")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Genetics and epigenetics of eating disorders
Authors Yilmaz Z, Hardaway A, Bulik C
Received 20 May 2014
Accepted for publication 18 July 2014
Published 10 March 2015 Volume 2015:5 Pages 131—150
DOI https://doi.org/10.2147/AGG.S55776
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Video abstract presented by Yilmaz Z, Hardaway JA, and Bulik CM.
Views: 2561
Zeynep Yilmaz,1 J Andrew Hardaway,1 Cynthia M Bulik1–3
1Department of Psychiatry, 2Department of Nutrition, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 3Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden
Abstract: Eating disorders (EDs) are serious psychiatric conditions influenced by biological, psychological, and sociocultural factors. A better understanding of the genetics of these complex traits and the development of more sophisticated molecular biology tools have advanced our understanding of the etiology of EDs. The aim of this review is to critically evaluate the literature on the genetic research conducted on three major EDs: anorexia nervosa, bulimia nervosa, and binge eating disorder. We will first review the diagnostic criteria, clinical features, prevalence, and prognosis of anorexia nervosa, bulimia nervosa, and binge eating disorder, followed by a review of family, twin, and adoption studies. We then review the history of genetic studies of EDs covering linkage analysis, candidate-gene association studies, genome-wide association studies, and the study of rare variants in EDs. Our review also incorporates a translational perspective by covering animal models of ED-related phenotypes. Finally, we review the nascent field of epigenetics of EDs and a look forward to future directions for ED genetic research.
Keywords: anorexia nervosa, binge eating disorder, bulimia nervosa, animal models, genome-wide association studies, high-throughput sequencing
Overview of eating disorders
Eating disorders (EDs) are serious psychiatric conditions with significant morbidity and mortality. That EDs have a genetic component may come as a surprise to many due to widespread misperception of them being disorders of volition. Research over the past decade has confirmed that genes do indeed play a role, and animal models of core related phenotypes are assisting with defining the underlying biology of these pernicious illnesses. In this review, we focus on three major EDs: anorexia nervosa (AN), bulimia nervosa (BN), and binge eating disorder (BED). Most of the genetic research has focused on AN and BN; less information is available for BED due to its status as a newly recognized ED diagnosis.
Anorexia nervosa
AN (International Classification of Diseases, 10th revision [ICD-10]: F50.00) is a serious ED with substantial morbidity and the highest lifetime mortality among psychiatric disorders.1–3 Low weight or body mass index (BMI) is the sine qua non of AN and the primary target of initial treatment.4,5 Symptoms of AN include persistent restriction of food intake, an intense fear of gaining weight or persistent behavior that interferes with weight gain, and a distorted body image.6 There are two subtypes of AN: restricting subtype (ICD-10: F50.01) and binge/purge subtype (ICD-10: F50.02).6 Although it is not uncommon for postmenarcheal female AN patients to present with amenorrhea or oligomenorrhea, menstrual dysfunction is associated with illness severity7 and not required for an AN diagnosis.
The lifetime prevalence of AN is 0.3%–0.9%,8–10 and it is estimated that 90% of afflicted individuals are female. Although onset of the illness typically occurs in adolescence, prepubescent onset is not uncommon,11 and AN and other EDs are also diagnosed in women in midlife and older adulthood.12 Crossover between ED diagnoses and subtypes is frequent: for instance, more than half of individuals with AN restricting subtype develop AN binge–purge subtype.13 Diagnostic migration from AN to BN may be as high as 36%, with higher premorbid and lifetime BMIs being predictors of crossover.14 Crossover from BN to AN may be less common, with the estimates ranging from 4% to 27%.14,15 However, studies also report a decline in crossover rates after 5 years,14,16 with most transitions occurring during the first 3–5 years of illness.13
AN, especially in adults, is difficult to treat and is associated with disturbingly high morbidity and mortality.5,17 Family-based therapy is one of the first-line treatments for adolescent AN patients.18 Medication trials have yet to identify drugs with clear benefit that target the core pathology of the disorder, rigorously controlled psychotherapy studies are sparse for adults,4 and the need for ongoing intervention after completion of a treatment program is the norm rather than the exception.19 In specialist settings, less than 50% of AN patients achieve full recovery, roughly a third of the patients improve, and about 21% develop a chronic course.20 A 12-year outcome study reported more concerning statistics, with 27.5% of those with AN having a good outcome, 25.3% having an intermediate outcome, 39.6% having a poor outcome, and close to 8% having been deceased at the end of 12 years.21 However, research in community samples suggests a much better long-term prognosis for AN compared to clinical samples,22 suggesting that treatment-seeking individuals may present with increased severity and chronicity, thus contributing to poorer outcome in clinical settings.
Bulimia nervosa
Individuals with BN (ICD-10: F50.2) present with recurrent episodes of binge eating – consumption of a large amount of food in a short period of time, accompanied by a sense of loss of control over eating – and compensatory behaviors such as self-induced vomiting, laxative or diuretic abuse, fasting outside of binge episodes, and excessive driven exercise. From a diagnostic perspective, whereas individuals with AN binge/purge subtype meet the low-weight criterion for AN, patients with BN do not and can present in the normal weight, overweight, or obese range. Two subtypes of BN exist: purging subtype and non-purging subtype.6 The lifetime prevalence of BN is 0.8%–2.9%,8–10,23,24 and similar to AN, the majority of those who suffer from BN are women.10,25 Binge eating behavior is relatively common in the general population.9 Bulimic behaviors often have their onset during adolescence and early adult years, typically somewhat later than AN, and as many as 13% of North American college students display varying degrees of bulimic symptoms.26,27 However, regular binge eating associated with characteristic psychopathology with purging (as in BN) or without purging (as in BED, discussed below) are less prevalent and require psychiatric attention.
Cognitive behavior therapy and selective serotonin reuptake inhibitors are the first-line treatments for BN.5,28 Long-term outcome studies have consistently shown that about 55%–70% of BN patients fully or partially recover, whereas 23%–30% of the cases either become chronic or cross over to another ED.3,24,29,30 Over a decade, while 51% of those with BN will meet criteria for good outcome following psychotherapy,31 11% will still meet full diagnostic criteria, and over an additional 18.5% will still suffer from subclinical disordered eating.30 Although effective treatments for BN are available, their use among primary care clinicians may be limited.32 Furthermore, due to the secrecy and shame associated with binge eating and purging, many individuals with BN refrain from seeking treatment.33
Binge eating disorder
BED (ICD-10: F50.8) is marked by recurrent episodes of binge eating accompanied by a sense of loss of control over eating, but differs from BN, in that recurrent inappropriate compensatory behaviors are absent.6 Other characteristics of BED include distress about the binge eating, eating alone during a binge episode because of embarrassment about overconsumption, and feelings of guilt or shame after a binge episode.6 Although overvaluation of the importance of shape and weight is not a diagnostic feature of BED,6 it is commonly present.34–36 The prevalence of BED is estimated to be between 2% and 3.5%,8,9,23 and the majority of individuals with BED are either overweight or obese.37 Unlike AN and BN, the age of onset for BED tends to be later (ie, early- to mid-20s instead of adolescence),23 and the prevalence of BED is distributed more evenly across sexes.9
Similar to BN, cognitive behavior therapy, selective serotonin reuptake inhibitors, and other medications have been shown to be effective in the treatment of BED.5,38 Studies on evidence-based treatments for BED have reported a posttreatment remission rate ranging from 25% to 80%, which suggests significantly better outcome compared to AN or BN.39 However, many individuals with BED often seek treatment at weight loss clinics instead of receiving psychological or psychiatric interventions.40 It is also not uncommon for individuals with BED to cross over to BN over time,8,39 whereas BED-to-AN crossover is rare.8,41
In summary, full-syndrome and subsyndromal EDs are relatively common in the general population and are associated with a significant increase in mortality, especially in young women in the case of AN and BN. There are no treatments with strong empirical support for adult patients with AN, and it is not uncommon for patients with BN and BED to relapse in the long run; thus, it is important to focus research efforts on the biological etiology of EDs in order to gain a better understanding of risk factors and develop more effective treatment strategies.
Genetic epidemiology of EDs: family and twin studies
EDs are strongly familial. The relative risk for AN susceptibility is elevated fourfold in family members of AN probands,42 and female relatives of AN patients are up to eleven times more likely to develop AN than individuals who do not have relatives with AN.43 Furthermore, individuals who have a relative with AN or BN are at elevated risk for developing either disorder,43,44 which suggests some genetic correlation between AN and BN. Of note, however, no cases of BN were found in relatives of individuals who had the restrictive subtype of AN in one study,45 suggesting limited genetic overlap between diagnostically stable restricting AN and BN. Similar to AN and BN, BED is also found to aggregate within families46 and in a manner independent of obesity.47
The heritability estimate for AN obtained from twin studies ranges from 0.48 to 0.74, meaning that up to 74% of phenotypic variation can be explained by additive genetic factors.48–54 The heritability of strictly defined AN (ie, individuals meeting all diagnostic criteria for the disorder) is usually higher than the reported heritability of broadly defined AN that includes subsyndromal cases.52,53 heritability of BN is estimated to be between 0.55 and 0.62,50,52,55,56 and most of the variance in core BN symptoms (especially vomiting) is due to additive genetic factors.57 Interestingly, co-twins of individuals who are preoccupied with weight and shape, and/or have AN-spectrum symptoms, are more likely to develop AN, whereas co-twins of individuals with bulimic symptoms are shown to be at a greater risk for developing BN.58 One twin study identified the genetic correlation between AN and BN to be 0.79,52 which may explain the high crossover rates between the two presentations. As for BED, twin studies, using varying definitions of illness, have estimated its heritability to be between 0.39 and 0.45.59,60
Genetic factors also contribute to disordered eating behaviors and dysfunctional eating attitudes that are associated with EDs. A large population-based twin study has shown that 43% of variance in individual differences in weight- and shape-related concern, and 49% of the variance in individual differences in binge eating, can be explained by additive genetic factors.61 In addition, shared environmental factors do not have a significant effect on disordered eating in adoptive siblings, and the heritability of disordered eating is high for twins reared apart,62 further highlighting the importance of genetic factors.
Molecular genetic studies of EDs
Linkage studies
Linkage studies aim to identify genomic regions that have an increased likelihood of containing genes that are associated with a disorder or trait.63 Linkage analysis is conducted on samples of related individuals (eg, parent-offspring trios, affected sibling pairs, dense pedigrees) and does not require a priori hypotheses based on biological function or prior data.
The first genome-wide linkage analysis in EDs detected a signal at chromosome 1p34.2, with D1S3721 as a possible susceptibility locus for restricting subtype AN.64 Chromosome 1 was also implicated in AN by a Japanese group as a part of a genome-wide microsatellite study.65 A later follow-up study on this signal identified serotonin 1D (HTR1D) and opioid delta 1 receptor (OPRD1) loci to be associated with AN in 191 cases,66 and similar results were reported in 226 female AN patients in a different study.67 A subsequent linkage analysis in 196 multiplex families that incorporated covariates detected signals in regions on chromosome 2 for obsessionality, chromosome 13 for drive for thinness, and chromosome 1 for the combined drive for thinness–obsessionality trait in AN.68 For BN, chromosome 10p was reported as a potential risk locus in 308 samples obtained from multiplex families.69 Since the publication of these studies, additional loci have been identified for ED-related behaviors and phenotypes in BN, including the involvement of 4q21 for lowest illness-related BMI.70
Despite the initial wave of linkage analyses of AN and BN, the findings reported by investigators were not followed up by rigorous replication studies. One key limitation of linkage analysis is its inability to narrow down regions to an experimentally feasible number of genes. In many instances, potential susceptibility loci could span multiple megabases in size. As a genome-wide approach, linkage analysis is currently reserved for instances in which dense pedigrees are studied, and more sophisticated approaches such as genome-wide association studies (GWAS) have replaced this method.
