")
Back to Journals » The Application of Clinical Genetics » Volume 14
Genetic Profile of the Dystrophin Gene Reveals New Mutations in Colombian Patients Affected with Muscular Dystrophinopathy
Authors Triana-Fonseca P , Parada-Márquez JF , Silva-Aldana CT, Zambrano-Arenas D, Arias-Gomez LL, Morales-Fonseca N, Medina-Méndez E, Restrepo CM , Silgado-Guzmán DF , Fonseca-Mendoza DJ
Received 12 May 2021
Accepted for publication 31 July 2021
Published 1 October 2021 Volume 2021:14 Pages 399—408
DOI https://doi.org/10.2147/TACG.S317721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Paula Triana-Fonseca,1 Juan Fernando Parada-Márquez,1 Claudia T Silva-Aldana,2 Daniela Zambrano-Arenas,1 Laura Lucia Arias-Gomez,2 Natalia Morales-Fonseca,3 Esteban Medina-Méndez,2 Carlos M Restrepo,4 Daniel Felipe Silgado-Guzmán,2 Dora Janeth Fonseca-Mendoza4
1Facultad de Medicina, Universidad del Bosque, Bogotá, DC, Colombia; 2Department of Molecular Diagnosis, Genética Molecular de Colombia SAS, Bogotá, DC, Colombia; 3Department of Medicine, Instituto Colombiano de Neurociencias, Bogotá, DC, Colombia; 4Center for Research in Genetics and Genomics – CIGGUR, GENIUROS Research Group, School of Medicine and Health Sciences, Universidad Del Rosario, Bogotá, DC, Colombia
Correspondence: Dora Janeth Fonseca-Mendoza; Daniel Felipe Silgado Guzmán Email [email protected]; [email protected]
Background: Duchenne and Becker muscular dystrophies (DMD/BMD) are the most common human dystrophinopathies with recessive X-linked inheritance. Dystrophin gene deletions and duplications are the most common mutations, followed by point mutations. The aim of this study is to characterize the mutational profile of the dystrophin gene in Colombian patients with DMD/BMD.
Material and Methods: Mutational profiling was determined in 69 affected patients using Sanger sequencing, next-generation sequencing (NGS) and/or multiplex ligation dependent-probes amplification (MLPA). Genetic variants were classified according to molecular consequence and new variants were determined through database and literature analysis.
Results: Mutational profile in affected patients revealed that large deletions/duplications analyzed by MLPA accounted for 72.5% of all genetic variations. By using Sanger sequencing or NGS, we identified point mutations in 15.9% and small deletions in 11.6% of the patients. New mutations were found, most of them were point mutations or small deletions (10.1%).
Conclusion: Our results described the genetic profile of the dystrophin gene in Colombian patients with DMD and contribute to efforts to identify molecular variants in Latin American populations. For our population, 18.8% of cases could be treated with FDA or MDA approved molecular therapies based on specific mutations. These data contribute to the establishment of appropriate genetic counseling and potential treatment.
Keywords: Duchenne–Becker muscular dystrophy, DMD, MLPA, next-generation sequencing, target molecular therapy, exon skipping, mutation
Introduction
Duchenne Muscular Dystrophy (DMD-OMIM #310200) and Becker Muscular Dystrophy (BMD-OMIM #300376), are the most common hereditary muscular dystrophies around the world.1 DMD and BMD occur with a frequency of 1/3.300 and 1/6.000 newborn males, respectively.2 These dystrophinopathies are caused by alterations in the DMD gene and have an X-linked recessive inheritance pattern.3 Changes in dystrophin protein synthesis is correlated with clinical manifestations, characterized by progressive muscle necrosis, loss of independent early walking ability, cardiomyopathy respiratory failure and premature death in the affected patients.4
The DMD gene encodes for a 427 kDa protein, called dystrophin, which is part of the dystrophin-associated glycoprotein complex. Dystrophin main function is binding actin to the protein complex in the sarcolemma plasma membrane and consequently adds stability to the muscle cell.5 Dystrophin protein has four structural domains, each one of them, related to specific functions: the N-terminal contains an actin-binding region, the central rod domain is formed by 24 spectrin repeats which provide flexibility, the cysteine-rich domain binds to dystroglycan and the C-terminal region establishes an association with dystrobevin and synthropin, which are cytosolic dystrophin-binding proteins.5,6 Through the use of independent promoters, three isoforms of the protein have been characterized, which have a tissue-specific expression.7
The DMD gene is molecularly complex and to date, more than 7000 causal mutations have been reported worldwide. The most frequent mutations are one or more exon deletions and duplications (large mutations), which account for about 80% of the total DMD gene alterations.8 The remaining 20% of DMD/BMD alterations are represented by small mutations (deletions or insertions of <1 exon and point mutations). Interestingly, the frequency of mutations has been shown to differ by geographic region. Large deletions are more commonly observed in Africa (88%); large duplications are more frequent in Europe, and small mutations have been associated with Oceania.8–10
