")
Back to Journals » Clinical Ophthalmology » Volume 17
Impact of Modifying Abicipar Manufacturing Process in Patients with Neovascular Age-Related Macular Degeneration: MAPLE Study Results
Authors Callanan D , Khurana RN , Maturi RK, Patel S, Wykoff CC , Eichenbaum D, Khanani AM , Hassan T, Badger H, Mehta S, Le G, Attar M, Seal J, Li XY
Received 26 January 2023
Accepted for publication 6 April 2023
Published 11 May 2023 Volume 2023:17 Pages 1367—1384
DOI https://doi.org/10.2147/OPTH.S405994
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
David Callanan,1 Rahul N Khurana,2 Raj K Maturi,3,4 Sunil Patel,5 Charles C Wykoff,6 David Eichenbaum,7,8 Arshad M Khanani,9,10 Tarek Hassan,11 Hanh Badger,12 Shraddha Mehta,12 Grace Le,12 Mayssa Attar,13 Jennifer Seal,13 Xiao-Yan Li12,14
1Texas Retina Associates, Arlington, TX, USA; 2Northern California Retina Vitreous Associates, Mountain View, CA, USA; 3Midwest Eye Institute, Indianapolis, IN, USA; 4Department of Ophthalmology, Indiana University School of Medicine, Indianapolis, IN, USA; 5West Texas Retina, Abilene, TX, USA; 6Retina Consultants of Houston, Retina Consultants of America, Blanton Eye Institute, Houston Methodist Hospital, Houston, TX, USA; 7Retina Vitreous Associates of Florida, St. Petersburg, FL, USA; 8Morsani College of Medicine, University of South Florida, Tampa, FL, USA; 9Sierra Eye Associates, Reno, NV, USA; 10University of Nevada, Reno School of Medicine, Reno, NV, USA; 11Associated Retinal Consultants, Royal Oak, MI, USA; 12Allergan plc, Irvine, CA, USA, at the time of this work; 13Allergan, an AbbVie company, Irvine, CA, USA; 14VivaVision Biotech, Inc, Shanghai, People’s Republic of China
Correspondence: David Callanan, Texas Retina Associates, 801 West Randol Mill Road, Suite 101, Arlington, TX, USA, 76012, Tel +1 817-261-9625, Fax +1 817-261-9586, Email [email protected]
Purpose: To evaluate the impact of modifying the abicipar pegol (abicipar) manufacturing process on the safety and treatment effect of abicipar in patients with neovascular age-related macular degeneration (nAMD).
Methods: A new process for manufacturing abicipar was developed to reduce host cell impurities. In a prospective, Phase 2, multicenter, open-label, 28-week clinical trial, patients (n=123) with active nAMD received intravitreal injections of abicipar 2 mg at baseline (day 1) and weeks 4, 8, 16, and 24. Outcome measures included proportion of patients with stable vision (< 15-letter loss from baseline; primary endpoint), change from baseline in best-corrected visual acuity (BCVA) and central retinal thickness (CRT), and adverse events.
Results: Overall, 8.9% (11/123) of patients experienced intraocular inflammation (IOI) and discontinued treatment. IOI cases were assessed as mild (2.4% [3/123]), moderate (4.9% [6/123]), or severe (1.6% [2/123]) and resolved with steroid treatment. Visual acuity in most patients with IOI (8 of 11) recovered to baseline BCVA or better by study end. No cases of endophthalmitis or retinal vasculitis were reported. Stable vision was maintained for ≥ 95.9% (≥ 118/123) of patients at all study visits. At week 28, treatment-naïve patients showed a greater mean improvement from baseline in BCVA compared with previously treated patients (4.4 vs 1.8 letters) and a larger mean CRT reduction from baseline (98.5 vs 45.5 μm).
Conclusion: Abicipar produced using a modified manufacturing process showed a moderately lower incidence and severity of IOI compared with Phase 3 abicipar studies. Beneficial effects of treatment were demonstrated.
Keywords: abicipar, age-related macular degeneration, inflammation
Introduction
Vascular endothelial growth factor-A (VEGF-A) is implicated in the pathophysiology of subretinal neovascularization that occurs in neovascular age-related macular degeneration (nAMD).1–4 Current therapies for nAMD use antibody-based inhibition of VEGF-A to target abnormal blood vessel growth. Four approved anti-VEGF therapies—pegaptanib sodium, ranibizumab, aflibercept, and brolucizumab—are currently used for the treatment of nAMD. Off-label use of a fifth anti-VEGF agent for nAMD, bevacizumab, has become common worldwide. In addition, recently approved faricimab5 has both anti-VEGF and anti-angiopoietin-2 activity, although the contribution of angiopoietin-2 inhibition to the treatment effect and clinical response in nAMD has yet to be established.6 Anti-VEGF intravitreal injections are currently the preferred treatment for nAMD,7,8 but their frequency of use poses a great burden for patients, physicians, and health care systems.9,10 Thus, there is an urgent need for novel therapies to improve treatment outcomes and provide longer duration of action to reduce treatment burden.11
DARPin® molecules, a class of engineered binding proteins, contain highly stable ankyrin repeat domains and selectively bind target proteins with high affinity.12 Their small molecular mass facilitates improved tissue penetration in comparison with conventional antibodies and antigen-binding fragments (Fabs). Abicipar pegol (abicipar) is a novel DARPin® molecule targeted against human VEGF-A.12,13 Abicipar offers a unique therapeutic profile with high binding affinity for VEGF-A (486 fM versus ranibizumab: 42.5 pM), long intraocular half-life (≥13 days14 versus ranibizumab: 7.2 days15), low molecular weight (34 kDa versus ranibizumab: 48 kDa), and the potential advantage of less frequent dosing (10 quarterly injections vs 25 monthly with ranibizumab at week 104).16 To date, it is the only anti-VEGF agent to demonstrate noninferiority to ranibizumab in two Phase 3 trials using fixed quarterly dosing.17 Preclinical characterization of abicipar demonstrated its effectiveness in blocking angiogenesis and vascular leakage in cell-based and in vivo models.18
In the Phase 3 CEDAR and SEQUOIA studies, patients with nAMD were treated with 2 mg abicipar every 8 weeks (Q8) or 12 weeks (Q12) following 3 loading doses or with 0.5 mg ranibizumab monthly (Q4). In the pooled analysis of CEDAR and SEQUOIA, both abicipar Q8 and Q12 achieved noninferiority to ranibizumab Q4 for the primary efficacy endpoint (proportion of patients with stable vision: abicipar Q8, 93.2%; abicipar Q12, 91.3%; and ranibizumab Q4, 95.8%) and secondary endpoints of best-corrected visual acuity (BCVA) and central retinal thickness (CRT) at week 52 with 6–8 injections of abicipar versus 13 injections of ranibizumab.17 The overall incidence of treatment-emergent adverse events (TEAEs) was similar among the three treatment arms (abicipar Q8, 76.0%; abicipar Q12, 79.6%; and ranibizumab Q4, 73.6%). However, while the incidence of intraocular inflammation (IOI) events in the two abicipar treatment arms was similar (abicipar Q8, 15.1% and Q12, 15.4%), this compared with only 0.3% in the ranibizumab Q4 group. Severe IOI including endophthalmitis (1.8%; Q8) and retinal vasculitis (1.8%; Q8) were also reported. In order to minimize the patient safety impact, further investigations were conducted to understand the IOI cause. To address the higher rate of IOI in the abicipar arms in the Phase 3 studies, the manufacturing process for abicipar was optimized and the drug substance was purified using a proprietary modified process.