Candidate-gene association studies
Following the initial wave of linkage studies and technological advancements, candidate-gene studies that focused on single nucleotide polymorphisms (SNPs) with a priori hypotheses based on biological function (as demonstrated in in vitro, in vivo, or animal studies) became a popular method to study the genetics of EDs. However, candidate-gene findings did not replicate due to small sample sizes, lack of rigorous correction for multiple testing, methodological heterogeneity, and potential population stratification. Although it should be noted that the field of psychiatric genetics has moved past candidate-gene methodology, this section will briefly summarize the highlights from candidate-gene studies in EDs in order to provide a historical backdrop for research in the field (Table 1). More comprehensive reviews of the candidate-gene findings in EDs are available elsewhere.71–73
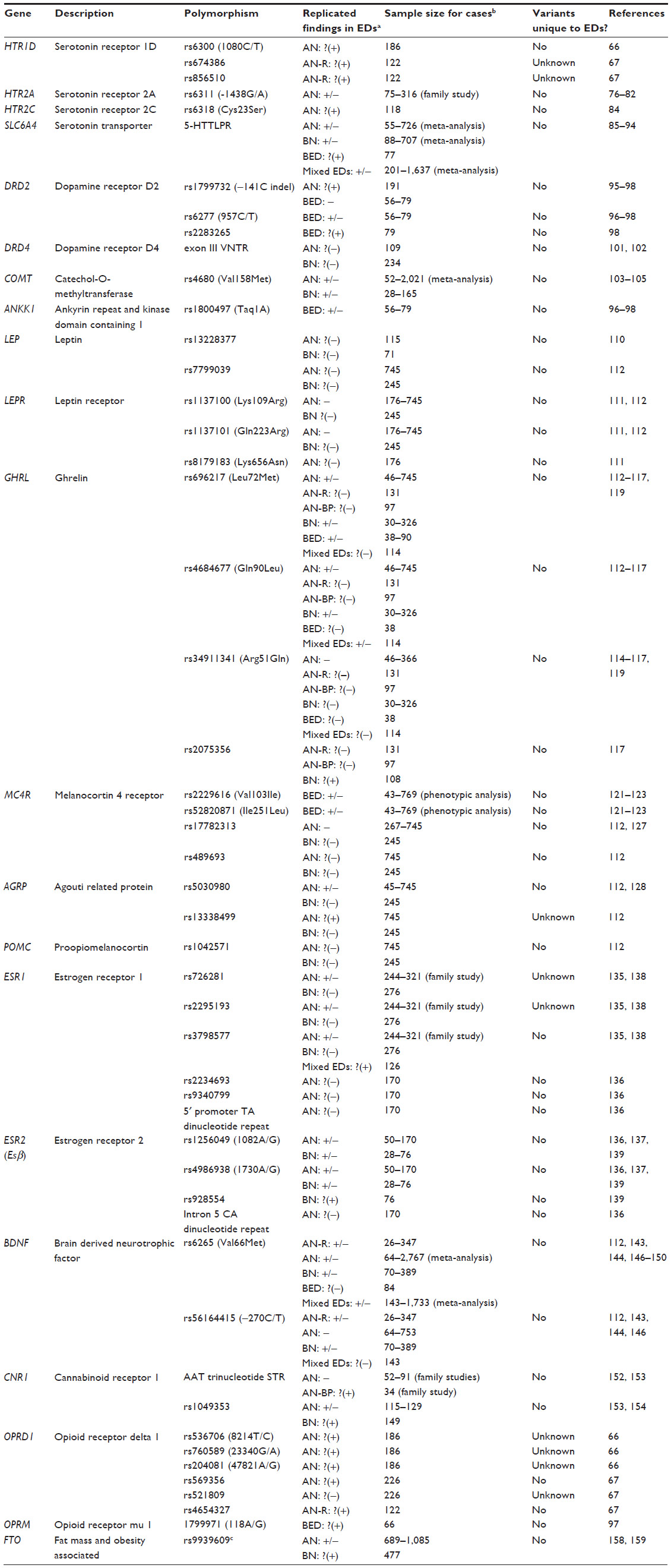 | Table 1 Overview of candidate-gene findings in eating disorders |
Because of the important role serotonin plays in eating behavior and the documented serotonergic abnormalities in EDs,74,75 serotonergic genes and their involvement in EDs have been studied extensively. As mentioned in the previous section, positive findings involving HTR1D SNPs in AN were reported by two preliminary studies.66,67 The serotonin 2A receptor (HTR2A) polymorphisms have yielded mixed findings in AN and BN,76–82 whereas other serotonin receptors have been studied in small sample sizes and not been followed up.83,84 Conflicting findings on the involvement of the 43-basepair insertion/deletion polymorphism in the promoter region of serotonin transporter gene (SLC6A4; also known as 5-HTTLPR) in AN have been published.85–89 In the case of BN, although meta-analyses have failed to replicate the previously reported association between 5-HTTLPR and BN diagnosis,89,90 the short variant has otherwise been associated with impulsivity, novelty seeking, and trauma in pilot studies of BN and BN-spectrum disorders.91–93 Additionally, one study found overrepresentation of the long-allele of 5-HTTLPR in 77 BED cases compared to 61 normal-weight controls.94
Dopamine is an important monoamine neurotransmitter that is involved in a large variety of brain functions, including feeding behavior, motor activity, and reward. Because of these connections, dopaminergic genes have been broadly studied in the context of EDs. In AN, DRD2 rs1799732 polymorphism has yielded positive findings in 191 cases,95 but no replication studies have been carried out. DRD2 and neighboring ankyrin repeat and kinase domain containing 1 (ANKK1) SNPs have been implicated in binge eating.96–98 The seven-repeat variant of the dopamine receptor D4 (DRD4) exon III variable number tandem repeats (VNTR) polymorphism has been linked to maximum lifetime BMI99,100 and a history of childhood attention-deficit/hyperactivity disorder in BN.101 In the case of AN, no association between DRD4 and BMI has been reported.102 In BN, studies have yielded mixed results regarding the overtransmission of catechol-O-methyltransferase (COMT) rs4680 variants,103,104 and a meta-analysis of eight case-control studies failed to find an association between rs4680 and AN susceptibility.105
Since the discovery of the role leptin receptor gene (Lepr) disruptions play in obesity and hyperphagia in mice, the leptinergic system has been one of the earliest targets for the genetic studies of obesity and weight regulation.106 Patients with acute AN have low plasma leptin levels,107,108 and serum leptin levels have also been correlated with BMI in patients with EDs.109 However, research on the role of the leptin gene (LEP) and LEPR has yielded variable findings in AN and BN.110–112 Ghrelin, the natural leptin antagonist often referred to as the hunger hormone, has also been studied in the context of EDs. Similar to LEP and LEPR, conflicting findings have been reported with regards to the relationship between the ghrelin gene (GHRL) polymorphisms and AN or BN;113–118 however, GHRL has yielded preliminary findings in a pilot study with 90 individuals with BED.119
Stimulation of the melanocortin system leads to a reduction in food intake and weight, and details of this pathway are discussed in more detail in the “Animal models” sections. Defects in the melanocortin 4 receptor gene (MC4R) have been a known cause of autosomal dominant obesity, accounting for 6% of all obesity cases;120 however, the role of MC4R mutations in BED remains controversial.121–123 MC4R haploinsufficiency was associated with maximum lifetime BMI in BN in one case report.124 Two separate studies failed to find an association between obesity-related MC4R common genetic variants125,126 and AN112,127 or BN.112 The research on the agouti related protein gene (AGRP), encoding the inverse agonist of melanocortin receptors, has mostly been limited to rs5030980, located in the coding region. Although two studies reported positive preliminary associations involving this polymorphism in AN,113,128 another study found a preliminary association with rs13338499 with minimum illness-related BMI in 745 individuals with AN, but no association with BN in 245 cases.112 In the case of pro-opiomelanocortin (POMC), missense mutations have been documented in a patient with AN,129 but no significant associations with common variants have been reported in AN or BN.112
The role of sex hormones in the development of AN and BN has recently garnered significant research attention. An inverse correlation between age of menarche and ED susceptibility has been reported, and since potential overlap between genetic factors linked to an earlier age of menarche and ED risk has been documented,130 there is some evidence for at least partial involvement for sex hormones in EDs. Although an intrauterine masculinization effect has been proposed for female twins with EDs who have a male co-twin,131 other twin studies reported no relationship between sex of the twin pair and disordered eating.132,133 Conversely, males with a female twin are reportedly at a higher risk of developing AN than females with a female twin,134 which suggests a possible role for intrauterine female hormone exposure and increased ED risk in men. Thus far, the focus on the investigation of genes encoding sex hormones has been on the estrogen receptor 1 (ESR1) and estrogen receptor 2 (ESR2; also known as ERβ) genes in the context of AN, and the studies have yielded variable findings.135–138 One study reported a positive association between ERβ and BN in 76 women.139 More recently, high-throughput sequencing projects (reviewed below in detail) have also implicated estrogen-related genes.140,141
Brain derived neurotrophic factor (BDNF) is a protein that supports the growth, survival, differentiation, and assigned function of neurons. In terms of eating behavior, BDNF is involved in appetite suppression by downstream regulation of melanocortin signaling in the hypothalamus.142 BDNF rs6265 (commonly referred to as Val66Met) has been extensively researched and implicated in AN and BN by various studies,99,143–148 but the nature of the findings has not been consistent.112,113,149,150 One study reported an association involving this polymorphism and binge eating frequency in 84 women with BED,148 which has yet to be replicated. The BDNF rs56164415 polymorphism (commonly referred to as –270C/T) and its role in EDs has been more controversial,99,112,143–146,151 especially due to the challenges associated with studying this low minor allele frequency SNP in small study samples.
Other genes have also been studied in the context of EDs. For instance, mixed findings involving the overtransmission of cannabinoid receptor 1 (CNR1) variants in AN have been reported,152,153 and preliminary case-control findings need replication.154 As mentioned in the “Linkage studies” section, preliminary results involving the OPRD1 gene have been reported in AN,66,67 whereas opioid receptor mu 1 gene (OPRM1) has been implicated in hedonic eating in one study with 300 participants.155 The fat mass and obesity-associated gene (FTO), which has been identified as an obesity locus in multiple GWAS publications,156,157 has yielded mixed findings regarding a possible association with AN or BN,127,158,159 and studies have yet to be conducted on its possible role in BED. Thus far, the most comprehensive candidate-gene study of AN has investigated the role of 182 genes in 1,085 AN cases and 677 healthy controls, in which none of the markers reached statistical significance following correction.160
In summary, various gene systems and polymorphisms – due to their known biological function or previous significant findings in other psychiatric disorders – have been studied in AN, BN, and to a smaller extent in BED; however, conclusive evidence on their role in EDs is uncertain due to small sample size, potential population stratification, and lack of replication. Considering these important limitations, any positive association reported in the literature should be interpreted with extreme caution.
GWAS
In contrast to candidate-gene studies that rely on biology and prior findings to select a small number of genes and devise a priori hypotheses, the GWAS method consists of a scan of the entire genome in a hypothesis-free manner.161 Recent advances in the field allow for millions of loci to be genotyped on the same genotyping chip, and the assays are designed in a way that utilizes high linkage disequilibrium between markers in order to ensure dense coverage of the genome. However, due to the inclusion of millions of markers in the same analysis, it is important to apply stringent statistical cutoffs to effectively control for potential false-positive findings that may be an artifact of multiple testing.162
The first GWAS of AN – carried out in 1,033 AN cases – reported a large and rare copy number variant on 13q12 present in two individuals but did not find any genome-wide significant loci in the case-control analysis.163 A GWAS collaboration on a twin sample also failed to find genome-wide significant susceptibility loci but provided some evidence for the possible involvement of eight loci with various ED-related phenotypes such as drive for thinness, body dissatisfaction, bulimia, weight fluctuations, as well as childhood obsessive-compulsive personality disorder trait.164 Similarly, another GWAS on disordered eating in female twins did not report any genome-wide significant findings, but a number of genes were implicated in AN- and BN-spectrum disorder phenotypes.165
The largest and most rigorous GWAS in AN – part of the Wellcome Trust Case-Control Consortium 3 – included 2,907 AN cases of European ancestry and 14,860 ancestry-matched female controls in the discovery meta-analysis.166 Although there were no genome-wide significant findings in the discovery dataset, 72 independent markers with the lowest P-values were selected for replication, and approximately 76% of these markers produced results in the same direction as the discovery sample.166 This was a promising indication that the prioritized set of genomic variants likely contained true positive signals for increased AN risk; however, the study was unable to detect effects due to lack of statistical power.166 Further replication studies, larger sample sizes, and experiments on expression and function are required to gain a better understanding of the role of these genes in AN. Thus far, there have not been any GWAS conducted in BN or BED.