In accordance with the wide mutational heterogeneity, multiple molecular strategies have been used for the identification of DMD gene mutations. Identification of large deletions and large duplications has been performed using multiplex-ligation dependent probes amplification (MLPA) or multiplex PCR. Sanger or Next-generation sequencing (NGS) have been used to identify point mutations and small insertions or duplications.10−12 Okubo et al reported that, through NGS, it is possible to identify about 92% of the dystrophin mutations.25 Given the complexity of the gene and its mutational spectrum, both Western blot and RNA analysis are required to achieve the best sensitivity.13,14
The importance of identifying mutations in affected patients is highlighted, given the existence of genetic therapy strategies that are applicable to specific types of mutations, offering adequate genetic counseling and to identify female carriers. Little is known about the genetic profile of Colombian patients affected with DMD/BMD. The aim of this study is to contribute to the knowledge gap on this field. We analyzed 69 Colombian patients with DMD/BMD, through NGS, Sanger sequencing and/or MLPA analysis. Our data showed that the distribution of the molecular mutations profile is similar to that of other populations but, interestingly, several mutations have not been previously reported worldwide.
Materials and Methods
Study Population
We studied molecular data of 69 Colombian DMD/BMD patients with identified mutation in the dystrophin gene. Despite the selection bias, the molecular information of these patients makes it possible to identify the mutational profile of the dystrophin gene. During the last five years patients underwent the DNA analysis in Genetics Molecular de Colombia, a private molecular laboratory. These patients are referred exclusively to the molecular study and the treating physician established the clinical diagnosis of dystrophinopathy. The patient’s clinical data were not available, and these data were not considered for the analysis.
All participants provided informed consent to take part in this study and for clinical attention in the private laboratory Genética Molecular de Colombia. Molecular diagnosis was performed following the procedures and quality controls established by the molecular laboratory. All experimental procedures were approved by the Ethics Committee of the Universidad del Rosario. The study followed the guidelines of the Declaration of Helsinki (Approved CEI-AMH002-000185).
Dystrophin Gene Molecular Analysis by Sequencing
Peripheral blood samples were obtained for DNA extraction using the Salting-out method. Point mutations and small deletions were analyzed by the Sanger method or NGS (recently introduced into the laboratory). For Sanger sequencing, 79 Dystrophin gene coding exons and intron-exon boundaries were amplified by PCR using specific primers designed with Primer Blastn (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and Primer3 software (https://primer3plus.com/cgi-bin/dev/primer3plus.cgi). Primer sequences and PCR conditions are available upon request. PCR products were purified and then sequenced using the ABI-3500 genetic analyzer. The patient´s sequences were aligned and compared using ClustalW software. The wild-type sequences are available in the Ensembl database (NM_004006). For NGS, library preparation and sequencing were carried out by CGC Genetics (Portugal). The libraries were generated using the SureSelectQXT Reagent Kit (Agilent Technologies). The DNA was fragmented (180–280pb), adaptor-tagged in the ends, purified and PCR amplified. Then, the prepared DNA library amplicons were hybridized and captured through the biotin-streptavidin coated magnetic beads system, followed by indexing and target enrichment. The samples were sequenced with the MiSeq platform (Illumina). Reads were aligned to the human reference genome hg19 (GRCh37) using the Burrows-Wheeler Aligner (BWA) and Genome Analysis Toolkit (GATK) softwares, followed by bioinformatic analysis. The quality data considered for optimal results were >Q30, 100% coverage of target regions and a depth of at least 30x. The final analysis was executed using Golden Helix software. For both, Sanger and NGS tools, the identified variants were chosen according to its potential molecular consequence and were classified as follows: missense, nonsense, frameshift and splice site. Regarding new variants, we revised the public databases (http://www.umd.be/, https://www.lovd.nl/, https://www.ncbi.nlm.nih.gov/clinvar/, https://www.ensembl.org/index.html) and the literature. We considered a new variant if it was not found in these resources.