Continuous manufacturing process improvements are common practice through the development and commercialization of biologics. Biologics are produced by living cells that can be the source of host-derived impurities that may be present in the drug product. These impurities in a drug product can lead to undesirable effects such as inflammation.19,20 The presence of innate immune response modulating impurities (IIRMIs), including host-cell proteins and other contaminants derived from the manufacturing and purification process, in preparations of therapeutic proteins is a safety and immunogenicity concern.21,22 Clearance of IIRMIs from the final product is a goal of the purification process. When a therapeutic protein is administered to a patient, residual IIRMIs may induce inflammation directly by inducing the release of cytokines or indirectly by acting as adjuvants to enhance potential immunogenicity of the therapeutic protein.23 Moreover, intravitreal administration poses unique challenges as compared with conventional systemic administration of biologics when considering impurity levels in a drug product. Ultra-low levels of impurities are required when biologics are administered intravitreally, because the impurities are diluted in a static compartment of relatively small volume, in close proximity to important tissues including the retina. By improving the manufacturing process through enhanced purification to remove host-derived impurities, undesirable effects can be reduced.24 In an effort to reduce the occurrence of adverse events (AEs) of inflammation, an optimized manufacturing process utilized high-resolution chromatography to clearly separate and remove E. coli host-derived proinflammatory impurities from the drug substance. This was combined with advancements in the analytical methods to detect and monitor impurities. This reduction in host-derived impurities is hypothesized to lead to a reduction in IOI. The MAPLE Phase 2, open-label, 28-week study evaluated the safety and efficacy of abicipar produced through this improved manufacturing process in patients with nAMD. The results showed only modest improvement in the safety of abicipar, and in June 2020 the FDA declined to approve abicipar for treatment of neovascular age-related macular degeneration, citing an unfavorable benefit–risk ratio resulting from the rate of IOI.
Materials and Methods
In Vitro Assessment of Modified Manufacturing Process
Healthy human peripheral blood mononuclear cells (PBMCs) from multiple donors were obtained and cultured in fresh Roswell Park Memorial Institute (RPMI) medium with 10% fetal bovine serum in 96-well plates. Two different abicipar drug product lots were evaluated at a final concentration of 1 mg/mL: Lot 1 was used in Phase 3 studies CEDAR and SEQUOIA whereas Lot 2 was used in the MAPLE study. Bevacizumab (Avastin, Genentech; 1 mg/mL) and lipopolysaccharide (LPS; 10 µg/mL from List Biological Laboratories) were used as negative and positive controls, respectively. Additional control cultures were treated with phosphate-buffered saline (PBS). Cultures were incubated at 37°C under 5% CO2, and supernatant samples were taken from cultures at 48 hours. Samples were analyzed for secretion of cytokines, including interleukin 6 (IL-6), tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β), using LUMINEX multiplex bead array assays and a custom Milliplex kit (MilliporeSigma).
Cytokine secretion by PBMCs obtained from all donors was analyzed. Results from PBMCs obtained from five donors were excluded due to their positive response at baseline (PBS control, ≥100 pg/mL) or positive response to bevacizumab. The stimulation index (SI) of each cytokine was calculated by dividing the drug-treated cytokine concentration by the PBS (control)-treated cytokine concentration from the same donor PBMCs (to provide the self-controlled fold change). Cytokines with an SI ≥2 were considered as exhibiting positive induction. The donor response rate for each cytokine was calculated by dividing the number of donor PBMC cell strains with positive induction by the total number of donors.
Clinical Study Design
The Phase 2 MAPLE study was a 28-week, open-label, single-arm, multicenter clinical trial conducted at 40 sites in the United States that evaluated the safety and treatment effect of 2 mg abicipar, produced by a proprietary modified manufacturing process with improved purification, in patients with nAMD (ClinicalTrials.gov identifier NCT01397409). MAPLE was conducted from May 23, 2018 to April 23, 2019 in compliance with the Declaration of Helsinki and was approved by the Copernicus Group IRB (Cary, NC) for each clinical site. All patients provided written informed consent prior to any study-related procedures or examinations.
Study Participants
The study enrolled patients 50 years of age or older, with active subfoveal and/or juxtafoveal choroidal neovascularization (CNV; within 200 μm of the center of the foveal avascular zone) secondary to AMD in the study eye. The presence of retinal fluid and/or leakage affecting the fovea was diagnosed by the investigator at screening using spectral-domain optical coherence tomography (SD-OCT) and fluorescein angiography. BCVA in the study eye was required to be between 78 and 24 Early Treatment Diabetic Retinopathy Study (ETDRS) letters (20/32 and 20/320 Snellen equivalents, respectively) and 34 or more ETDRS letters (approximately 20/200 or better) in the fellow eye. The area of CNV within the lesion was required to be >50% of the total lesion area as assessed by the investigator at screening. Patients who were anti-VEGF treatment-naïve (n=83) and previously treated (n=40) were included in the study.
Patients who received previous verteporfin photodynamic therapy or ocular anti-angiogenic therapy within 1 month (ranibizumab), 6 weeks (pegaptanib, bevacizumab), or 2 months (aflibercept) of baseline (day 1), or prior treatment with abicipar at any time, were excluded. Other key exclusion criteria included the presence of structural damage to the center of the macula likely to preclude improvement in BCVA (eg, macular hole stage 3 or 4, retinal pigment epithelium atrophy, subretinal fibrosis/scarring), vitreous hemorrhage, macular hemorrhage greater than 50% of the lesion area or greater than 1 disc area in size involving the center of fovea, history of vitrectomy or submacular surgery, treatment with fluocinolone acetonide implant within the past 36 months or with other ocular corticosteroid injections within the past 6 months, and spherical equivalent of the refractive error of ≤−8 diopters at screening. Patients with a history of uncontrolled glaucoma or ocular hypertension (≥25 mm Hg), recurrent or active systemic or ocular/intraocular infection or inflammation (eg, uveitis), and any medical condition that could prevent safe participation in the study or interfere with the injection procedure or evaluation of efficacy or safety were also excluded.
If both eyes qualified for study treatment, the eye with worse BCVA assessed at screening and confirmed at the baseline (day 1) visit was selected for treatment. If both eyes qualified and had identical BCVA values, the patient selected the non-dominant eye, or the right eye was selected as the study eye for treatment.
Study Treatment and Assessments
At the start of the study, enrolled patients received 2 mg abicipar administered intravitreally at baseline (day 1) and weeks 4, 8, 16, and 24. There was no control treatment, and no randomization was performed. Abicipar produced using a modified manufacturing process was used in this trial. Abicipar injection volume of 50 μL was administered to the study eye. There was a total of 9 scheduled visits during the study that included screening (days −21 to −2), baseline (day 1), and weeks 4, 8, 12, 16, 20, 24, and 28/early exit (Figure 1). For patients at selected sites who participated in pharmacokinetic (PK) blood sampling, there was 1 additional visit on day 3.
 |
Figure 1 Schematic of study design. Abbreviation: BL, baseline (day 1). |
Safety measures included adverse events (AEs), ophthalmic examination findings, BCVA by the ETDRS method, post-injection assessment (eg, intraocular pressure [IOP] and status of retinal artery), vital signs, and laboratory values. Efficacy measures included BCVA by the ETDRS method and CRT assessed by SD-OCT. PK measures included serum levels of free abicipar determined using an enzyme-linked immunosorbent assay, and immunogenicity measures included presence of anti-abicipar antibodies, anti-polyethylene glycol (PEG) antibodies, and neutralizing antibodies.
