HB. The impact of curcumin derived polyphenols on the structure and flexibility COVID-19 main protease binding pocket: a molecular dynamics simulation study. PeerJ. 2021;9:e11590. doi:10.7717/peerj.11590
30. Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibition, which causes 50% inhibition (IC_< 50>) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi:10.1016/0006-2952(73)90196-2
31. Ul Qamar MT, Alqahtani SM, Alamri MA, Chen L-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020;10:313–319. doi:10.1016/j.jpha.2020.03.009
32. Maowa J, Hosen M, Alam A, et al. Pharmacokinetics and molecular docking studies of uridine derivatives as SARS-COV-2 Mpro inhibitors. Phys Chem Res. 2021;9:385–412.
33. Duque-Soto C, Borrás-Linares I, Quirantes-Piné R, et al. Potential antioxidant and antiviral activities of hydroethanolic extracts of selected Lamiaceae species. Foods. 2022;11:1862. doi:10.3390/foods11131862
34. Chattopadhyay D, Naik TN. Antivirals of ethnomedicinal origin: structure-activity relationship and scope. Mini Rev Med Chem. 2007;7:275–301. doi:10.2174/138955707780059844
35. Falcó I, Randazzo W, Gómez-Mascaraque L, Aznar R, López-Rubio A, Sánchez G. Effect of (−)-epigallocatechin gallate at different pH conditions on enteric viruses. LWT Food Sci Technol. 2017;81:250–257. doi:10.1016/j.lwt.2017.03.050
36. Ekins S, Waller CL, Swaan PW, Cruciani G, Wrighton SA, Wikel JH. Progress in predicting human ADME parameters in silico. J Pharmacol Toxicol Methods. 2000;44:251–272. doi:10.1016/S1056-8719(00)00109-X
37. Prasanth D, Murahari M, Chandramohan V, Panda SP, Atmakuri LR, Guntupalli C. In silico identification of potential inhibitors from Cinnamon against main protease and spike glycoprotein of SARS CoV-2. J Biomol Struct Dyn. 2021;39:4618–4632. doi:10.1080/07391102.2020.1779129
38. Cabrera-Pérez MÁ, Pham-The H. Computational modeling of human oral bioavailability: what will be next? Expert Opin Drug Discov. 2018;13:509–521. doi:10.1080/17460441.2018.1463988
39. Falcón-Cano G, Molina C, Cabrera-Pérez MÁ. ADME prediction with KNIME: in silico aqueous solubility consensus model based on supervised recursive random forest approaches. ADMET DMPK. 2020;8:251–273. doi:10.5599/admet.852
40. Khan MF, Nahar N, Rashid RB, Chowdhury A, Rashid MA. Computational investigations of physicochemical, pharmacokinetic, toxicological properties and molecular docking of betulinic acid, a constituent of Corypha taliera (Roxb.) with Phospholipase A2 (PLA2). BMC Complement Altern Med. 2018;18:1–15. doi:10.1186/s12906-018-2116-x
41. Alhazmi MI. Molecular docking of selected phytocompounds with H1N1 Proteins. Bioinformation. 2015;11:196. doi:10.6026/97320630011196
42. Lombardo F, Gifford E, Shalaeva MY. In silico ADME prediction: data, models, facts and myths. Mini Rev Med Chem. 2003;3:861–875. doi:10.2174/1389557033487629
43. Smith DA, Beaumont K, Maurer TS, Di L. Volume of distribution in drug design: miniperspective. J Med Chem. 2015;58:5691–5698. doi:10.1021/acs.jmedchem.5b00201
44. Hwang J, Youn K, Ji Y, et al. Biological and computational studies for dual cholinesterases inhibitory effect of zerumbone. Nutrients. 2020;12:1215. doi:10.3390/nu12051215
45. X-l. M, Chen C, Yang J. Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacol Sin. 2005;26:500–512. doi:10.1111/j.1745-7254.2005.00068.x
46. Guttman Y, Kerem Z. Computer-aided (in silico) modeling of cytochrome P450-mediated Food–Drug Interactions (FDI). Int J Mol Sci. 2022;23:8498. doi:10.3390/ijms23158498
47. El-Shamy NT, Alkaoud AM, Hussein RK, Ibrahim MA, Alhamzani AG, Abou-Krisha MM. DFT, ADMET and molecular docking investigations for the antimicrobial activity of 6, 6′-Diamino-1, 1′, 3, 3′-tetramethyl-5, 5′-(4-chlorobenzylidene) bis [pyrimidine-2, 4 (1H, 3H)-dione]. Molecules. 2022;27:620. doi:10.3390/molecules27030620
48. Alves VM, Muratov EN, Zakharov A, Muratov NN, Andrade CH, Tropsha A. Chemical toxicity prediction for major classes of industrial chemicals: is it possible to develop universal models covering cosmetics, drugs, and pesticides? Food Chem Toxicol. 2018;112:526–534. doi:10.1016/j.fct.2017.04.008
49. Toropov AA, Toropova AP, Raska JI, Leszczynska D, Leszczynski J. Comprehension of drug toxicity: software and databases. Comput Biol Med. 2014;45:20–25. doi:10.1016/j.compbiomed.2013.11.013
50. Drwal MN, Banerjee P, Dunkel M, Wettig MR, Preissner R. ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014;42:W53–W58. doi:10.1093/nar/gku401
51. El-Din HMA, Loutfy SA, Fathy N, et al. Molecular docking based screening of compounds against VP40 from Ebola virus. Bioinformation. 2016;12:192. doi:10.6026/97320630012192
52. Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today. 2004;1:337–341. doi:10.1016/j.ddtec.2004.11.007
53. Tumilaar SG, Fatimawali F, Niode NJ, et al. The potential of leaf extract of Pangium edule Reinw as HIV-1 protease inhibitor: a computational biology approach. J Appl Pharm Sci. 2021;11:101–110.
54. Shaji D. Molecular docking studies of human MCT8 protein with soy isoflavones in Allan-Herndon-Dudley syndrome (AHDS). J Pharm Anal. 2018;8:318–323. doi:10.1016/j.jpha.2018.07.001
55. Tallei TE, Tumilaar SG, Niode NJ, et al. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Scientifica. 2020;2020. doi:10.1155/2020/6307457
56. Benet LZ, Hosey CM, Ursu O, Oprea TI. BDDCS, the Rule of 5 and drugability. Adv Drug Deliv Rev. 2016;101:89–98. doi:10.1016/j.addr.2016.05.007
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.