Multiplex Ligation-Dependent Probe Amplification (MLPA)
MLPA was performed using SALSA MLPA probemix DMD P034 and P035 according to the manufacturer’s instructions. The probemixes contain together one probe for each of the 79 exons of the DMD gene transcript variant Dp427m (MRC-Holland, Amsterdam).
For the initial step, we used 5μL (10 ng/μL) of DNA and was denatured during 5 min at 98°C. For the hybridization reaction the probemix and the MLPA buffer were added to the denatured samples which were incubated, first during 1 minute at 95°C and then during 16–20 hours at 60°C. In the ligation reaction the mix was incubated at 54°C with the ligase-65, followed by heating at 98°C for 5 minutes. For the final PCR step, we used exon-specific probes with universal-tagged primers. Amplicons were separated by capillary gel electrophoresis on the ABI3500 (Applied Biosystems) genetic analyzer with GeneScan 500 LIZ as the dye size standard. The Copy Number Variation (CNV) status was analyzed in the Coffalyser.Net software, according to the dosage quotient (DQs) between the patient samples and reference samples from males and females. These values are described in the SALSA MLPA DMD product description (MCR Holland) (www.mlpa.com).
Data Analysis
We used the molecular diagnostic data obtained with a genetic study of the dystrophin gene ordered by the treating physician. For all mutations, we identified the DNA change, establishing the molecular consequence on the protein. We classified the mutations into out-of-frame, in-frame, nonsense, splice site and missense. Frequencies were established for each type of mutations identified through sequencing or MLPA analysis. To determine the pathogenic consequences of splice site mutations, we performed in silico analysis using the bioinformatic tool Human Splicing Finder (www.http://www.umd.be/HSF3/). Taken together, these results allowed us to know the percentage of Colombian patients who could benefit from molecular therapies based on specific mutations.
Results
Through Sanger sequencing, NGS or MLPA, we identified mutations in 69 patients affected with DMD/BMD (Supplementary Table 1). The most frequent genetic mutations were large deletions (58%), followed by large duplications (14.5%), small deletions (11.6%), nonsense (11.6%) and splicing site mutations (4.3%). In the analyzed samples, missense mutations were not found (Table 1).
 |
Table 1 Frequency of Mutations and Molecular Consequence |
Regarding the consequence on the reading frame in patients with large deletions, we found a) 31 of 40 patients (77.5%) had out-of-frame mutations and b) 7 of 40 patients (17.5%) had in-frame mutations. In two cases, the large deletion involved the first methionine, and it was not possible to establish the molecular consequence (Table 1).
For large duplications, 60% of patients (6 of 10) had out-of-frame mutations and 40% in-frame mutations. Finally, all small deletions identified (8 patients) generated out-of-frame mutations.
In three patients, splicing site mutations were found (4.3%) (Supplementary Table 1). In silico analysis of these variants revealed that these mutations modify the donor splice site, affecting the normal splicing of the messenger RNA.
In this study, 7 (10.1%) new mutations were found, of which 4 (57.1%) corresponded to small deletion, 1 (14.2%) nonsense mutation and 2 (28.6%) were large deletions. All the small deletions caused out-of-frame mutations and the large deletions in-frame mutations (Table 2).
 |
Table 2 Description of New Mutations in Dystrophin Gene |
The mutational distribution based on exons showed that most of the described mutations were within the worldwide described hot spots; exons from 2 to 20 and 45 to 55. In exons 58, 59, 60, 61, 63, 68, 69, 70, 71 and 72 no mutations were found (Figure 1).
 |
Figure 1 Mutational frequency by exons in the analyzed population. |
Regarding patients with mutations involved in current pharmacologic therapies, we found that 18.8% meet the criteria. A priori, eight patients with nonsense mutations could be treated with Ataluren, two patients affected with large deletions in exons 48 to 50 and 50 are candidates for Eteplirsen treatment and three for Golodirsen with deletions in exons 49 to 52, 45 to 52 and 52 (Table 3).