Study of rare variants
With the recent advancements in high-throughput sequencing, it is now much easier to detect rare variants (minor allele frequency of less than 1%) and significantly cheaper to sequence a larger number of cases than ever before. While common variants are easier to detect and may confer risk for a larger number of individuals in the population, they also have small effect sizes by themselves. Rare variants, on the other hand, are more likely to have higher penetrance compared to common variants but may only be present in a smaller number of cases. Currently, the most commonly used high-throughput sequencing methods are targeted gene sequencing, exome sequencing, and whole genome sequencing. Targeted gene sequencing allows researchers to focus their efforts on capturing a select number of genes in a manner similar to candidate-gene association studies, whereas exome sequencing consists of sequencing all exons in the genome, and although more costly, whole genome sequencing offers a complete view of an individual’s entire DNA sequence. More recently, exome genotype chips that cover an extensive set of novel, rare, or putative functional exonic variants selected based on previous sequencing projects have been developed.
To date, only one high-throughput sequencing study in unrelated ED cases has been published. While the sequencing of 152 candidate genes in a sample of 261 AN cases and 73 controls did not yield any genome-wide significant findings, epoxide hydrolase 2 (EPHX2) – one of the top hits in the sequence analysis – was associated with depression and anxiety scores in a small independent AN replication sample and was further linked to BMI and elevated cholesterol measures in healthy controls from a larger longitudinal population study.141 Another preliminary study combined linkage analysis, exome sequencing, and whole genome sequencing efforts in the examination of two densely affected ED pedigrees. The study reported a missense mutation in estrogen-related receptor alpha (ESRRA) in the first pedigree, whereas a potentially deleterious mutation in histone deacetylase 4 (HDAC4) was found in the second pedigree.140 As mentioned in the “Candidate-gene association studies” section of this review, these genes are deemed to be of biological interest because of their possible role in the estrogen system;140 however, the results of this study need to be replicated, and furthermore, it is important to consider the possibility of any identified rare mutations being private to each pedigree and thus not necessarily explaining common genetic risk for EDs.
Whole genome and exome sequencing projects have yet to be carried out in unrelated AN cases and controls, and considering the rare nature of the sequencing variants, samples sizes in the tens of thousands are required to obtain meaningful data for ED case-control studies. Thus far, there are no published high-throughput sequencing studies in BN or BED.
Gene regulation related to EDs: animal studies
Over the past 20 years, there has been rapid progress in using laboratory animals to model EDs. A significant number of these studies have used behavioral manipulations in wild-type strains of rodents to produce distinct ED-related phenotypes, a topic that has been comprehensively reviewed by others.167–169 In this section, we restrict our discussion to experiments using model organisms with either spontaneous or induced genetic variations. Globally, these studies have revealed a complex network of genes functioning within anatomically and genetically defined neuronal ensembles to regulate different modalities of feeding. We discuss how these genes function within evolutionarily conserved neural circuits and how maladaptive alterations of neural-circuit activity may explain ED pathology (summarized in Table 2).
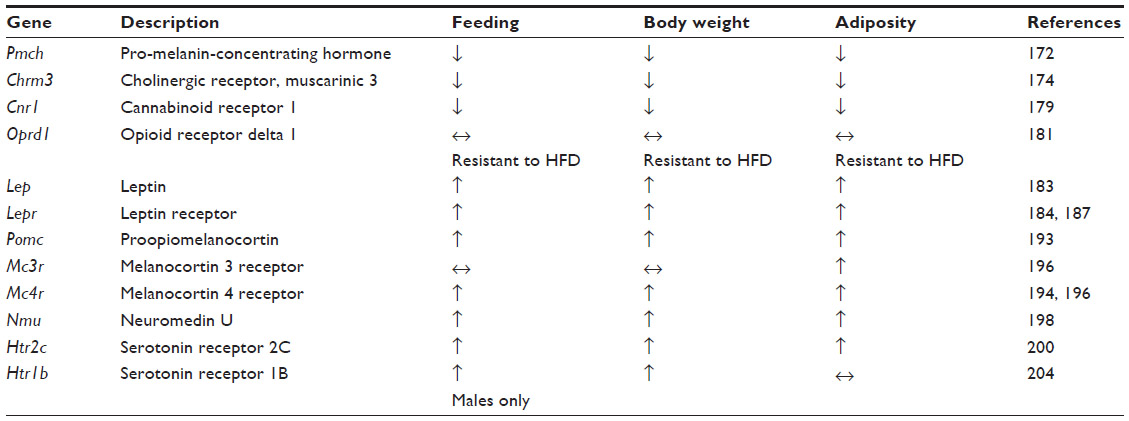 | Table 2 Eating disorder phenotypes in mouse knockout models |
Animal models of AN
One of the most long-standing AN mouse models is the anx/anx mouse. This line contains an allele that arose by spontaneous mutation wherein homozygous carriers display reduced growth and an emaciated appearance. These animals have a significantly reduced body weight that is apparent by 10 days of age, is due to reduced food intake,170 and leads to death approximately 22 days postnatal. These animals also have other neurological abnormalities like body tremors, uncoordinated gait, hyperactivity, and head weaving. By virtue of their decreased body weight and food consumption, the anx/anx mice possess strong face validity as a model of AN. More recent studies, however, suggest that the hypophagia is due to mitochondrial dysfunction and oxidative stress in hypothalamic circuits. This gross abnormality in central nervous system (CNS) function may ultimately derive from a mutation of the NADH dehydrogenase (ubiquinone) 1α-subcomplex, assembly factor 1 (Ndufaf1) gene which is located in a previously mapped genomic interval of the anx/anx allele.171 As no genomic studies in AN patients to date have implicated CNS mitochondrial protein dysfunction, the construct validity of the anx/anx mouse as a model AN is still in question.
Gene knockout studies using homologous recombination in mice have revealed a host of genes involved in feeding and metabolism. Although the clinical validity of these models is not always apparent due to a lack of genome-wide associations in AN patients, these experiments have nonetheless helped reveal the fundamental gene networks and neural circuitry necessary for feeding. Among reverse genetic knockout studies in mice, there are specific genes that produce hypophagic and lean phenotypes when altered. The loss of the neuropeptide melanin-concentrating hormone (MCH), encoded by the pro-melanin-concentrating hormone gene (Pmch–/– mice), results in reduced body weight, body adiposity, and food intake.172 Conversely, overexpression of MCH through intracerebroventricular injection of this peptide is known to increase food intake.173 MCH is highly expressed in hypothalamic nuclei like the lateral hypothalamus (LH), but the role of endogenous MCH released from LH neurons in regulating feeding is still unclear.
Hypophagia and reduced body weight and fat are triggered by loss of the M3 muscarinic receptor (Chrm3–/– mice). They also display a reduced capacity for feeding, induced by injection of the orexigenic agent AGRP, but an intact response to MCH,174 suggesting that M3 acts downstream of AGRP, but either upstream or in a pathway parallel to MCH. The comprehensive mechanism by which the M3 muscarinic receptor promotes feeding and normal adiposity is still unknown, but it may accomplish its orexigenic role through signaling in the nucleus accumbens, where cholinergic signaling is known to modulate feeding.175–178
One of the most robust lean-inducing knockouts is the cannabinoid receptor type 1 (CB1) null mouse (Cnr1–/– mice). Cnr1–/– mice display a highly significant reduction in body weight when fed standard chow, are resistant to the obesogenic effects of a high-fat diet, and show a reduction in food intake of either standard or high-fat diet.179 Recently, it was shown that CB1s are expressed on glutamatergic neurons that project to inhibitory granule cells in the medial olfactory bulb and normally dampen excitatory synaptic transmission at these synapses to drive food intake.180 The authors elegantly demonstrated that expression of CB1s in the anterior olfactory nucleus is both necessary and sufficient for fasting-induced hyperphagia. Thus the phenotypes apparent in the Cnr1–/– mice are likely due to its function in the anterior olfactory nucleus to medial olfactory bulb circuit.
Lastly, loss of the delta opioid receptors (Oprd1–/– mice) can produce leanness, resistance to high fat diet-induced weight gain, and an increase in thermogenesis.181 Although the current mechanism by which delta opioid receptors promote feeding is unknown, it is highly expressed in the mouse olfactory bulb and anterior olfactory nucleus.182 Thus, one hypothesis is that these receptors may act in a pathway that contains CB1s. Furthermore, Oprd1–/– mice may have construct validity as a model of AN as point mutations in the homologous human OPRD1 locus have been associated with AN in preliminary studies, as discussed previously.66
Animal models of BED and BN
One of the most clinically relevant phenotypes for BED and BN that can be modeled in rodents is binge eating. BED is commonly comorbid with obesity, which is also a prevalent hallmark in mouse models of binge eating.37 The most well-known models of obesity in mice are the Lepob/ob or Leprdb/db mice, which arose due to spontaneous mutations. Similar to alterations in humans, a loss of function mutation in these genes results in hyperphagia and obesity.183–190 Since the discovery of leptin and its cognate receptor, dozens of studies have revealed how leptin is released in the periphery from adipose tissue and acts on leptin receptors that are expressed in the brain. Their expression is highly enriched on neurons in the arcuate nucleus (ARC) of the hypothalamus,191,192 which also expresses POMC. POMC is then processed and released as alpha melanocyte-stimulating hormone and binds downstream melanocortin receptors expressed in other hypothalamic nuclei like the paraventricular nucleus of the hypothalamus and LH. These genes are critical for feeding and energy balance, as loss of Pomc, Mc3r, or Mc4r are each sufficient to produce hyperphagia and obesity.193–196 Selective restoration of MC4R in the paraventricular nucleus and a population of central amygdala neurons expressing the transcription factor Sim1 incompletely rescues the hyperphagia and obesity of the Mc4r–/– mice.197 These data are consistent with the hypothesis that the anorexigenic properties of melanocortin signaling may derive from its release at the paraventricular nucleus and central amygdala.
In addition to the POMC and AGRP pathways, the ARC expresses additional neuropeptides that may represent critical nodes for feeding. Neuromedin U (NMU) is a highly conserved neuropeptide that is expressed throughout the body including the brain and the gut. Within the brain, the expression of Nmu mRNA is enriched in the ARC as well as the dorsomedial and ventromedial hypothalamic nuclei. Nmu–/– mice display increased body weight, adiposity, and hyperphagia.198 These results suggest that NMU normally mediates anorexia, and subsequent research has shown that the NMU 2 receptor is critical in the reduction of feeding mediated by central application of NMU.199 However, to date, neither NMU nor its receptor have been reported in genetic associations with ED patients.
One of the first genetically induced models of binge feeding to be developed was mice lacking the serotonin 2C receptor (5-HT2C; Htr2c–/– mice). These mice are overweight relative to their wild-type littermates, display elevated adiposity, are hyperphagic, and display a blunted response to the anorexigenic properties of the serotonin receptor agonist meta-Chlorophenylpiperazine.200 Recent research suggests that the functional significance of 5-HT2C in regulating food intake and metabolism may derive from its expression in the ARC POMC neurons, as selective restoration of 5-HT2C in POMC neurons in the Htr2c–/– mice rescues hyperphagia, obesity, adiposity, and elevated circulating leptin levels.201 The regulation of ARC POMC neurons by serotonin may be more complex, however, as these neurons also express serotonin 1B receptors (encoded by the Htr1b gene), the activation of which can disinhibit POMC neurons to mediate its anorexic effects.202,203 Interestingly, Htr1b–/– mice display increased body weight without causing obesity, but the gene’s cell autonomous role in the ARC POMC neurons has not been explored.204
One valid critique of each of these models is how closely they resemble the disordered eating phenotypes of ED patients. The presence of hyperphagia, for example, in the aforementioned mouse models of BED only partially captures the construct of binge eating in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Moreover, the majority of the BED mouse models are also obese, and although there is a significant comorbidity between obesity and BED, not all patients with BED are obese.37,205
Study of epigenetic changes and gene regulation linked to EDs
Overview of epigenetic mechanisms
Epigenetics refers broadly to the regulation of gene function without a change in the DNA coding sequence. Epigenetic modulation can occur through several discrete mechanisms that include DNA methylation, changes in chromatin structure through modification of histone N-termini with different chemical moieties, and gene expression changes through the actions of small non-protein encoding RNAs.206,207 The majority of the published studies that examine epigenetic alterations in the context of EDs focus on the role of DNA methylation (as discussed in the next section and summarized in Table 3), so we will limit our discussion to how DNA methylation occurs and how it influences gene expression.