A protocol amendment was made in October 2018 to discontinue study treatment in the event of an IOI in the study eye, and patients remained in the study and were followed through unscheduled safety follow-up visits until the IOI resolved and anti-inflammatory treatment was no longer required, at which time they were exited from the study. After the amendment, no retreatments were allowed.
Treatment Regimen Adjustments
Treatment regimen adjustments were made if the study eye developed a TEAE during the study. In the event of IOI, the treatment was discontinued, and the patient was observed in the study and exited from the study following resolution of the IOI. In the event of increased IOP to ≥30 mm Hg, treatment was suspended until IOP returned to <30 mm Hg (either spontaneously or after IOP-lowering treatment). Study treatment was withheld in the event of a new retinal break or retinal detachment and resumed following successful treatment of the retinal break or detachment. Following the occurrence of an extraocular or periocular infection, dosing was suspended until the infection resolved. Investigators could withhold or discontinue study treatment for other safety reasons at their discretion.
Outcome Measures
Safety evaluations of AEs (including treatment-emergent changes in ocular and nonocular parameters) were graded by the investigator as mild (awareness of a sign or symptom, but easily tolerated), moderate (discomfort enough to cause interference with usual activity), or severe (incapacitating with inability to work or do usual activity). Events of special interest included any event related to IOI, increased IOP, decreased BCVA, events potentially related to systemic VEGF inhibition (eg, arterial thromboembolic events, hypertension, nonocular hemorrhage, and proteinuria), and any systemic AEs potentially related to immunogenicity (eg, hypersensitivity reaction, arthritis, and vasculitis).
Efficacy outcomes included the proportion of patients with stable vision (primary endpoint) defined as loss of <15 letters in BCVA from baseline. Secondary efficacy endpoints included the mean change from baseline in BCVA and CRT (as assessed with SD-OCT).
Statistical Analysis
The statistical analysis occurred after all patients either completed week 28 or exited early. A sample size of 100 was planned to provide 95% power to estimate the IOI rate within approximately 3% of the actual rate, assuming an actual IOI rate of 2–3%. While the number of patients enrolled (N=123) exceeded the planned enrollment, the sample size was appropriate for the planned assessments and allowed for additional safety observations. Statistical analyses were conducted using SAS Version 9.3 or newer. The analysis population included the safety population, which consisted of patients who received at least one study treatment. Immunogenicity data were not included in the primary database lock but were analyzed separately.
Safety Analyses
The Medical Dictionary for Regulatory Activities (MedDRA) version 20.1 nomenclature was used to code all AEs. The incidence of TEAEs was calculated and presented as the number and percent of patients experiencing the TEAE during the reporting period.
Biomicroscopy and ophthalmoscopy findings with a severity grade change from the baseline value for the same MedDRA-preferred terms were analyzed for patients at each follow-up visit. In addition, a separate analysis for findings of the most severe ocular inflammation based on biomicroscopy and ophthalmoscopy data were tabulated by preferred terms and severity grade. Anterior chamber cells, anterior chamber flare, vitreous chamber cells, and vitreous chamber haze were evaluated and graded according to previously published criteria.14
Efficacy Analyses
Summaries for efficacy variables (proportion of patients with stable vision, mean change from baseline in BCVA, and mean change from baseline in CRT) were based on observed data and are presented for the study eye only. The proportion of patients with stable vision at each post-baseline visit was calculated, and the 95% confidence interval (CI) for the proportion was calculated using the exact method based on binomial distribution. For continuous variables (eg, change from baseline in BCVA and in CRT), the mean and 95% CI were reported. The 95% CI for the mean change was calculated based on t-distribution. In additional analyses, missing data for stable vision and mean change from baseline in BCVA were imputed using the last observation carried forward (LOCF). Data after the first use of prohibited medications were also replaced by LOCF. Ad hoc analyses of patients with and patients without prior anti-VEGF treatment were performed following database lock.
PK and Immunogenicity Analyses
Free serum abicipar concentrations in the subset of patients who participated in PK blood sampling were described using descriptive statistics. Blood samples for PK analyses were collected from patients at selected sites on day 1 (predose, n=46), day 3 (n=41), and week 8 (n=42).
A cumulative summary of immunogenicity based on positive findings at any visit was done. Antidrug antibody titers for anti-abicipar and anti-PEG were generated for each visit that collected immunogenicity data. Blood samples for immunogenicity analysis were collected from all patients on day 1 and weeks 4, 8, 12, 20 and 28. An enzyme-linked immunosorbent assay method was used to detect antibodies directed against abicipar and the pegol moiety of abicipar. This method involved an initial screening and confirmation of positive samples with competitive binding assays using abicipar or PEG. Those samples positive for anti-abicipar antibodies were assessed in a cell-based assay to determine the presence of neutralizing antibodies against abicipar. Immunogenicity results were reported as positive or negative and are presented as number and percentage of patients for each treatment arm. In addition, antibody titers against abicipar and against PEG were summarized. To evaluate the impact of immunogenicity responses on safety, AEs, IOI AEs, and serious IOI AEs were analyzed based on antidrug antibody response.
Results
In Vitro Assessment of Modified Manufacturing Process
An in vitro assay with human PBMCs was used to monitor the effects of IIRMIs and compare the potential for abicipar manufactured with different processes to induce an inflammatory response. The donor response rate for each measured cytokine was reduced for abicipar Lot 2 (used in MAPLE) compared with abicipar Lot 1 (used in CEDAR and SEQUOIA). Donor response rates are presented in Table 1.
 |
Table 1 Human Donor PBMC Cytokine Response Rates to Abicipar Manufactured Using 2 Different Processes and to Positive and Negative Controls |
Baseline Demographics and Characteristics
The MAPLE study enrolled 123 patients. The study population included 71 females (57.7%) and 52 males (42.3%) with a mean age of 78.3 years. The majority of patients were white (96.7%) and pseudophakic (61.8%). Of the 123 patients enrolled and treated with abicipar 2 mg, 83 (67.5%) were anti-VEGF treatment-naïve and 40 (32.5%) were previously treated. Baseline demographic and ocular characteristics are presented in Table 2.
 |
Table 2 Demographic and Baseline Characteristics of Patients and Study Eyes (Safety Population) |
Safety Outcomes
A total of 106 (86.1%) patients completed the 28-week study. Overall, 13.8% (17/123) discontinued from the study treatment: 11.4% (14/123) due to an AE (including 1.6% [2/123] due to death not related to the study treatment), 0.8% (1/123) due to lost to follow-up, 0.8% (1/123) due to progressive disease, and 0.8% (1/123) due to “other” (protocol deviation – visit out of window). Overall, 59.3% (73/132) of patients experienced TEAEs (Table 3): 36.6% (45/123) ocular and 44.7% (55/123) nonocular. The most common TEAEs (reported for ≥4% of the patients) were hypertension (5.7% [7/123]), conjunctival hemorrhage (4.9% [6/123]), vitreous detachment (4.9% [6/123]), neovascular age-related macular degeneration (4.1% [5/123]), upper respiratory infection (4.1% [5/123]), and IOP increase (4.1% [5/123]). TEAEs potentially related to systemic VEGF inhibition were reported for 10.6% (13/123) of the patients (Table 4). Treatment-related TEAEs were reported for 17.1% (21/123) of the patients (Table 5). IOP change from baseline ≥10 mm Hg was reported for 5 patients; and IOP ≥35 mm Hg was reported for 2 of these patients. Serious AEs, including fatal serious AEs, were reported for 13.0% (16/123) of patients, with 4.9% (6/123) from neoplasms. The percentage of patients who discontinued due to AEs was 11.4% (14/123), with 8.9% (11/123) discontinued due to IOI. No cases of endophthalmitis or retinal vasculitis were reported.