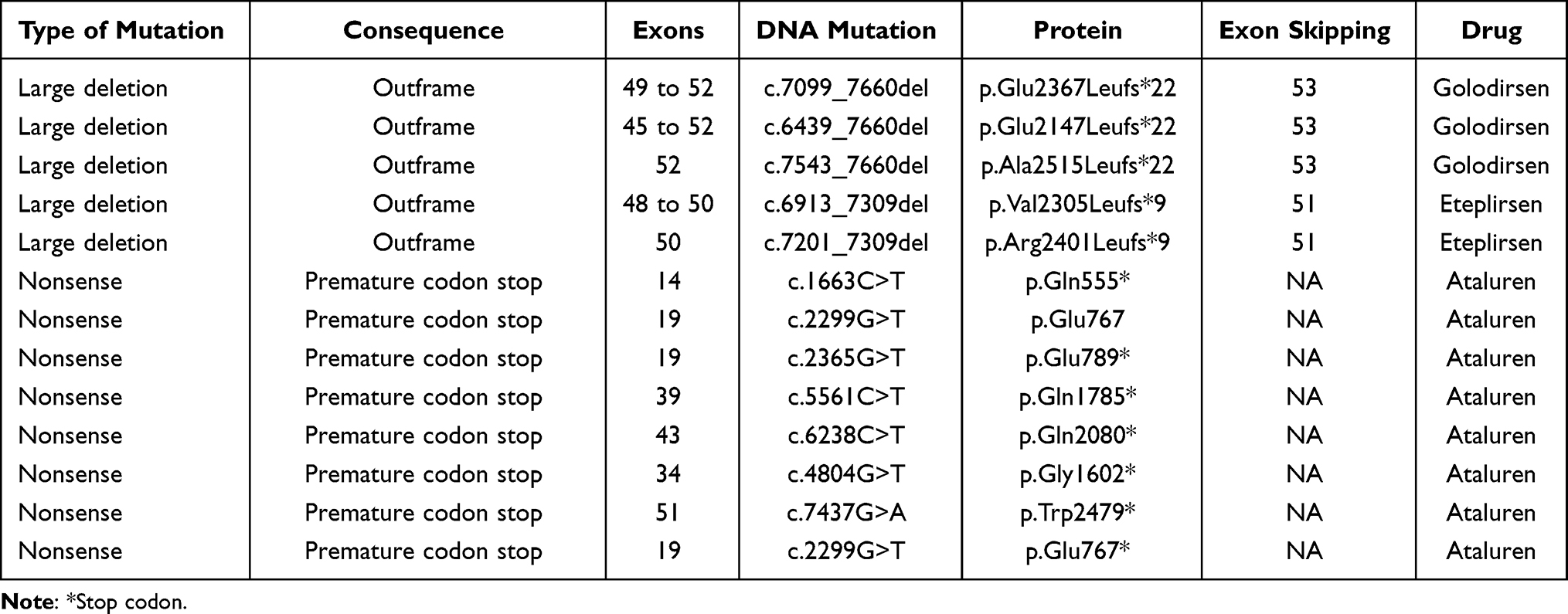 |
Table 3 Patients Who Expect to Benefit from Specific-Mutations Treatment |
Discussion
Duchenne muscular dystrophy is a progressive neuromuscular disorder characterized by muscle degeneration due to pathogenic mutations in the dystrophin gene. Affected patients lose normal ambulation, which leads to wheelchair dependency and in the final stages of the disease, respiratory and cardiac failure.15 Worldwide, several dystrophin gene mutations have been described; most of them involve large deletions and duplications and others correspond to point mutations and small rearrangements.8 Identification of genetic variation in DMD/BMD patients has enabled precision medicine treatments based on specific mutations and genetic counseling prevention.16
In Colombia there is no registry of DMD cases, which generates an underreporting of the real incidence in the country. Bernal Villegas et al estimated the cases of genetic diseases according to the number of births and determined that by the year 2021–2025 our country could have 1030 cases of DMD.2 In this context, the cohort of patients analyzed represents only a proportion of the cases in the country and contributes to the knowledge of the molecular characterization of dystrophin variants. Recently, Luce et al conducted a systematic review of literature showing that only 6 Latin American countries have reported dystrophin genetic profiles based on an algorithm that detects CNVs and sequencing variants, reflecting the knowledge gap with respect to European and North American countries.17
In this study, we analyzed the mutational profile of the dystrophin gene in 69 Colombian patients affected with DMD/BMD. The results revealed mutational heterogeneity in the DMD gene, as expected, with 57 different mutations in 69 patients. Large deletions were the genetic cause of 58% of the cases. Similar results have been described for other populations, where this type of mutation accounts for at least 50% of the cases.8,13,18–20
In our study, 80% of deletions were located in the mutational hot spots described in the DMD gene (exons 2–20 and exons 45–55). Precisely, 35.7% of them were in the distal region, the same proportion in the proximal region and in 5% of cases, the extent of the deletion involved exons from both regions. Deletions outside the exons considered hot spots accounted for 20% of the total. A previous publication about mutational hotspots (31 to 79 exons) in Colombian patients showed a lower frequency of deletions (33%).21 The difference in the exons analyzed and the methodology could affect the results reported between both studies, indicating that the molecular approach using only exons located on hot spots significantly reduces the detection of deletions. These findings highlight the importance of complete gene analysis, all exons sequencing and deletion/duplication analysis, for molecular diagnosis of DMD.