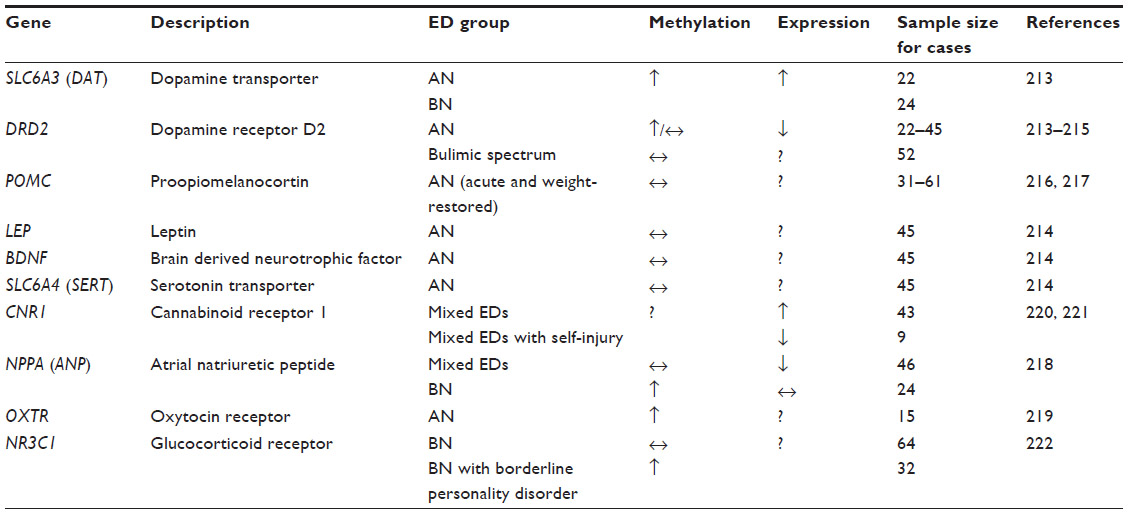 | Table 3 Genes associated with epigenetic changes in eating disorders |
DNA methylation occurs at the 5′ end of cytosine nucleotides that occur almost exclusively in 5′-CpG-3′ islands (CGIs). Methylation of these cytosines is common throughout the genome and is necessary to repress the expression of specific genes throughout development.208 CpG sequences are not evenly distributed throughout the genome and are preferentially enriched in putative gene promoter regions. Methylation of cytosine nucleotides at CGIs occurs through the actions of DNA methyltransferases, which results in the addition of a methyl group that sits in the DNA major groove. Methylation of CGIs in gene promoter regions normally acts to recruit repressive factors like histone deacetylases and histone methyltransferases, thereby repressing transcription of the methylated gene target through subsequent chromatin modifications.209 It is estimated that 6%–8% of the CGIs are methylated in human tissues like blood, brain, and spleen, and that most methylated CGIs are associated with known gene promoter regions. Interestingly, certain transcription factors that govern cell fate choice show tissue-specific methylation patterns.210,211 From an ED perspective, de novo or maintenance DNA methylation provides a mechanism by which to alter gene expression patterns and molecular physiology within defined cell types like CNS neurons, and within peripheral tissues like adipose tissue and the gut, to alter feeding behavior and metabolism.
Epigenetic studies of EDs
In recent years, the study of epigenetics has risen in popularity. It has been proposed that in the case of obesity, environmental factors such as an abundance of high-fat and high-carbohydrate foods may increase the expression of adiposity-related genes in vulnerable individuals;212 in a similar fashion, it is also possible that an environment that promotes thinness can lead to the overexpression of genes that suppress appetite and/or weight in individuals who are already genetically vulnerable to weight suppression. In accordance with these points, although there may not necessarily be any differences in nucleotide sequence between AN cases and controls, it is possible that gene expression or methylation patterns may significantly vary in individuals with EDs. Thus far, most epigenetic studies have focused on the promoter-specific methylation of the candidate genes that have been studied previously in EDs (discussed in detail below and summarized in Table 3). The majority of the studies comprised AN cases, whereas a small number of studies included BN or BN-spectrum individuals (summarized in Table 3). To date, no epigenetic studies in BED have been published.
Due to their association with reward, dopaminergic genes have been especially of interest to epigenetic researchers. It was first reported that AN patients (n=22) may present with elevated dopamine transporter (SLC6A3; also referred to as DAT) mRNA expression due to hypermethylation of the gene’s promoter region, as well as DRD2 promoter hypermethylation.213 The DAT hypermethylation was also observed in 24 BN cases.213 However, another study failed to find significant differences in the promoter-specific DNA methylation for DRD2, LEP, BDNF, and serotonin transporter (SLC6A4, also referred to as SERT) genes in AN, and similarly, no association between BMI and DNA methylation was observed.214 In the case of BN and related psychopathology, no difference between women with bulimic spectrum disorder and healthy controls was observed in DRD2 promoter methylation; however, a subgroup of participants with bulimic spectrum disorders with comorbid psychopathology had increased DRD2 promoter methylation in this preliminary study.215
POMC has also garnered attention due to its important role in appetite regulation. Expression of the functionally relevant long POMC mRNA was linked to leptin levels in 31 individuals with acute AN.216 In a different study by the same group of researchers, no association between BMI or AN status (acute versus weight recovered) and POMC promoter methylation was observed, but hypomethylation was associated with cigarette smoking,217 suggesting a possible role for environmental factors rather than AN diagnosis in POMC expression.
Various other candidate genes have been the subjects of epigenetic studies. For instance, decreased atrial natriuretic peptide (NPPA, also referred to as ANP) mRNA levels have been reported in 46 patients with mixed-diagnosis EDs, whereas hypermethylation of the ANP promoter was observed in 24 BN cases.218 Another pilot study detected a number of CpG sites in the oxytocin receptor gene (OXTR) with higher than average methylation levels in 15 AN patients, which was negatively associated with BMI.219 Significantly higher levels of CNR1 (also referred to as CB1) receptor mRNA in the blood of 43 patients with either AN or BN compared to 26 healthy controls were also reported, but paradoxically, expression was inversely correlated with ED psychopathology,220 which highlights the need for replication of findings reported by studies with small sample sizes in general. Another small-scale study reported a downregulation of CB1 receptor mRNA in nine ED patients who engage in self-injury.221 In the case of BN, increased methylation of the glucocorticoid receptor gene (NR3C1) promoter was observed in 32 BN patients with comorbid borderline personality disorder or a history of suicidality, whereas no other case-control differences were significant between women with BN and healthy controls.222 Taken all together, these results involving various candidate genes need to be replicated in much larger sample sizes in order for researchers to be able to reach more informed conclusions regarding their possible involvement in EDs.
When considering global methylation rather than specific candidate-gene promoter regions, a pattern of significant global DNA hypomethylation was reported in 22 AN patients compared to 30 healthy controls, and a similar trend was observed in 24 BN cases.223 Similar – albeit modest – reduction in whole-blood global DNA methylation was also reported in 32 adolescents with AN, which was independent of ED-related psychopathology and illness severity.224 However, another study failed to replicate these findings and reported no alterations in global or gene-specific DNA methylation in AN compared to controls.225
In summary, epigenetics is a growing research area that holds the potential to make important contributions to our understanding of the role of elements outside of DNA coding sequence in ED susceptibility. Although a number of preliminary findings have emerged, whether these associations will be replicated is yet to be seen. Furthermore, a number of important limitations akin to those described in the section on “Candidate-gene association studies” (including small sample sizes, almost exclusive focus on candidate genes selected based on a priori hypotheses, and methodological heterogeneity) also merit consideration when reviewing the literature. Additionally, EDs are, at least in part, brain disorders, and considering the tissue specificity of epigenetic modifications, use of blood and buccal cells as proxies to brain tissue is another limitation of the published studies. In the case of candidate-gene promoter methylation studies, no control genes were included to determine whether the methylation changes are specific to the regions of interest or of global nature.
Future directions
In the last few decades, significant advances have emerged in our understanding of the etiology of AN, BN, and BED. Our knowledge of genetics and its contribution to the etiology of psychiatric disorders has greatly increased, and with the development of more sophisticated laboratory and bioinformatic tools, researchers are now able to make more meaningful connections between genomic variants and complex disorders.
Currently, the greatest barrier to identification of genetic variants for EDs is sample size. The largest GWAS in AN conducted to date had over 5,000 samples in the discovery and replication datasets combined,166 and yet the study was underpowered to produce genome-wide significant results. The only way to reach the statistical power required to find risk variants in AN and other EDs without sacrificing statistical rigor is to focus research efforts on the recruitment of large case-control cohorts. For instance, the schizophrenia analysis conducted by the Psychiatric Genomics Consortium (PGC) initially yielded promising findings but failed to produce any genome-wide significant results before reaching around 10,000 samples.226,227 Since that threshold has been met, the PGC GWAS projects have yielded many significant associations that were successfully replicated, thus making enormous contributions to our understanding of genetic risk factors for numerous psychiatric disorders.162,227–234 In a similar fashion, there is no reason to doubt that GWAS in EDs can also achieve this outcome once statistical power is attained. While boosting sample size, future GWAS efforts should also include BN, BED, and analysis of large male cohorts alongside females in order to make meaningful discoveries about the genetic risk factors for EDs in men.
As epigenetic investigations gain popularity in EDs, researchers should consider the complex nature of epigenetic regulation and focus their efforts at a genomic level as opposed to the study of select candidate-gene promoters. In addition, the role of environmental and individual risk factors further complicate epigenetic research, and sample sizes as large as those required for GWAS (if not larger) may be warranted to discover meaningful and replicable epigenetic risk factors. Future research should also explore epigenetic mechanisms other than DNA methylation, including but not limited to histone modification, chromatin remodeling, and microRNA studies. Finally, it will be crucial for research to tie any epigenetic changes to actual protein expression in disease-relevant tissues (such as the brain), so future studies should include relevant biomarkers in research design as well.
It is also important to consider rare variants alongside common variants for conferring ED genetic risk, which may give researchers a better chance to explain the genetic contribution to EDs.235–237 Indeed, exome sequencing has contributed to improving our understanding of the role of rare and de novo variants in psychiatric disorders such as autism, bipolar disorder, and schizophrenia.238–243 In the future, exploration of rare exonic variants with rigorous hypothesis-free methodology in large samples could yield important new information on the effects of rare variants in EDs.
As for the study of ED-relevant animal models, the past 5 years have seen tremendous growth in novel strategies to manipulate model system genomes like zinc finger nucleases, clustered regulatory interspaced palindromic repeats (CRISPR)/Cas9, and transcription activator-like effector nucleases (TALENs).244 These strategies offer improved efficiency for gene targeting and speed at generating new rodent strains in one step245 and can even be applied to genome-wide screens in human cancer and stem cell models.246 Moreover, these approaches can be used in a blue light inducible fashion to control the regulation of gene function temporally and spatially.247 This is an important consideration to avoid lethality or compensatory effects that may derive from loss of a gene constitutively and at an organism-wide level.
Many of the previously described animal models measure body adiposity and feeding, but do not examine the role of particular genes in construct valid models of ED like the activity-based-anorexia model for AN and a limited-access binge eating paradigm for BED.167 As these genes function in distinct neural circuits, circuit level manipulations like optogenetics and designer receptors exclusively activated by designer drugs (DREADDs) that allow for the selective activation or inhibition of genetically defined cell types will be necessary to make causal statements about the role of that circuit in an ED-relevant behavior. With regards to feeding, there are a growing number of studies that describe the induction of feeding behavior following activation of mostly hypothalamic-centric circuits.248–256 As discussed in the Animal models sections, these studies suggest that there exist many parallel, redundant circuits that are sufficient to induce voracious feeding. These studies do not, however, show that these circuits are necessary under normal, non-artificial conditions to promote binge eating or satiety. We argue that a more comprehensive mechanistic investigation will involve the use of inhibitory opsins and DREADDs, temporally and spatially induced genetic modifications, and behavioral models that more closely resemble EDs. Lastly, the comorbidity of EDs with conditions like anxiety and substance abuse suggests there may exist shared, overlapping genetic and neural networks that regulate feeding, reward, and anxiety. Experimental limitations notwithstanding, the use of construct valid ED animal models to determine genetic influences on feeding and metabolism in parallel with other ED-related phenotypes promises a more complete view of genes that might represent novel, specific pharmacotherapeutic targets for ED.