 |
Table 3 TEAE Summary |
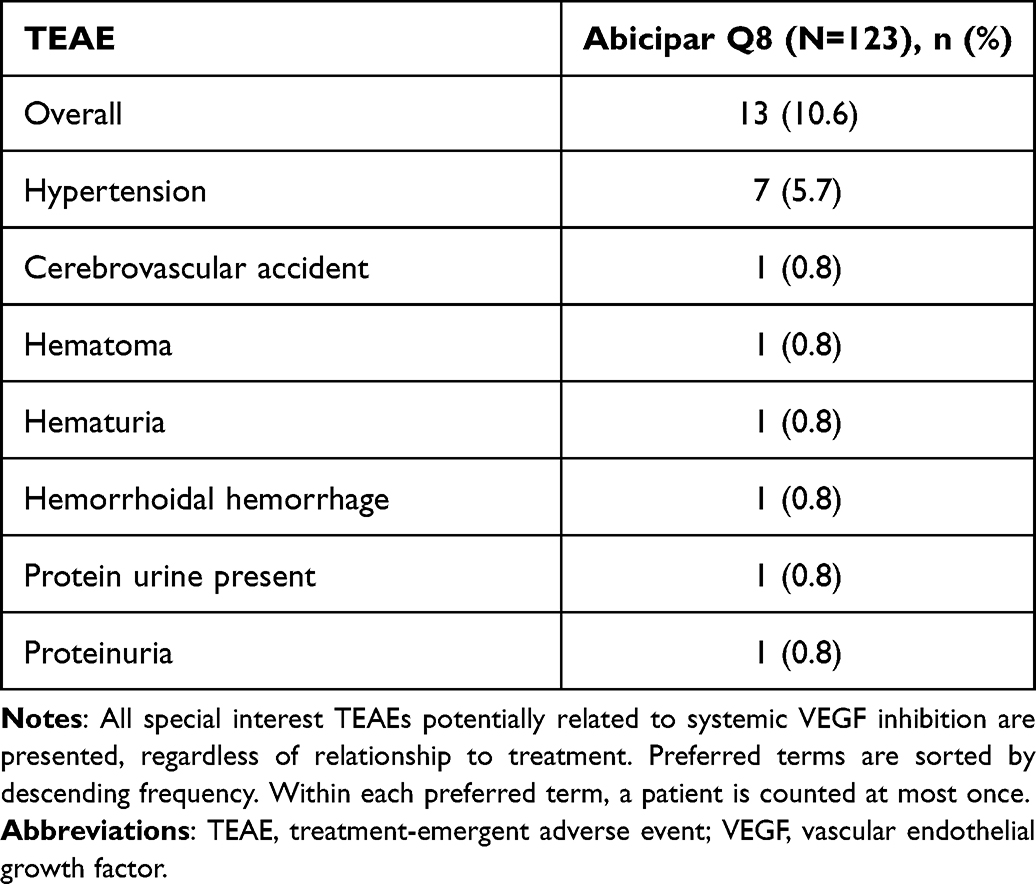 |
Table 4 TEAEs Potentially Related to Systemic VEGF Inhibition (Safety Population) |
 |
Table 5 Treatment-Related TEAEs (Safety Population) |
IOI in the study eye was reported in 8.9% (11/123) of the patients (Table 6). The majority of patients with IOI were treatment-naïve (9 of 11); two patients (Patients 4 and 11) were previously treated. IOI was diagnosed after one injection in three study eyes, two injections in two study eyes, and four injections in six study eyes (Table 7). Nine cases were reported by the investigators as mild (3, 2.4%) or moderate (6, 4.9%). Severe IOIs were reported in 1.6% (2/123) of study eyes. Ten patients with IOI were treated with topical corticosteroids upon presentation of the AE; four cases with moderate and/or severe IOI also received oral or intraocular steroids (Table 6). One patient with IOI did not receive any treatment for the IOI, and the IOI fully resolved by study end. Of the two patients with severe IOI, one patient (Patient 10; Table 6) with iritis achieved full resolution with topical steroid treatment by study end with BCVA recovered to baseline levels. The other patient (Patient 11; Table 6) with panuveitis achieved full resolution with topical and oral steroids after study end with BCVA recovered to baseline levels. Of the 11 patients with reported events of IOI, the event was considered resolved for 9 patients and ongoing for 2 patients (Patients 6 and 11; Table 6) by study end. Both patients with ongoing IOI were treated with topical and oral steroids, were followed for safety reasons after completing the study exit visit, and achieved full resolution of IOI within 2 months post exit. One patient (Patient 1; Table 6) with IOI also had reported retinal hemorrhage, considered to be related to the study drug and not related to the study procedure, and vitreous hemorrhage, which was not considered to be related to the study drug or the procedure. At the week 28 exit visit, this patient’s BCVA dropped to 25 letters as a result of the hemorrhage and subsequent vitrectomy in the study eye. Visual acuity in the majority of patients with IOIs (8/11) recovered to baseline levels or better by study end. Four out of five patients with increased IOP had concurrent events of IOI. After study completion, all IOI cases completely resolved with IOP returned to the normal range, and overall vision in the majority of these patients recovered to slightly better than baseline.
 |
Table 6 Summary of Patients with IOI After Intravitreal Abicipar Injection |
 |
Table 7 First IOI Relative to Abicipar Injection Cycle |
To evaluate the potential for abicipar to inhibit systemic VEGF action and elicit production of anti-abicipar/pegol antibodies, PK and immunogenicity testing were conducted. Serum concentrations of free abicipar were below the limit of quantification (BLQ) at baseline (day 1) in patients included in the PK analysis. Free abicipar concentrations were generally measurable in serum on day 3 following abicipar administration with mean concentrations of 0.813 ± 0.664 nM. At week 8 (predose), free abicipar concentrations were BLQ in all patients.
Serum samples were analyzed for antidrug antibodies in all 123 patients. The cumulative incidence of anti-abicipar antibodies detectable in blood samples from patients receiving 2 mg abicipar was 30.1% (37/123). Titers following a single injection were low at week 4, peaked by week 12, and declined thereafter. Among patients with anti-abicipar antibodies, the median maximal titer over the duration of the study was 640 (range: 10–81,920). Following a single intravitreal injection of abicipar, the incidence of neutralizing antibodies was low (1, 0.8%). This incidence increased after 3 injections and then plateaued; the cumulative number of patients developing neutralizing antibodies at any visit was 18.7% (23/123). The incidence of anti-PEG antibodies was low and did not vary significantly throughout the course of the study. The cumulative number of patients developing anti-PEG antibodies at any visit was 2.4% (3/123). Titers were low throughout the study, and the median maximal titer over the duration of the study among patients with anti-PEG antibodies was 20 (range: 20–80). Serum samples taken from the study participants before abicipar treatment served as a negative control; no patient with an evaluable baseline serum sample tested positive for pre-existing anti-abicipar or anti-PEG antibodies.
Evaluation of the impact of antidrug antibodies on AEs within this study showed no correlation between overall incidence of AEs and antidrug antibody response. The majority of patients with IOI AEs had positive anti-abicipar antibody responses postbaseline, but the majority of patients with antidrug antibodies did not develop IOI, and not all patients who developed IOI had antidrug antibodies. The relationship between immunogenicity and IOI was analyzed further. Of the patients who were positive for anti-abicipar antibodies postbaseline, 75.7% (28/37) did not have IOI. However, the majority (81.8% [9/11]) of patients with IOI AEs were positive for anti-abicipar antibodies postbaseline. In these patients, maximum titers at any visit ranged from 20 to >80,000. A higher percentage of patients (6 of 8, 75%) who had high (≥10,000) anti-abicipar titers at any visit during the study also developed IOI. However, the magnitude of the anti-abicipar response to abicipar treatment did not appear to be associated with the severity of the IOI AEs, and inflammation in patients with high titers was not necessarily deemed severe. Of the eight patients with high anti-abicipar titers (≥10,000), five patients developed IOI: one patient had severe inflammation, four patients had moderate inflammation, and three patients had no inflammation. The onset of these events and positive anti-abicipar antibody status did not appear to have a consistent temporal pattern in that an anti-abicipar response did not consistently precede, coincide with, or follow the AE. Moreover, there was no trend for a relationship between the severity of the AE and the magnitude of the anti-abicipar antibody response. Thus, a causal relationship between AEs, including IOI, and antidrug antibody response could not be established.
Safety measures did not indicate any safety concerns apart from IOI. Mean changes from baseline in all laboratory test results were minimal, and a potentially clinically significant postbaseline laboratory value was reported for no more than 1 patient for each hematology and blood chemistry parameter. Similarly, there were no clinically meaningful mean changes in systolic or diastolic blood pressure or pulse rate. Findings on biomicroscopy and ophthalmoscopy generally were related to the injection procedure (eg, conjunctival hemorrhage), underlying nAMD (eg, macular edema), or IOI (eg, keratic precipitates).
Vision Outcomes
Stable vision was consistent and maintained for ≥95.9% (118/123) of patients at all study visits (Figure 2). The proportion of patients with stable vision at week 28 (primary endpoint) was 97.6% (120/123). Vision improvement, as assessed by the proportion of patients who gained at least 10 or 15 letters from baseline, showed a similar trend of consistent results from week 4 through week 28. The proportion of patients with BCVA ≥70 letters (20/40 Snellen equivalent) also remained consistent from weeks 4 to 28. In general, mean change from baseline BCVA in number of letters demonstrated numerical improvement from weeks 4 through 28 (Figure 3). The mean baseline BCVA value was 62 letters. At week 28, the mean change in BCVA from baseline was +3.6 letters (95% CI, 2.24 to 5.01). Additionally, the mean change in BCVA from baseline was numerically greater at week 28 in patients without prior anti-VEGF treatment (+4.4 letters; 95% CI, 2.67 to 6.19) compared with previously treated patients (+1.8 letters; 95% CI, −0.36 to 3.86).
 |
Figure 2 Proportion of patients with stable vision (<15-letter loss in best-corrected visual acuity from baseline) in the safety population. Missing values were imputed with the last-observation-carried-forward method. |
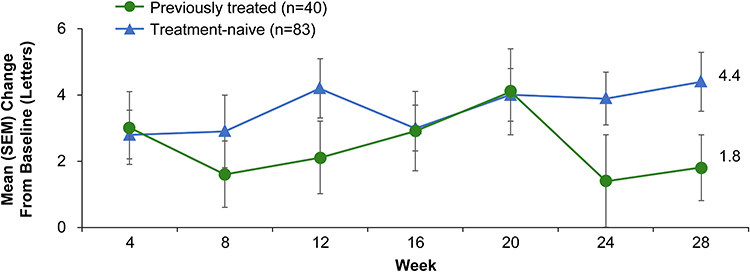 |
Figure 3 Mean ± standard error of the mean (SEM) change in best-corrected visual acuity (BCVA) from baseline to study end in treatment-naïve and previously treated patients who received 2 mg abicipar. |
Anatomic Outcomes
The CRT in study eyes decreased in both previously treated and treatment-naïve eyes, and the improvement in CRT after the initial doses was maintained through week 28 (Figure 4). Mean reduction in CRT was generally consistent across visits and ranged from −52.3 to −82.5 μm. Mean CRT change from baseline at week 28 was −82.5 µm (95% CI, −102.14 to −62.80). At week 28, the mean change in CRT from baseline showed greater reductions for patients without prior anti-VEGF treatment (−98.5 μm; 95% CI, −121.65 to −75.26) compared with previously treated patients (−45.5 μm; 95% CI, −81.02 to −9.98).
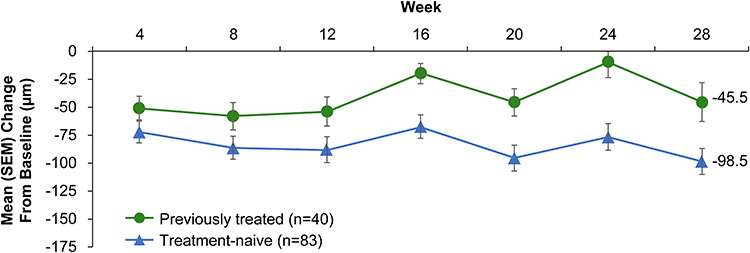 |
Figure 4 Mean ± standard error of the mean (SEM) change in central retinal thickness (CRT) from baseline to study end in treatment-naïve and previously treated patients who received 2 mg abicipar. |
Discussion
To improve the safety profile of abicipar, the drug substance manufacturing process was modified to reduce overall host-derived impurities. In vitro IIRMI assessment using PBMCs demonstrated a marked reduction in the inflammatory marker signal for the revised manufacturing process when compared with the manufacturing process employed for the investigational product used in the CEDAR and SEQUOIA studies. The MAPLE study evaluated 2 mg abicipar, produced through the modified manufacturing process, which achieved stable vision at week 28 with an improved safety profile in both treatment-naïve and previously treated patients with nAMD.