Additionally, it is possible that comorbid psychopathology and traits linked to EDs – such as obsessionality in AN and impulsivity in BN – may have important implications for human genetic studies. For instance, the prevalence of obsessive compulsive disorder is significantly elevated among the first-degree relatives of ED probands compared to controls,44 and a pilot study reported shared genetic etiology between AN and obsessive compulsive disorder.257 This possible etiological link suggests perfectionistic and obsessive traits as factors in the disordered eating behavior, suppression of body weight, and maintenance of low weight in AN. In the case of BN, impulsivity could be among the reasons for the inability to suppress body weight despite the drive for thinness. Indeed, prevalence of childhood attention-deficit/hyperactivity disorder – in which impulsivity is often a key symptom – is considerably high among BN cases compared to the general population.104,258,259 Furthermore, there may be important clinical, neurobiological, and genetic overlap between BN and alcohol/substance use disorders, which are conditions also characterized by impulse dysregulation.260,261 In future ED genetic research, these differences in comorbidities and key psychopathology should be carefully considered in relation to potentially different genetic etiologies for AN, BN, and BED.
Finally, there is merit in stepping beyond the diagnostic criteria of full-syndromal EDs to explore the important phenotypes in a cross-disorder manner. Indeed, recent work by the PGC highlights the importance of shared genetic risk loci in the etiology of five separate psychiatric disorders,232 and with the addition of AN to the PGC working groups, future directions include the study of important ED-related phenotypes (eg, highest and lowest illness-related BMIs, anxiety, anhedonia, obsessionality, impulsivity, etc) in a cross-disorder manner in order to maximize sample size and explore the possibility of common genetic etiology for these overlapping phenotypes across psychiatric diagnoses.
Acknowledgment
Drs Yilmaz and Hardaway are supported by the National Institutes of Health (NIH) Grant T32MH076694 (PI: Bulik).
Disclosure
Dr Bulik is a consultant for Shire Pharmaceuticals. Other authors report no conflicts of interest in this work.
References
Hoang U, Goldacre M, James A. Mortality following hospital discharge with a diagnosis of eating disorder: national record linkage study, England, 2001–2009. Int J Eat Disord. 2014;47(5):507–515. | |
Suokas JT, Suvisaari JM, Gissler M, et al. Mortality in eating disorders: a follow-up study of adult eating disorder patients treated in tertiary care, 1995–2010. Psychiatry Res. 2013;210(3):1101–1106. | |
Smink FR, van Hoeken D, Hoek HW. Epidemiology, course, and outcome of eating disorders. Curr Opin Psychiatry. 2013;26(6):543–548. | |
Attia E. Anorexia nervosa: current status and future directions. Annu Rev Med. 2010;61:425–435. | |
American Psychiatric Association. Treatment of patients with eating disorders, third edition. American Psychiatric Association. Am J Psychiatry. 2006;163(Suppl 7):4–54. | |
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA: American Psychiatric Association; 2013. | |
Attia E, Roberto CA. Should amenorrhea be a diagnostic criterion for anorexia nervosa? Int J Eat Disord. 2009;42(7):581–589. | |
Stice E, Marti CN, Rohde P. Prevalence, incidence, impairment, and course of the proposed DSM-5 eating disorder diagnoses in an 8-year prospective community study of young women. J Abnorm Psychol. 2013;122(2):445–457. | |
Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry. 2007;61(3):348–358. | |
Swanson SA, Crow SJ, Le Grange D, Swendsen J, Merikangas KR. Prevalence and correlates of eating disorders in adolescents. Results from the national comorbidity survey replication adolescent supplement. Arch Gen Psychiatry. 2011;68(7):714–723. | |
Halmi KA. Anorexia nervosa: an increasing problem in children and adolescents. Dialogues Clin Neurosci. 2009;11(1):100–103. | |
Gagne DA, Von Holle A, Brownley KA, et al. Eating disorder symptoms and weight and shape concerns in a large web-based convenience sample of women ages 50 and above: results of the Gender and Body Image (GABI) study. Int J Eat Disord. 2012;45(7):832–844. | |
Eddy KT, Keel PK, Dorer DJ, Delinsky SS, Franko DL, Herzog DB. Longitudinal comparison of anorexia nervosa subtypes. Int J Eat Disord. 2002;31(2):191–201. | |
Tozzi F, Thornton LM, Klump KL, et al. Symptom fluctuation in eating disorders: correlates of diagnostic crossover. Am J Psychiatry. 2005;162(4):732–740. | |
Fichter MM, Quadflieg N. Six-year course of bulimia nervosa. Int J Eat Disord. 1997;22(4):361–384. | |
van Son GE, van Hoeken D, van Furth EF, Donker GA, Hoek HW. Course and outcome of eating disorders in a primary care-based cohort. Int J Eat Disord. 2010;43(2):130–138. | |
Arcelus J, Mitchell AJ, Wales J, Nielsen S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry. 2011;68(7):724–731. | |
Bulik CM, Berkman ND, Brownley KA, Sedway JA, Lohr KN. Anorexia nervosa treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40(4):310–320. | |
Long CG, Fitzgerald KA, Hollin CR. Treatment of chronic anorexia nervosa: a 4-year follow-up of adult patients treated in an acute inpatient setting. Clin Psychol Psychother. 2012;19(1):1–13. | |
Steinhausen HC. The outcome of anorexia nervosa in the 20th century. Am J Psychiatry. 2002;159(8):1284–1293. | |
Fichter MM, Quadflieg N, Hedlund S. Twelve-year course and outcome predictors of anorexia nervosa. Int J Eat Disord. 2006;39(2):87–100. | |
Keski-Rahkonen A, Hoek HW, Susser ES, et al. Epidemiology and course of anorexia nervosa in the community. Am J Psychiatry. 2007;164(8):1259–1265. | |
Kessler RC, Berglund PA, Chiu WT, et al. The prevalence and correlates of binge eating disorder in the World Health Organization World Mental Health Surveys. Biol Psychiatry. 2013;73(9):904–914. | |
Keski-Rahkonen A, Hoek HW, Linna MS, et al. Incidence and outcomes of bulimia nervosa: a nationwide population-based study. Psychol Med. 2009;39(5):823–831. | |
Allen KL, Byrne SM, Oddy WH, Crosby RD. DSM-IV-TR and DSM-5 eating disorders in adolescents: prevalence, stability, and psychosocial correlates in a population-based sample of male and female adolescents. J Abnorm Psychol. 2013;122(3):720–732. | |
Halmi KA, Falk JR, Schwartz E. Binge-eating and vomiting: a survey of a college population. Psychol Med. 1981;11(4):697–706. | |
Rush CC, Becker SJ, Curry JF. Personality factors and styles among college students who binge eat and drink. Psychol Addict Behav. 2009; 23(1):140–145. | |
Shapiro JR, Berkman ND, Brownley KA, Sedway JA, Lohr KN, Bulik CM. Bulimia nervosa treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40(4):321–336. | |
Steinhausen HC, Weber S. The outcome of bulimia nervosa: findings from one-quarter century of research. Am J Psychiatry. 2009;166(12):1331–1341. | |
Keel PK, Mitchell JE, Miller KB, Davis TL, Crow SJ. Long-term outcome of bulimia nervosa. Arch Gen Psychiatry. 1999;56(1):63–69. | |
Clausen L. Review of studies evaluating psychotherapy in bulimia nervosa: the influence of research methods. Scand J Psychol. 2004;45(3):247–252. | |
Jaite C, Hoffmann F, Glaeske G, Bachmann CJ. Prevalence, comorbidities and outpatient treatment of anorexia and bulimia nervosa in German children and adolescents. Eat Weight Disord. 2013;18(2):157–165. | |
Hepworth N, Paxton SJ. Pathways to help-seeking in bulimia nervosa and binge eating problems: a concept mapping approach. Int J Eat Disord. 2007;40(6):493-504. | |
Mond JM, Hay PJ, Rodgers B, Owen C. Recurrent binge eating with and without the “undue influence of weight or shape on self-evaluation”: implications for the diagnosis of binge eating disorder. Behav Res Ther. 2007;45(5):929–938. | |
Grilo CM, Hrabosky JI, White MA, Allison KC, Stunkard AJ, Masheb RM. Overvaluation of shape and weight in binge eating disorder and overweight controls: refinement of a diagnostic construct. J Abnorm Psychol. 2008;117(2):414–419. | |
Hrabosky JI, Masheb RM, White MA, Grilo CM. Overvaluation of shape and weight in binge eating disorder. J Consult Clin Psychol. 2007;75(1):175–180. | |
de Zwaan M. Binge eating disorder and obesity. Int J Obes Relat Metab Disord. 2001;25 Suppl 1:S51–S55. | |
Brownley KA, Berkman ND, Sedway JA, Lohr KN, Bulik CM. Binge eating disorder treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40(4):337–348. | |
Castellini G, Lo Sauro C, Mannucci E, et al. Diagnostic crossover and outcome predictors in eating disorders according to DSM-IV and DSM-V proposed criteria: a 6-year follow-up study. Psychosom Med. 2011;73(3):270–279. | |
Kalman D, Cascarano H, Krieger DR, Incledon T, Woolsey M. Frequency of binge eating disorder in an outpatient weight loss clinic. J Am Diet Assoc. 2002;102(5):697–699. | |
Fichter MM, Quadflieg N. Long-term stability of eating disorder diagnoses. Int J Eat Disord. 2007;40(Suppl):S61–S66. | |
Steinhausen HC, Jakobsen H, Helenius D, Munk-Jørgensen P, Strober M. A nation-wide study of the family aggregation and risk factors in anorexia nervosa over three generations. Int J Eat Disord. Epub April 29, 2014. | |
Strober M, Freeman R, Lampert C, Diamond J, Kaye W. Controlled family study of anorexia nervosa and bulimia nervosa: evidence of shared liability and transmission of partial syndromes. Am J Psychiatry. 2000;157(3):393–401. | |
Lilenfeld LR, Kaye WH, Greeno CG, et al. A controlled family study of anorexia nervosa and bulimia nervosa: psychiatric disorders in first-degree relatives and effects of proband comorbidity. Arch Gen Psychiatry. 1998;55(7):603–610. | |
Grigoroiu-Serbanescu M, Magureanu S, Milea S, Dobrescu I, Marinescu E. Modest familial aggregation of eating disorders in restrictive anorexia nervosa with adolescent onset in a Romanian sample. Eur Child Adolesc Psychiatry. 2003;12(Suppl 1):I47–153. | |
Fowler SJ, Bulik CM. Family environment and psychiatric history in women with binge-eating disorder and obese controls. Behav Change. 1997;14(2):106–112. | |
Hudson JI, Lalonde JK, Pindyck LJ, et al. Binge-eating disorder as a distinct familial phenotype in obese individuals. Arch Gen Psychiatry. 2006;63(3):313–319. | |
Kipman A, Gorwood P, Mouren-Simeoni MC, Ades J. Genetic factors in anorexia nervosa. Eur Psychiatry. 1999;14(4):189–198. | |
Klump KL, Miller KB, Keel PK, McGue M, Iacono WG. Genetic and environmental influences on anorexia nervosa syndromes in a population-based twin sample. Psychol Med. 2001;31(4):737–740. | |
Kortegaard LS, Hoerder K, Joergensen J, Gillberg C, Kyvik KO. A preliminary population-based twin study of self-reported eating disorder. Psychol Med. 2001;31(2):361–365. | |
Bulik CM, Sullivan PF, Tozzi F, Furberg H, Lichtenstein P, Pedersen NL. Prevalence, heritability, and prospective risk factors for anorexia nervosa. Arch Gen Psychiatry. 2006;63(3):305–312. | |
Bulik CM, Thornton LM, Root TL, Pisetsky EM, Lichtenstein P, Pedersen NL. Understanding the relation between anorexia nervosa and bulimia nervosa in a Swedish national twin sample. Biol Psychiatry. 2010;67(1):71–77. | |
Dellava JE, Thornton LM, Lichtenstein P, Pedersen NL, Bulik CM. Impact of broadening definitions of anorexia nervosa on sample characteristics. J Psychiatr Res. 2011;45(5):691–698. | |
Wade TD, Bulik CM, Neale M, Kendler KS. Anorexia nervosa and major depression: shared genetic and environmental risk factors. Am J Psychiatry. 2000;157(3):469–471. | |