IOI is a well-recognized risk of anti-VEGF injections. Optimal manufacturing, formulation, handling, and delivery of anti-VEGF biologic drug products are complex and require a commitment to continuous process improvement to ensure safety and effectiveness. Intrinsic process impurities such as E. coli host cell proteins, extrinsic impurities such as endotoxin- or syringe-associated silicone droplets, and the formulation have been implicated as causes of IOI.25–29 In the Phase 1/2 FOCUS study, ranibizumab had an IOI rate of 38.1%. The incidence of IOIs dramatically decreased for ranibizumab following adoption of a solubilized formulation used in the ANCHOR and MARINA studies and derived from the lyophilized formulation used in FOCUS. In recent Phase 3 studies, the rate of IOI reported with ranibizumab was 8.4% (but only 0.8% reported as possibly drug-related) in the 48-week COLUMBUS-AMD study,30 and the rate of iritis/uveitis reported with aflibercept was <1% in the 48-week HAWK and HARRIER studies.31
In the MAPLE study, reduction in the incidence and severity of IOI was achieved consistent with removal of pro-inflammatory impurities from the drug, but the rate of IOI (8.9%) remained higher than has been reported in recent Phase 3 studies of approved anti-VEGF therapies. The modified manufacturing process used for abicipar production, enabled by high-resolution chromatography purification methodology and sophisticated analytical methodology, allowed the removal of host-derived impurities, specifically host cell proteins. The overall IOI rate in MAPLE was 8.9% (11 of 123 patients)—a reduction from the 13.1% (96 of 625 patients on Q8 regimen) IOI rate reported in the Phase 3 pivotal studies; the rate of severe IOI TEAEs in MAPLE was 1.6% (2 of 123 patients) compared with 3.4% (21 of 625 patients on Q8 dosing schedule) through 28 weeks of follow-up in the combined Phase 3 trials. The majority of IOI TEAEs reported in MAPLE were mild to moderate (9 of 11) and treated with topical and/or oral corticosteroids upon presentation of the AE. One patient did not receive any treatment and had full resolution of the IOI. All 11 IOI cases in MAPLE completely resolved, and overall vision recovered to better than baseline; 9 cases resolved by study end with the remaining 2 achieving full resolution within 2 months after study end. No cases of endophthalmitis or retinal vasculitis were reported in the MAPLE study. However, further reduction of IOI is necessary for abicipar to achieve an IOI incidence in line with other anti-VEGF biologics. The cause of IOI is likely multifactorial for any drug. The different considerations for potential causes are depicted in Figure 5. Emerging evidence indicates that the syringe, needle, and silicone oil content can contribute to eliciting IOI and was recently reviewed by Melo et al.32 Thus, in addition to the drug manufacturing process, the product presentation components must also be carefully considered to minimize IOI.
 |
Figure 5 Multifactorial considerations for the potential causes of intraocular inflammation. Images created with Biorender.com. Abbreviation: CMC, Chemistry, Manufacturing and Controls. |
Vision and anatomic outcomes in the MAPLE study showed a clinically meaningful improvement in BCVA and CRT, in line with previous results in the CEDAR and SEQUOIA studies. In MAPLE, stable vision was maintained for ≥95.9% of patients at all study visits, the mean BCVA improved by 3.6 letters and the mean CRT decreased by 82.5 µm from baseline to week 28 with 86.2% of patients receiving 5 injections. The difference in vision and anatomic outcomes at week 28 in MAPLE and the CEDAR and SEQUOIA Phase 3 trials can be explained by differences in baseline BCVA and CRT. The mean baseline BCVA and CRT of patients in MAPLE was 62 letters and 353.1 µm compared with 56.8 letters and 382.5 µm in the CEDAR and SEQUOIA abicipar Q8 population. Patients in MAPLE may have experienced a ceiling effect, as patients with higher initial BCVA and thinner CRT typically achieve less BCVA gain and CRT reduction than individuals with more significant BCVA loss and thicker CRT (eg, patients in CEDAR/SEQUOIA). Despite having better baseline vision and anatomic characteristics, study eyes in MAPLE showed clinically relevant improvements in the mean change from baseline BCVA and CRT in both treatment-naïve and previously treated populations (3.6 letters gain in BCVA and 82.5 µm reduction in CRT).
Limitations of the MAPLE study come from its open-label, single-arm design, which did not include comparator or placebo arms. Differences in the study populations between the MAPLE and CEDAR/SEQUOIA studies potentially could also have contributed to the observed reduction in the rate of IOI. Additionally, patient assessments were not confirmed by an independent reading center, but patients were examined by the same investigator at the screening visit and at every treatment and evaluation visit. MAPLE included both treatment-naïve and previously treated study eyes, making it difficult to compare results from this study to results from other clinical trials that only enrolled treatment-naïve patients. However, the patient population in MAPLE is more representative of a real clinic setting, allowing critical insights for retina specialists on the management of nAMD in patients with prior anti-VEGF treatment.
Conclusions
The MAPLE study evaluated abicipar, produced through a modified manufacturing process, in both treatment-naïve and previously treated patients with nAMD and demonstrated a moderate reduction in IOI rate compared with the Phase 3 CEDAR and SEQUOIA trials, with an IOI rate of 8.9% observed in MAPLE. Use of abicipar produced through a modified manufacturing process was associated with favorable functional and anatomical outcomes, while lessening the treatment burden for patients maintained on a Q8 treatment regimen or those on a treat-and-extend regimen that may require more visits than a predictable fixed dosing schedule. To fully realize the potential of abicipar as a treatment for nAMD, further reduction of IOI should be achieved.
Abbreviations
Abicipar, abicipar pegol; AE, adverse event; BCVA, best-corrected visual acuity; BLQ, below the limit of quantification; CI, confidence interval; CNV, choroidal neovascularization; CRT, central retinal thickness; ETDRS, Early Treatment Diabetic Retinopathy Study; IIRMI, innate immune response modulating impurity; IL-1β, interleukin-1 beta; IL-6, interleukin 6; IOI, intraocular inflammation; IOP, intraocular pressure; LOCF, last observation carried forward; LPS, lipopolysaccharide; MedDRA, Medical Dictionary for Regulatory Activities; nAMD, neovascular age-related macular degeneration; PBMC, peripheral blood mononuclear cell; PBS, phosphate-buffered saline; PEG, polyethylene glycol; Q4, monthly; Q8, every 8 weeks following three loading doses; Q12, every 12 weeks following three loading doses; RPMI, Roswell Park Memorial Institute; SD-OCT, spectral-domain optical coherence tomography; TEAE, treatment-emergent adverse event; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor.
Data Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (eg, protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvieclinicaltrials.com/hcp/data-sharing/.html.
Acknowledgments
AbbVie and the authors thank the investigators and patients who participated in the MAPLE study, and Dr Swati Gupta (AbbVie) and Dr Joe Zhou (former employee of Allergan) for conducting the in vitro PBMC studies. Nayna Sanathara, PhD, of AbbVie Inc., provided medical writing assistance for the development of this publication. Editorial support was provided by Evidence Scientific Solutions, Inc. (Philadelphia, PA) and funded by AbbVie. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship. DARPin is a registered trademark of Molecular Partners (Zurich, Switzerland).
This work was presented in part at the following congresses: American Society of Retina Specialists, July 30, 2019, Chicago, Illinois; Bascom Palmer Eye Institute – Angiogenesis, February 8, 2020, Miami, Florida; Macula Society, February 21, 2020, San Diego, California; Association for Research in Vision and Ophthalmology, June 4, 2020 (virtual meeting); and American Society of Retina Specialists, July 24, 2020 (virtual meeting).
Funding
This study was sponsored by Allergan plc (Dublin, Ireland; prior to acquisition by AbbVie Inc). Allergan/AbbVie participated in the design of the study, data management, data analysis, interpretation of the data, and preparation, review, and approval of the manuscript.