Bulik CM, Sullivan PF, Kendler KS. heritability of binge-eating and broadly defined bulimia nervosa. Biol Psychiatry. 1998;44(12):1210–1218. | |
Trace SE, Thornton LM, Baker JH, et al. A behavioral-genetic investigation of bulimia nervosa and its relationship with alcohol use disorder. Psychiatry Res. 2013;208(3):232–237. | |
Mazzeo SE, Mitchell KS, Bulik CM, Aggen SH, Kendler KS, Neale MC. A twin study of specific bulimia nervosa symptoms. Psychol Med. 2010;40(7):1203–1213. | |
Bulik CM, Sullivan PF, Kendler KS. An empirical study of the classification of eating disorders. Am J Psychiatry. 2000;157(6):886–895. | |
Javaras KN, Laird NM, Reichborn-Kjennerud T, Bulik CM, Pope HG Jr, Hudson JI. Familiality and heritability of binge eating disorder: results of a case-control family study and a twin study. Int J Eat Disord. 2008;41(2):174–179. | |
Mitchell KS, Neale MC, Bulik CM, Aggen SH, Kendler KS, Mazzeo SE. Binge eating disorder: a symptom-level investigation of genetic and environmental influences on liability. Psychol Med. 2010;40(11):1899–1906. | |
Munn MA, Stallings MC, Rhee SH, et al. Bivariate analysis of disordered eating characteristics in adolescence and young adulthood. Int J Eat Disord. 2010;43(8):751–761. | |
Klump KL, Suisman JL, Burt SA, McGue M, Iacono WG. Genetic and environmental influences on disordered eating: An adoption study. J Abnorm Psychol. 2009;118(4):797–805. | |
Devlin B, Jones BL, Bacanu SA, Roeder K. Mixture and linear models for linkage analysis with covariates. Genet Epidemiol. 2002;23:449–455. | |
Grice DE, Halmi KA, Fichter MM, et al. Evidence for a susceptibility gene for anorexia nervosa on chromosome 1. Am J Hum Genet. 2002;70(3):787–792. | |
Nakabayashi K, Komaki G, Tajima A, et al. Identification of novel candidate loci for anorexia nervosa at 1q41 and 11q22 in Japanese by a genome-wide association analysis with microsatellite markers. J Human Genet. 2009;54(9):531–537. | |
Bergen AW, van den Bree MB, Yeager M, et al. Candidate genes for anorexia nervosa in the 1p33–36 linkage region: serotonin 1D and delta opioid receptor loci exhibit significant association to anorexia nervosa. Mol Psychiatry. 2003;8(4):397–406. | |
Brown KM, Bujac SR, Mann ET, Campbell DA, Stubbins MJ, Blundell JE. Further evidence of association of OPRD1 and HTR1D polymorphisms with susceptibility to anorexia nervosa. Biol Psychiatry. 2007;61(3):367–373. | |
Devlin B, Bacanu SA, Klump KL, et al. Linkage analysis of anorexia nervosa incorporating behavioral covariates. Hum Mol Genet. 2002;11(6):689–696. | |
Bulik CM, Devlin B, Bacanu SA, et al. Significant linkage on chromosome 10p in families with bulimia nervosa. Am J Hum Genet. 2003;72(1):200–207. | |
Bacanu SA, Bulik CM, Klump KL, et al. Linkage analysis of anorexia and bulimia nervosa cohorts using selected behavioral phenotypes as quantitative traits or covariates. Am J Med Genet B Neuropsychiatr Genet. 2005;139B(1):61–68. | |
Clarke TK, Weiss AR, Berrettini WH. The genetics of anorexia nervosa. Clin Pharmacol Ther. 2012;91(2):181–188. | |
Trace SE, Baker JH, Penas-Lledo E, Bulik CM. The genetics of eating disorders. Annu Rev Clin Psychol. 2013;9:589–620. | |
Hinney A, Scherag S, Hebebrand J. Genetic findings in anorexia and bulimia nervosa. Prog Mol Biol Trans Sci. 2010;94:241–270. | |
Kaye WH, Bailer UF, Frank GK, Wagner A, Henry SE. Brain imaging of serotonin after recovery from anorexia and bulimia nervosa. Physiol Behav. 2005;86(1–2):15–17. | |
Kaye WH, Frank GK, Bailer UF, et al. Serotonin alterations in anorexia and bulimia nervosa: new insights from imaging studies. Physiol Behav. 2005;85(1):73–81. | |
Ricca V, Nacmias B, Boldrini M, et al. Psychopathological traits and 5-HT2A receptor promoter polymorphism (-1438 G/A) in patients suffering from Anorexia Nervosa and Bulimia Nervosa. Neurosci Lett. 2004;365(2):92–96. | |
Martásková D, Slachtová L, Kemlink D, Záhoráková D, Papezová H. Polymorphisms in serotonin-related genes in anorexia nervosa. The first study in Czech population and metaanalyses with previously performed studies. Folia Biol (Praha). 2009;55(5):192–197. | |
Enoch MA, Kaye WH, Rotondo A, Greenberg BD, Murphy DL, Goldman D. 5-HT2A promoter polymorphism -1438G/A, anorexia nervosa, and obsessive-compulsive disorder. Lancet. 1998;351(9118):1785–1786. | |
Kiezebrink K, Mann ET, Bujac SR, Stubbins MJ, Campbell DA, Blundell JE. Evidence of complex involvement of serotonergic genes with restrictive and binge purge subtypes of anorexia nervosa. World J Biol Psychiatry. 2010;11(6):824–833. | |
Gorwood P, Adès J, Bellodi L, et al. The 5-HT(2A) -1438G/A polymorphism in anorexia nervosa: a combined analysis of 316 trios from six European centres. Mol Psychiatry. 2002;7(1):90–94. | |
Hinney A, Ziegler A, Nothen MM, Remschmidt H, Hebebrand J. 5-HT2A receptor gene polymorphisms, anorexia nervosa, and obesity. Lancet. 1997;350(9087):1324–1325. | |
Nishiguchi N, Matsushita S, Suzuki K, Murayama M, Shirakawa O, Higuchi S. Association between 5HT2A receptor gene promoter region polymorphism and eating disorders in Japanese patients. Biol Psychiatry. 2001;50(2):123–128. | |
Lim SW, Ha J, Shin DW, Woo HY, Kim KH. Associations between the serotonin-1A receptor C(-1019)G polymorphism and disordered eating symptoms in female adolescents. J Neural Transm. 2010;117(6):773–779. | |
Hu X, Giotakis O, Li T, Karwautz A, Treasure J, Collier DA. Association of the 5-HT2c gene with susceptibility and minimum body mass index in anorexia nervosa. Neuroreport. 2003;14(6):781–783. | |
Hinney A, Barth N, Ziegler A, et al. Serotonin transporter gene-linked polymorphic region: allele distributions in relationship to body weight and in anorexia nervosa. Life Sci. 1997;61(21):PL 295–303. | |
Castellini G, Ricca V, Lelli L, et al. Association between serotonin transporter gene polymorphism and eating disorders outcome: A 6-year follow-up study. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(5):491–500. | |
Urwin RE, Bennetts BH, Wilcken B, Beumont PJ, Russell JD, Nunn KP. Investigation of epistasis between the serotonin transporter and norepinephrine transporter genes in anorexia nervosa. Neuropsychopharmacology. 2003;28(7):1351–1355. | |
Calati R, De Ronchi D, Bellini M, Serretti A. The 5-HTTLPR polymorphism and eating disorders: a meta-analysis. Int J Eat Disord. 2011;44(3):191–199. | |
Lee Y, Lin PY. Association between serotonin transporter gene polymorphism and eating disorders: a meta-analytic study. Int J Eat Disord. 2010;43(6):498–504. | |
Polsinelli GN, Levitan RN, De Luca V. 5-HTTLPR polymorphism in bulimia nervosa: a multiple-model meta-analysis. Psychiatr Genet. 2012;22(5):219–225. | |
Steiger H, Fichter M, Bruce KR, et al. Molecular-genetic correlates of self-harming behaviors in eating-disordered women: findings from a combined Canadian-German sample. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):102–106. | |
Steiger H, Richardson J, Joober R, et al. The 5HTTLPR polymorphism, prior maltreatment and dramatic-erratic personality manifestations in women with bulimic syndromes. J Psychiatry Neurosci. 2007;32(5):354–362. | |
Thaler L, Groleau P, Joober R, et al. Epistatic interaction between 5HTTLPR and TPH2 polymorphisms predicts novelty seeking in women with bulimia nervosa spectrum disorders. Psychiatr Res. 2013;208(1):101–103. | |
Monteleone P, Tortorella A, Castaldo E, Maj M. Association of a functional serotonin transporter gene polymorphism with binge eating disorder. Am J Med Genet B Neuropsychiatr Genet. 2006; 141B(1):7–9. | |
Bergen AW, Yeager M, Welch RA, et al. Association of multiple DRD2 polymorphisms with anorexia nervosa. Neuropsychopharmacology. 2005;30(9):1703–1710. | |
Davis C, Levitan RD, Kaplan AS, et al. Reward sensitivity and the D2 dopamine receptor gene: A case-control study of binge eating disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(3):620–628. | |
Davis CA, Levitan RD, Reid C, et al. Dopamine for “wanting” and opioids for “liking”: a comparison of obese adults with and without binge eating. Obesity (Silver Spring). 2009;17(6):1220–1225. | |
Davis C, Levitan RD, Yilmaz Z, Kaplan AS, Carter JC, Kennedy JL. Binge eating disorder and the dopamine D2 receptor: genotypes and sub-phenotypes. Prog Neuropsychopharmacol Biol Psychiatry. 2012; 38(2):328–335. | |
Kaplan AS, Levitan RD, Yilmaz Z, Davis C, Tharmalingam S, Kennedy JL. A DRD4/BDNF gene-gene interaction associated with maximum BMI in women with bulimia nervosa. Int J Eat Disord. 2008;41(1):22–28. | |
Levitan RD, Kaplan AS, Davis C, Lam RW, Kennedy JL. A season-of-birth/DRD4 interaction predicts maximal body mass index in women with bulimia nervosa. Neuropsychopharmacology. 2010;35(8):1729–1733. | |
Yilmaz Z, Kaplan AS, Levitan RD, Zai CC, Kennedy JL. Possible association of the DRD4 gene with a history of attention-deficit/hyperactivity disorder in women with bulimia nervosa. Int J Eat Disord. 2012;45(4):622–625. | |
Hinney A, Schneider J, Ziegler A, et al. No evidence for involvement of polymorphisms of the dopamine D4 receptor gene in anorexia nervosa, underweight, and obesity. Am J Med Genet. 1999;88(6):594–597. | |
Mikolajczyk E, Smiarowska M, Grzywacz A, Samochowiec J. Association of eating disorders with catechol-o-methyltransferase gene functional polymorphism. Neuropsychobiology. 2006;54(1):82–86. | |
Yilmaz Z, Kaplan AS, Zai CC, Levitan RD, Kennedy JL. COMT Val158Met variant and functional haplotypes associated with childhood ADHD history in women with bulimia nervosa. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(4):948–952. | |
Brandys MK, Slof-Op’t Landt MC, van Elburg AA, et al. Anorexia nervosa and the Val158Met polymorphism of the COMT gene: meta-analysis and new data. Psychiatr Genet. 2012;22(3):130–136. | |
Grayson BE, Seeley RJ. Deconstructing obesity: the face of fatness before and after the discovery of leptin. Diabetologia. 2012;55(1):3–6. | |
Janeckova R. The role of leptin in human physiology and pathophysiology. Physiol Res. 2001;50(5):443–459. | |
Moriya J, Takimoto Y, Yoshiuchi K, Shimosawa T, Akabayashi A. Plasma agouti-related protein levels in women with anorexia nervosa. Psychoneuroendocrinology. 2006;31(9):1057–1061. | |
Ferron F, Considine RV, Peino R, Lado IG, Dieguez C, Casanueva FF. Serum leptin concentrations in patients with anorexia nervosa, bulimia nervosa and non-specific eating disorders correlate with the body mass index but are independent of the respective disease. Clin Endocrinol (Oxf). 1997;46(3):289–293. | |
Hinney A, Bornscheuer A, Depenbusch M, et al. No evidence for involvement of the leptin gene in anorexia nervosa, bulimia nervosa, underweight or early onset extreme obesity: identification of two novel mutations in the coding sequence and a novel polymorphism in the leptin gene linked upstream region. Mol Psychiatry. 1998;3(6):539–543. | |
Quinton ND, Meechan DW, Brown K, Eastwood H, Blakemore AI. Single nucleotide polymorphisms in the leptin receptor gene: studies in anorexia nervosa. Psychiatr Genet. 2004;14(4):191–194. | |
Yilmaz Z, Kaplan AS, Tiwari AK, et al. The role of leptin, melanocortin, and neurotrophin system genes on body weight in anorexia nervosa and bulimia nervosa. J Psychiatr Res. 2014;55:77–86. | |
Dardennes RM, Zizzari P, Tolle V, et al. Family trios analysis of common polymorphisms in the obestatin/ghrelin, BDNF and AGRP genes in patients with Anorexia nervosa: association with subtype, body-mass index, severity and age of onset. Psychoneuroendocrinology. 2007;32(2):106–113. | |
Cellini E, Nacmias B, Brecelj-Anderluh M, et al. Case-control and combined family trios analysis of three polymorphisms in the ghrelin gene in European patients with anorexia and bulimia nervosa. Psychiatr Genet. 2006;16(2):51–52. | |
Monteleone P, Tortorella A, Castaldo E, Di Filippo C, Maj M. No association of the Arg51Gln and Leu72Met polymorphisms of the ghrelin gene with anorexia nervosa or bulimia nervosa. Neurosci Lett. 2006;398(3):325–327. | |
Kindler J, Bailer U, de Zwaan M, et al. No association of the neuropeptide Y (Leu7Pro) and ghrelin gene (Arg51Gln, Leu72Met, Gln90Leu) single nucleotide polymorphisms with eating disorders. Nord J Psychiatry. 2011;65(3):203–207. | |
Ando T, Komaki G, Naruo T, et al. Possible role of preproghrelin gene polymorphisms in susceptibility to bulimia nervosa. Am J Med Genet B Neuropsychiatr Genet. 2006;141(8):929–934. | |
Miyasaka K, Hosoya H, Sekime A, et al. Association of ghrelin receptor gene polymorphism with bulimia nervosa in a Japanese population. J Neural Transm. 2006;113(9):1279–1285. | |
Monteleone P, Tortorella A, Castaldo E, Di Filippo C, Maj M. The Leu72Met polymorphism of the ghrelin gene is significantly associated with binge eating disorder. Psychiatr Genet. 2007;17(1):13–16. | |
Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348(12):1085–1095. | |
Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF. Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003;348(12):1096–1103. | |
Hebebrand J, Geller F, Dempfle A, et al. Binge-eating episodes are not characteristic of carriers of melanocortin-4 receptor gene mutations. Mol Psychiatry. 2004;9(8):796–800. | |
Lubrano-Berthelier C, Dubern B, Lacorte JM, et al. Melanocortin 4 receptor mutations in a large cohort of severely obese adults: prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating. J Clin Endocrinol Metab 2006;91(5):1811–1818. | |
Hebebrand J, Fichter M, Gerber G, et al. Genetic predisposition to obesity in bulimia nervosa: a mutation screen of the melanocortin-4 receptor gene. Mol Psychiatry. 2002;7(6):647–651. | |
Loos RJ, Lindgren CM, Li S, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40(6):768–775. | |
Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. | |
Brandys MK, van Elburg AA, Loos RJ, et al. Are recently identified genetic variants regulating BMI in the general population associated with anorexia nervosa? Am J Med Genet B Neuropsychiatr Genet. 2010;153B(2):695–699. | |
Vink T, Hinney A, van Elburg AA, et al. Association between an agouti-related protein gene polymorphism and anorexia nervosa. Mol Psychiatry. 2001;6(3):325–328. | |
Hinney A, Becker I, Heibult O, et al. Systematic mutation screening of the pro-opiomelanocortin gene: identification of several genetic variants including three different insertions, one nonsense and two missense point mutations in probands of different weight extremes. J Clin Endocrinol Metab 1998;83(10):3737–3741. | |
Baker JH, Thornton LM, Bulik CM, Kendler KS, Lichtenstein P. Shared genetic effects between age at menarche and disordered eating. J Adolesc Health. 2012;51(5):491–496. | |
Culbert KM, Breedlove SM, Burt SA, Klump KL. Prenatal hormone exposure and risk for eating disorders: a comparison of opposite-sex and same-sex twins. Arch Gen Psychiatry. 2008;65(3):329–336. | |
Lydecker JA, Pisetsky EM, Mitchell KS, et al. Association between co-twin sex and eating disorders in opposite sex twin pairs: evaluations in North American, Norwegian, and Swedish samples. J Psychosom Res. 2012;72(1):73–77. | |
Raevuori A, Kaprio J, Hoek HW, Sihvola E, Rissanen A, Keski-Rahkonen A. Anorexia and bulimia nervosa in same-sex and opposite-sex twins: lack of association with twin type in a nationwide study of Finnish twins. Am J Psychiatry. 2008;165(12):1604–1610. | |
Procopio M, Marriott P. Intrauterine hormonal environment and risk of developing anorexia nervosa. Arch Gen Psychiatry. 2007;64(12):1402–1407. | |
Versini A, Ramoz N, Le Strat Y, et al. Estrogen receptor 1 gene (ESR1) is associated with restrictive anorexia nervosa. Neuropsychopharmacology. 2010;35(8):1818–1825. | |
Eastwood H, Brown KM, Markovic D, Pieri LF. Variation in the ESR1 and ESR2 genes and genetic susceptibility to anorexia nervosa. Mol Psychiatry. 2002;7(1):86–89. | |
Rosenkranz K, Hinney A, Ziegler A, et al. Systematic mutation screening of the estrogen receptor beta gene in probands of different weight extremes: identification of several genetic variants. J Clin Endocrinol Metab 1998;83(12):4524–4527. | |
Slof-Op’t Landt MC, van Furth EF, Meulenbelt I, et al. Association study of the estrogen receptor I gene (ESR1) in anorexia nervosa and eating disorders: no replication found. Int J Eat Disord. 2014;47(2):211–214. | |
Nilsson M, Naessen S, Dahlman I, Linden Hirschberg A, Gustafsson JA, Dahlman-Wright K. Association of estrogen receptor beta gene polymorphisms with bulimic disease in women. Mol Psychiatry. 2004;9(1):28–34. | |
Cui H, Moore J, Ashimi SS, et al. Eating disorder predisposition is associated with ESRRA and HDAC4 mutations. J Clin Invest. 2013;123(11):4706–4713. | |
Scott-Van Zeeland AA, Bloss CS, Tewhey R, et al. Evidence for the role of EPHX2 gene variants in anorexia nervosa. Mol Psychiatry.2014;19(6):724–732. | |
Xu B, Goulding EH, Zang K, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6(7):736–742. | |
Ribases M, Gratacos M, Armengol L, et al. Met66 in the brain-derived neurotrophic factor (BDNF) precursor is associated with anorexia nervosa restrictive type. Mol Psychiatry. 2003;8(8):745–751. | |
Ribasés M, Gratacòs M, Fernández-Aranda F, et al. Association of BDNF with anorexia, bulimia and age of onset of weight loss in six European populations. Hum Mol Genet. 2004;13(12):1205–1212. | |
Ribasés M, Gratacòs M, Fernández-Aranda F, et al. Association of BDNF with restricting anorexia nervosa and minimum body mass index: a family-based association study of eight European populations. Eur J Hum Genet. 2005;13(4):428–434. | |
Dmitrzak-Weglarz M, Skibinska M, Slopien A, et al. BDNF Met66 allele is associated with anorexia nervosa in the Polish population. Psychiatr Genet. 2007;17(4):245–246. | |
Gratacos M, Gonzalez JR, Mercader JM, de Cid R, Urretavizcaya M, Estivill X. Brain-derived neurotrophic factor Val66Met and psychiatric disorders: meta-analysis of case-control studies confirm association to substance-related disorders, eating disorders, and schizophrenia. Biol Psychiatry. 2007;61(7):911–922. | |
Monteleone P, Zanardini R, Tortorella A, et al. The 196G/A (val66met) polymorphism of the BDNF gene is significantly associated with binge eating behavior in women with bulimia nervosa or binge eating disorder. Neurosci Lett. 2006;406(1–2):133–137. | |
Brandys MK, Kas MJ, van Elburg AA, et al. The Val66Met polymorphism of the BDNF gene in anorexia nervosa: New data and a meta-analysis. World J Biol Psychiatry. 2011;14(6):441–451. | |
de Krom M, Bakker SC, Hendriks J, et al. Polymorphisms in the brain-derived neurotrophic factor gene are not associated with either anorexia nervosa or schizophrenia in Dutch patients. Psychiatr Genet. 2005;15(2):81. | |
Mercader JM, Ribases M, Gratacos M, et al. Altered brain-derived neurotrophic factor blood levels and gene variability are associated with anorexia and bulimia. Genes Brain Behav. 2007;6(8):706–716. | |
Siegfried Z, Kanyas K, Latzer Y, et al. Association study of cannabinoid receptor gene (CNR1) alleles and anorexia nervosa: differences between restricting and binging/purging subtypes. Am J Med Genet B Neuropsychiatr Genet. 2004;125B(1):126–130. | |
Müller TD, Reichwald K, BrÖnner G, et al. Lack of association of genetic variants in genes of the endocannabinoid system with anorexia nervosa. Child Adolesc Psychiatry Ment Health. 2008;2(1):33. | |
Monteleone P, Bifulco M, Di Filippo C, et al. Association of CNR1 and FAAH endocannabinoid gene polymorphisms with anorexia nervosa and bulimia nervosa: evidence for synergistic effects. Genes Brain Behav. 2009;8(7):728–732. | |
Davis C, Zai C, Levitan RD, et al. Opiates, overeating and obesity: a psychogenetic analysis. Int J Obes (Lond). 2011;35(10):1347–1354. | |
Loos RJ, Bouchard C. FTO: the first gene contributing to common forms of human obesity. Obes Rev. 2008;9(3):246–250. | |
Luan J, Kerner B, Zhao JH, et al. A multilevel linear mixed model of the association between candidate genes and weight and body mass index using the Framingham longitudinal family data. BMC Proc. 2009;3 Suppl 7:S115. | |
Muller TD, Greene BH, Bellodi L, et al. Fat mass and obesity-associated gene (FTO) in eating disorders: evidence for association of the rs9939609 obesity risk allele with bulimia nervosa and anorexia nervosa. Obes Facts. 2012;5(3):408–419. | |
Jonassaint CR, Szatkiewicz JP, Bulik CM, et al. Absence of association between specific common variants of the obesity-related FTO gene and psychological and behavioral eating disorder phenotypes. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(4):454–461. | |
Pinheiro AP, Bulik CM, Thornton LM, et al. Association study of 182 candidate genes in anorexia nervosa. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(5):1070–1080. | |
Corvin A, Craddock N, Sullivan PF. Genome-wide association studies: a primer. Psychol Med. 2010;40(7):1063–1077. | |
Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13(8):537–551. | |
Wang K, Zhang H, Bloss CS, et al. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol Psychiatry. 2011;16(9):949–959. | |
Boraska V, Davis OS, Cherkas LF, et al. Genome-wide association analysis of eating disorder-related symptoms, behaviors, and personality traits. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(7):803–811. | |
Wade TD, Gordon S, Medland S, et al. Genetic variants associated with disordered eating. Int J Eat Disord. 2013;46(6):594–608. | |
Boraska V, Franklin CS, Floyd JA, et al. A genome-wide association study of anorexia nervosa. Mol Psychiatry. Epub February 11, 2014. | |
Bocarsly ME, Avena NM. Animal Models of Binge Eating Palatable Foods: Emergence of Addiction-Like Behaviors and Brain Changes in the Rat. In: Avena NM, editor. Animal Models of Eating Disorders. New York, NY: Humana Press; 2012:179–191. | |
Mathes WF, Brownley KA, Mo X, Bulik CM. The biology of binge eating. Appetite. 2009;52(3):545–553. | |
Gutierrez E. A rat in the labyrinth of anorexia nervosa: contributions of the activity-based anorexia rodent model to the understanding of anorexia nervosa. Int J Eat Disord. 2013;46(4):289–301. | |
Maltais LJ, Lane PW, Beamer WG. Anorexia, a recessive mutation causing starvation in preweanling mice. J Hered. 1984;75(6):468–472. | |
Lindfors C, Nilsson IA, Garcia-Roves PM, et al. Hypothalamic mitochondrial dysfunction associated with anorexia in the anx/anx mouse. Proc Natl Acad Sci U S A. 2011;108(44):18108–18113. | |
Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396(6712):670–674. | |
Qu D, Ludwig DS, Gammeltoft S, et al. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature. 1996;380(6571):243–247. | |
Yamada M, Miyakawa T, Duttaroy A, et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature. 2001;410(6825):207–212. | |
Pratt WE, Kelley AE. Nucleus accumbens acetylcholine regulates appetitive learning and motivation for food via activation of muscarinic receptors. Behav Neurosci. 2004;118(4):730–739. | |