Disclosure
David Callanan serves as a consultant for Allergan (an AbbVie company), Applied Genetic Technologies Corporation, EyePoint, Eyevensys, Graybug, Regenerative Patch Technologies, and Takeda; is on the Speaker’s Bureau for Allergan (an AbbVie company); has received research support from Aerie, Amgen, Diopsys, Eyevensys, Genentech/Roche, Gilead, Ionis, and Regeneron; and is an equity holder and employee of Aviceda Therapeutics. Rahul N. Khurana serves as a consultant for Apellis, Bausch + Lomb, Genentech, NGM Biopharmaceuticals, and Ophthea; and has received grant support from Apellis, Chengdu Kanghong, Clearside Biomedical, Eyepoint, Genentech, NGM Biopharmaceuticals, Ophthea, Oxurion, and RegenXBio. Raj Maturi serves as a consultant to and has received grants from Aerpio, AiViva, Allegro, Allergan (an AbbVie company), Allgenesis, Astellas, Boehringer Ingelheim, Clearside, Dutch Ophthalmic, Eli Lilly, Genentech, Gemini, GlaxoSmithKline, Graybug, Gyroscope, Jaeb Center for Health Research, Kalvista, NGM Biopharmaceuticals, Neurotech, Novartis, Ophthea, Oxurion, Ribomic, Roche, Samsung, Santen, Senju, ThromboGenics and Unity. Sunil Patel serves as a consultant for AiViva, Allergan (an AbbVie company), Allgenesis, Genentech-Roche, Kala, Kodiak Sciences; is on an advisory board for Allergan (an AbbVie company), Genentech-Roche, and Kodiak Sciences; has received research support from Aerie, Aerpio, Allergan, Allgenesis, Apellis, Boehringer Ingelheim, Chengdu Kanghong, Clearside, Eyepoint, Genentech-Roche, Ionis Pharmaceuticals, Iveric Bio, KalVista, Kodiak Sciences, Mylan, Novartis, Oculis, Opthea, Ophthotech, Ora, Oxurion, Regeneron, Samsung, SmileBiotek, Stealth Biotherapeutics, ThromboGenics, and Xbrane Biopharma; and has received personal fees from AiViva, Kala, Ocugenix, and RegenxBio, outside the submitted work; he is also the Chief Medical Officer and stock owner of Allgenesis Biotherapeutics, Inc. Charles C. Wykoff is a consultant for 4DMT, Adverum, Aerie Pharmaceuticals, AGTC, Alcon, Alimera, Allergan (an AbbVie company), Allgenesis, Alnylam, Annexon, Apellis, Arrowhead Pharmaceuticals, Bausch + Lomb, Bayer, Bionic Vision Technologies, Boehringer Ingelheim, Cholgene, Clearside, Curacle, Chengdu Kanghong Biotechnologies, Clearside Biomedical, EyePoint Pharmaceuticals, Foresite, Frontera, Genentech/Roche, Gyroscope, IACTA, Iveric Bio, Janssen, Kato Pharmaceuticals, Kiora, Kodiak Sciences, Kriya, Merck, Nanoscope, NGM Biopharmaceuticals, Notal Vision, Novartis, OccuRx, Ocular Therapeutix, Ocuterra, OliX, ONL Therapeutics, Opthea Limited, Oxurion, Palatin, PerceiveBio, Perfuse, PolyPhotonix, Ray, RecensMedical, Regeneron, Resonance, REGENXBIO, Roche, SAI MedPartners, SciNeuro, Stealth, Surrozen, Takeda, THEA, TissueGen, Valo, and Verana Health; has received research support from 4DMT, Adverum, Aerie Pharmaceuticals, Aldeyra, Aerpio, AffaMed, Alexion, Alimera Sciences, Alkahest, Allergan (an AbbVie company), Allgenesis, Amgen, Annexin, Annexon, Apellis, Arctic Vision, Asclepix, Bayer, Boehringer Ingelheim, Chengdu Kanghong Biotechnologies, Clearside Biomedical, EyePoint, Gemini Therapeutics, Genentech/Roche, GlaxoSmithKline, Graybug Vision, Gyroscope, IONIS Pharmaceuticals, iRENIX, Iveric Bio, Kodiak Sciences, LMRI, Nanoscope, Neurotech Pharmaceuticals, NGM Biopharmaceuticals, Novartis, Ocular Therapeutix, Ocuphire, OcuTerra, Ophthotech, Opthea, Outlook Therapeutics, Oxurion, Oxular, Oyster Point, PerceiveBio, RecensMedical, Regeneron, REGENXBIO, Roche, SamChunDang Pharm, Sandoz, Senju, Taiwan Liposome Company, UNITY, Verily, and Xbrane BioPharma; and has stock options for ONL Therapeutics, PolyPhotonix, RecensMedical, TissueGen, and Visgenx. David Eichenbaum is a consultant for Alimera Sciences, Allergan (an AbbVie company), Apellis, Bausch + Lomb, Coherus, Crinetics, the Dutch Ophthalmic Research Center, EyePoint, Genentech, Gyroscope Therapeutics Limited, Iveric Bio, KKR, Kodiak Sciences, Novartis, Ocular Therapeutix, Opthea, Outlook, RecensMedical, Regeneron, Regenxbio, ReVive, US Retina, and Vial; has served as an investigator for 4DMT, Alexion, Alkahest, Allegenesis, Annexon, AsclepiX, Bayer, Chengdu Kanghong Biotechnologies, EyePoint, Gemini, Genentech, Gyroscope Therapeutics Limited, Ionis, Iveric Bio, Kodiak Sciences, Mylan, NGM Biopharmaceuticals, Novartis, Ocular Therapeutics, Opthea, RecensMedical, Regeneron, Regenxbio, and Unity; has received speaker fees from Allergan (an AbbVie company), Apellis, Bausch + Lomb, Bayer, the Dutch Ophthalmic Research Center, EyePoint, Genentech, and Novartis; has received personal fees from Alimera and Samsama; has received grants from 4DMT, Alexion, Alkahest, Allegenesis, Annexon, AsclepiX, Aviceda, Chengdu, EyePoint, Gemini, Genentech, Gyroscope, Ionis, IvericBio, Kodiak, Mylan, NGM, Novartis, Ocular Therapeutix, Opthea, RecensMedical, Regeneron, RegenxBio, and Unity, outside the submitted work; holds equity and/or stocks for Boston Image Reading Center, Clearside Biomedical, Hemera Biopharmaceuticals, Network Eye, ReVive, and US Retina; and founded Network Eye. Arshad Khanani is a consultant for Adverum, Aerpio, Allergan (an AbbVie company), Chengdu Kanghong, DORC International, Genentech, Kato, Kodiak, Novartis, Gemini Therapeutics, Gyroscope, Iveric Bio, Opthea, Oxurion, RecensMedical, Regenxbio, and Roche; has received research support from Adverum, Allergan (an AbbVie company), Chengdu Kanghong, Gemini Therapeutics, Genentech, Gyroscope, Iveric Bio, Kodiak, NGM Bio, Novartis, Opthea, Oxurion, RecensMedical, Regenxbio, and Roche; and has received speaker fees from Allergan (an AbbVie company) and Novartis. Tarek Hassan is a consultant for Allergan (an AbbVie company), Aviceda, Bayer, EyePoint, Genentech, Iveric Bio, Novartis, Regeneron, and Roche. Hanh Badger, Shraddha Mehta, Grace Le, and Xiao-Yan Li were employees of Allergan plc at the time of the study. Mayssa Attar and Jennifer Seal are employees of AbbVie and may hold stock/stock options. The authors report no other conflicts of interest in this work.