Pratt WE, Kelley AE. Striatal muscarinic receptor antagonism reduces 24-h food intake in association with decreased preproenkephalin gene expression. Eur J Neurosci. 2005;22(12):3229–3240. | |
Pratt WE, Spencer RC, Kelley AE. Muscarinic receptor antagonism of the nucleus accumbens core causes avoidance to flavor and spatial cues. Behav Neurosci. 2007;121(6):1215–1223. | |
Perry ML, Pratt WE, Baldo BA. Overlapping striatal sites mediate scopolamine-induced feeding suppression and mu-opioid-mediated hyperphagia in the rat. Psychopharmacology (Berl).2014;231(5):919–928. | |
Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrié P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord. 2004;28(4):640–648. | |
Soria-Gómez E, Bellocchio L, Reguero L, et al. The endocannabinoid system controls food intake via olfactory processes. Nat Neurosci. 2014;17(3):407–415. | |
Czyzyk TA, Romero-Picó A, Pintar J, et al. Mice lacking δ-opioid receptors resist the development of diet-induced obesity. FASEB J. 2012;26(8):3483–3492. | |
Allen Brain Atlas [webpage on the Internet]. Allen Institute for Brain Science; 2010. Available from: http://www.brain-map.org. Accessed June 29, 2014. | |
Ingalls AM, Dickie MM, Shell GD. Obese, a new mutation in the house mouse. J Hered. 1950;41:317–318. | |
Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153(3740):1127–1128. | |
Clément K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392(6674):398–401. | |
Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–908. | |
Chen H, Charlat O, Tartaglia LA, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84(3):491–495. | |
Lee GH, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379(6566):632–635. | |
Tartaglia LA, Dembski M, Weng X, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83(7):1263–1271. | |
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. | |
Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature. 1998;393(6686):684–688. | |
Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012;15(10):1350–1355. | |
Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5(9):1066–1070. | |
Huszar D, Lynch CA, Fairchild-Huntress V, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. | |
Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385(6612):165–168. | |
Butler AA, Kesterson RA, Khong K, et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141(9):3518–3521. | |
Balthasar N, Dalgaard LT, Lee CE, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. | |
Hanada R, Teranishi H, Pearson JT, et al. Neuromedin U has a novel anorexigenic effect independent of the leptin signaling pathway. Nat Med. 2004;10(10):1067–1073. | |
Peier A, Kosinski J, Cox-York K, et al. The antiobesity effects of centrally administered neuromedin U and neuromedin S are mediated predominantly by the neuromedin U receptor 2 (NMUR2). Endocrinology. 2009;150(7):3101–3109. | |
Tecott LH, Sun LM, Akana SF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374(6522):542–546. | |
Xu Y, Jones JE, Kohno D, et al. 5-HT2CRs expressed by pro-ppiomelanocortin neurons regulate energy homeostasis. Neuron. 2008;60(4):582–589. | |
Heisler LK, Jobst EE, Sutton GM, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51(2):239–249. | |
Doslikova B, Garfield AS, Shaw J, et al. 5-HT2C receptor agonist anorectic efficacy potentiated by 5-HT1B receptor agonist coapplication: an effect mediated via increased proportion of pro-opiomelanocortin neurons activated. J Neurosci. 2013;33(23):9800–9804. | |
Bouwknecht JA, van der Gugten J, Hijzen TH, Maes RA, Hen R, Olivier B. Male and female 5-HT(1B) receptor knockout mice have higher body weights than wildtypes. Physiol Behav. 2001;74(4–5):507–516. | |
Striegel-Moore RH, Franko DL. Epidemiology of binge eating disorder. Int J Eat Disord. 2003;34 Suppl:S19–S29. | |
Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70(5):813–829. | |
Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12(11):623–637. | |
Jones PA. The role of DNA methylation in mammalian epigenetics. Science. 2001;293(5532):1068–1070. | |
Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci. 2008;66(4):596–612. | |
Illingworth R, Kerr A, DeSousa D, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6(1):e22. | |
Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. | |
Rokholm B, Silventoinen K, Ängquist L, Skytthe A, Kyvik KO, Sørensen TI. Increased genetic variance of BMI with a higher prevalence of obesity. PLoS One. 2011;6(6):e20816. | |
Frieling H, Romer KD, Scholz S, et al. Epigenetic dysregulation of dopaminergic genes in eating disorders. Int J Eat Disord. 2010;43(7):577–583. | |
Pjetri E, Dempster E, Collier DA, et al. Quantitative promoter DNA methylation analysis of four candidate genes in anorexia nervosa: a pilot study. J Psychiatr Res. 2013;47(2):280–282. | |
Groleau P, Joober R, Israel M, et al. Methylation of the dopamine D2 receptor (DRD2) gene promoter in women with a bulimia-spectrum disorder: associations with borderline personality disorder and exposure to childhood abuse. J Psychiatr Res. 2014;48(1):121–127. | |
Ehrlich S, Weiss D, Burghardt R, et al. Promoter specific DNA methylation and gene expression of POMC in acutely underweight and recovered patients with anorexia nervosa. J Psychiatr Res. 2010;44(13):827–833. | |
Ehrlich S, Walton E, Roffman JL, et al. Smoking, but not malnutrition, influences promoter-specific DNA methylation of the proopiomelanocortin gene in patients with and without anorexia nervosa. Can J Psychiatry. 2012;57(3):168–176. | |
Frieling H, Bleich S, Otten J, et al. Epigenetic downregulation of atrial natriuretic peptide but not vasopressin mRNA expression in females with eating disorders is related to impulsivity. Neuropsychopharmacology. 2008;33(11):2605–2609. | |
Kim YR, Kim JH, Kim MJ, Treasure J. Differential methylation of the oxytocin receptor gene in patients with anorexia nervosa: a pilot study. PLoS One. 2014;9(2):e88673. | |
Frieling H, Albrecht H, Jedtberg S, et al. Elevated cannabinoid 1 receptor mRNA is linked to eating disorder related behavior and attitudes in females with eating disorders. Psychoneuroendocrinology. 2009;34(4):620–624. | |
Schroeder M, Eberlein C, de Zwaan M, Kornhuber J, Bleich S, Frieling H. Lower levels of cannabinoid 1 receptor mRNA in female eating disorder patients: Association with wrist cutting as impulsive self-injurious behavior. Psychoneuroendocrinology. 2012;37(12):2032–2036. | |
Steiger H, Labonte B, Groleau P, Turecki G, Israel M. Methylation of the glucocorticoid receptor gene promoter in bulimic women: associations with borderline personality disorder, suicidality, and exposure to childhood abuse. Int J Eat Disord. 2013;46(3):246–255. | |
Frieling H, Gozner A, Romer KD, et al. Global DNA hypomethylation and DNA hypermethylation of the alpha synuclein promoter in females with anorexia nervosa. Mol Psychiatry. 2007;12(3):229–230. | |
Tremolizzo L, Conti E, Bomba M, et al. Decreased whole-blood global DNA methylation is related to serum hormones in anorexia nervosa adolescents. World J Biol Psychiatry. 2013;15(4):327–333. | |
Saffrey R, Novakovic B, Wade TD. Assessing global and gene specific DNA methylation in anorexia nervosa: a pilot study. Int J Eat Disord. 2014;47(2):206–210. | |
International Schizophrenia Consortium, Purcell SM, Wray NR, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. | |
Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969–976. | |
Ruderfer DM, Fanous AH, Ripke S, et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol Psychiatry. 2014;19(9):1017–1024. | |
Wray NR, Pergadia ML, Blackwood DH, et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry. 2012;17(1):36–48. | |
Lee SH, DeCandia TR, Ripke S, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet. 2012;44(3):247–250. | |
Sullivan PF. Puzzling over schizophrenia: schizophrenia as a pathway disease. Nat Med. 2012;18(2):210–211. | |
Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371–1379. | |
Schork AJ, Thompson WK, Pham P, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9(4):e1003449. | |
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. | |
Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. | |
Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19(3):212–219. | |
Visscher PM, Goddard ME, Derks EM, Wray NR. Evidence-based psychiatric genetics, AKA the false dichotomy between common and rare variant hypotheses. Mol Psychiatry. 2012;17(5):474–485. | |
Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. | |
Timms AE, Dorschner MO, Wechsler J, et al. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry. 2013;70(6):582–590. | |
Liu L, Sabo A, Neale BM, et al. Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 2013;9(4):e1003443. | |
Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. | |
Kerner B, Rao AR, Christensen B, Dandekar S, Yourshaw M, Nelson SF. Rare genomic variants link bipolar disorder with anxiety disorders to CREB-regulated intracellular signaling pathways. Front Psychiatry. 2013;4:154. | |
Cukier HN, Dueker ND, Slifer SH, et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism. 2014;5(1):1. | |
Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405. | |
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154(6):1370–1379. | |
Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. | |
Konermann S, Brigham MD, Trevino A, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500(7463):472–476. | |
Stachniak TJ, Ghosh A, Sternson SM. Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus → midbrain pathway for feeding behavior. Neuron. 2014;82(4):797–808. | |
Betley JN, Cao ZF, Ritola KD, Sternson SM. Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell. 2013;155(6):1337–1350. | |
Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012;488(7410):172–177. | |
Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. 2011;14(3):351–355. | |
Krashes MJ, Shah BP, Madara JC, et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014;507(7491):238–242. | |
Krashes MJ, Shah BP, Koda S, Lowell BB. Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metabol. 2013;18(4):588–595. | |
Krashes MJ, Koda S, Ye C, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121(4):1424–1428. | |
Jennings JH, Sparta DR, Stamatakis AM, et al. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496(7444):224–228. | |
Land BB, Narayanan NS, Liu RJ, et al. Medial prefrontal D1 dopamine neurons control food intake. Nat Neurosci. 2014;17(2):248–253. | |
Mas S, Plana MT, Castro-Fornieles J, et al. Common genetic background in anorexia nervosa and obsessive compulsive disorder: preliminary results from an association study. J Psychiatr Res. 2013;47(6):747–754. | |
Seitz J, Kahraman-Lanzerath B, Legenbauer T, et al. The role of impulsivity, inattention and comorbid ADHD in patients with bulimia nervosa. PLoS One. 2013;8(5):e63891. | |
Bleck JR, DeBate RD, Olivardia R. The comorbidity of ADHD and eating disorders in a nationally representative sample. J Behav Health Serv Res. Epub July 10, 2014. | |
Yilmaz Z, Kaplan AS, Zawertailo LA. Bulimia nervosa and alcohol use disorder: evidence for shared etiology and neurobiology. Curr Psychiat Rev. 2012;8(4):69–81. | |
Baker JH, Mitchell KS, Neale MC, Kendler KS. Eating disorder symptomatology and substance use disorders: prevalence and shared risk in a population based twin sample. Int J Eat Disord. 2010;43(7):648–658. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.