References
1. Ding X, Patel M, Chan -C-C. Molecular pathology of age-related macular degeneration. Prog Retin Eye Res. 2009;28(1):1–18. doi:10.1016/j.preteyeres.2008.10.001
2. Cheung LK, Eaton A. Age-related macular degeneration. Pharmacotherapy. 2013;33(8):838–855. doi:10.1002/phar.1264
3. Ciulla TA, Rosenfeld PJ. Antivascular endothelial growth factor therapy for neovascular age-related macular degeneration. Curr Opin Ophthalmol. 2009;20(3):158–165. doi:10.1097/ICU.0b013e32832d25b3
4. Grisanti S, Tatar O. The role of vascular endothelial growth factor and other endogenous interplayers in age-related macular degeneration. Prog Retin Eye Res. 2008;27(4):372–390. doi:10.1016/j.preteyeres.2008.05.002
5. Shirley M. Faricimab: first approval. Drugs. 2022;82(7):825–830. doi:10.1007/s40265-022-01713-3
6. Vabysmo prescribing information. Available from: https://www.gene.com/download/pdf/vabysmo_prescribing.pdf.
7. Flaxel CJ, Adelman RA, Bailey ST, et al. Age-related macular degeneration Preferred Practice Pattern®. Ophthalmology. 2020;127(1):P1–P65. doi:10.1016/j.ophtha.2019.09.024
8. Schmidt-Erfurth U, Chong V, Loewenstein A, et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br J Ophthalmol. 2014;98(9):1144–1167. doi:10.1136/bjophthalmol-2014-305702
9. Prenner JL, Halperin LS, Rycroft C, et al. Disease burden in the treatment of age-related macular degeneration: findings from a time-and-motion study. Am J Ophthalmol. 2015;160(4):725–731.e1. doi:10.1016/j.ajo.2015.06.023
10. Brown MM, Brown GC, Lieske HB, et al. Societal costs associated with neovascular age-related macular degeneration in the United States. Retina. 2016;36(2):285–298. doi:10.1097/IAE.0000000000000717
11. Singh RP, Stone TW, eds. 2008 Global Trends in Retina Survey. Chicago, IL: American Society of Retina Specialists; 2018.
12. Smithwick E, Stewart MW. Designed ankyrin repeat proteins: a look at their evolving use in medicine with a focus on the treatment of chorioretinal vascular disorders. Antiinflamm Antiallergy Agents Med Chem. 2017;16(1):33–45. doi:10.2174/1871523016666170502115816
13. Plückthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol. 2015;55(1):489–511. doi:10.1146/annurev-pharmtox-010611-134654
14. Campochiaro PA, Channa R, Berger BB, et al. Treatment of diabetic macular edema with a designed ankyrin repeat protein that binds vascular endothelial growth factor: a phase I/II study. Am J Ophthalmol. 2013;155(4):697. doi:10.1016/j.ajo.2012.09.032
15. Krohne TU, Liu Z, Holz FG, Meyer CH. Intraocular pharmacokinetics of ranibizumab following a single intravitreal injection in humans. Am J Ophthalmol. 2012;154(4):682–686.e2. doi:10.1016/j.ajo.2012.03.047
16. Khurana RN, Kunimoto D, Yoon YH, et al. Two-year results of the phase 3 randomized controlled study of abicipar in neovascular age-related macular degeneration. Ophthalmology. 2020;128(7):1027–1038.
17. Kunimoto D, Yoon YH, Wykoff CC, et al. Efficacy and safety of abicipar in neovascular age-related macular degeneration: 52-week results of phase 3 randomized controlled study. Ophthalmology. 2020;127(10):1331–1344. doi:10.1016/j.ophtha.2020.03.035
18. Rodrigues GA, Mason M, Christie L-A, et al. Functional characterization of abicipar-pegol, an anti-VEGF DARPin therapeutic that potently inhibits angiogenesis and vascular permeability. Invest Ophthalmol Vis Sci. 2018;59(15):5836–5846. doi:10.1167/iovs.18-25307
19. Heier JS, Boyer DS, Ciulla TA, et al. Ranibizumab combined with verteporfin photodynamic therapy in neovascular age-related macular degeneration: year 1 results of the FOCUS study. Arch Ophthalmol. 2006;124(11):1532–1542. doi:10.1001/archopht.124.11.1532
20. Chong DY, Anand R, Williams PD, et al. Characterization of sterile intraocular inflammatory responses after intravitreal bevacizumab injection. Retina. 2010;30(9):1432–1440. doi:10.1097/IAE.0b013e3181dc04da
21. Verthelyi D, Wang V, Vij N. Trace levels of innate immune response modulating impurities (IIRMIs) synergize to break tolerance to therapeutic proteins. PLoS One. 2010;5(12):e15252. doi:10.1371/journal.pone.0015252
22. Holley CK, Dobrovolskaia MA. Innate immunity modulating impurities and the immunotoxicity of nanobiotechnology-based drug products. Molecules. 2021;26(23):7308. doi:10.3390/molecules26237308
23. Haile LA, Puig M, Polumuri SK, et al. In vivo effect of innate immune response modulating impurities on the skin milieu using a macaque model: impact on product immunogenicity. J Pharm Sci. 2017;106(3):751–760. doi:10.1016/j.xphs.2016.11.001
24. Spitzer MS, Ziemssen F, Bartz-Schmidt KU, et al. Treatment of age-related macular degeneration: focus on ranibizumab. Clin Ophthalmol. 2008;2(1):1–14. doi:10.2147/OPTH.S1959
25. Greenberg JP, Belin P, Butler J, et al. Aflibercept-related sterile intraocular inflammation outcomes. Ophthalmol Retina. 2019;3(9):753–759. doi:10.1016/j.oret.2019.04.006
26. Agrawal S, Joshi M, Christoforidis JB. Vitreous inflammation associated with intravitreal anti-VEGF pharmacotherapy. Mediators Inflamm. 2013;2013:943409. doi:10.1155/2013/943409
27. Ness T, Feltgen N, Agostini H, et al. Toxic vitreitis outbreak after intravitreal injection. Retina. 2010;30(2):332–338. doi:10.1097/IAE.0b013e3181baf691
28. Palmer JM, Amoaku WM, Kamali F. Quality of bevacizumab compounded for intravitreal administration. Eye. 2013;27(9):1090–1097. doi:10.1038/eye.2013.139
29. Liu L, Ammar DA, Ross LA, et al. Silicone oil microdroplets and protein aggregates in repackaged bevacizumab and ranibizumab: effects of long-term storage and product mishandling. Invest Ophthalmol Vis Sci. 2011;52(2):1023–1034. doi:10.1167/iovs.10-6431
30. Holz FG, Oleksy P, Ricci F, Kaiser PK, Kiefer J, Schmitz-Valckenberg S; COLUMBUS-AMD Study Group. Efficacy and safety of biosimilar FYB201 compared with ranibizumab in neovascular age-related macular degeneration. Ophthalmology. 2022;129(1):54–63. doi:10.1016/j.ophtha.2021.04.031
31. Dugel PU, Koh A, Ogura Y; HAWK and HARRIER Study Investigators. HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology. 2020;127(1):72–84. doi:10.1016/j.ophtha.2019.04.017
32. Melo GB, Cruz NFSD, Emerson GG, et al. Critical analysis of techniques and materials used in devices, syringes, and needles used for intravitreal injections. Prog Retin Eye Res. 2021;80:100862. doi:10.1016/j.preteyeres.2020.100862
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.