The second most frequent type of mutation corresponds to large duplications, identified in 14.5% of the cases. Similar findings have been reported in other populations such as China (16.4%), India (10%), Japan (16%) and Spain (19%).18,22–25 In 100% of our cases, large duplications involved exons from the mutation-prone regions, with a greater proportion (70%) in the proximal region and less (30%) in the distal part of the gene. This distribution is consistent with other studies, as described by Vieitez et al, who analyzed a Spanish population and reported a 67.9% of mutations in the proximal region and 28.6% in the distal region.19 Our data in agreement with those reported in the worldwide literature, supports that large deletions and large duplications are the most frequent molecular damage in affected patients with DMD/BMD. In this study, the frequency of these mutations was 72.5% (50 patients). Several mechanisms are related to large genomic rearrangements, however, the non-homologous end joining mechanism (NHEJ) and the Alu repeats crosslinking by the non-allelic homologous recombination (NAHR) are the most frequent molecular mechanism involved in the occurrence of deletions and duplications in DMD gene.13
Deletions and duplications (small and large) in the DMD gene can alter the open reading frame (ORF) generating out-of-frame mutations, which results in aberrant forms or absence of dystrophin protein. In other cases, there was no shift in the reading frame (in-frame mutations). These mutations are related to the severity of the phenotype.26 In our study, 77.6% of the cases had out-of-frame mutations and 18.9% in-frame mutations. The reading frame rule hypothesis referred by Monaco et al establish that Adjacent exons that can maintain an open reading frame (ORF) in the spliced mRNA despite a deletion event would give rise to the less severe BMD phenotype and predict the production of a lower molecular weight, semifunctional dystrophin protein. Adjacent exons that cannot maintain an ORF because of frame shifted triplet codons would give rise to the more severe DMD phenotype due to the production of a truncated, nonfunctional dystrophin protein.27 As a limitation of our study, we could not determine the genotype-phenotype correlation because clinical information to classify both DMD/BMD was not provided for all cases.
Regarding in-frame mutations, we found four patients with mutations mainly affecting the ROD domain of the protein, which is related to elasticity and flexibility. One of the patients had a mutation in the exon 73 or distal exon, within the C-terminal binding domain to α-syntrophin (ASD) and dystrobrevin (DBD). Two patients presented a mutation affecting the ABD domain required for the interaction of the protein with the cytoskeleton. Finally, four patients had a mutation involving both ROD and ABD domains. Although in-frame mutations, are less severe than out-of-frame, these could significantly alter the proper functioning of the protein.13
In two patients with large deletions, it was not possible to determine the molecular consequence (out-of-frame or in frame) due to their deletion involved the dystrophin gene start codon (first methionine), which affect the protein translation.
The scope of our study did not include functional studies (eg Western blot) that would allow us to predict a potential protein synthesis due to translation restart, which is a potential rescue mechanism of this type of mutations.28 Given the extension of the deletion described for these patients (exons 1 to 43 and 1 to 93, Supplementary Table 1), it is possible to hypothesize its clinical implication. In skeletal and cardiac muscle, dystrophin binds via an N-terminal domain to filamentous actin. This actin-binding domain involved the CH1 and CH2 domains, which are located at amino acids p.Arg13 to p.Val120 (CH1) and p.Asn135 to p.Val238 (CH2) that are lost in both patients with the large deletion,28 therefore, it may possibly affect the protein´s function.
Small deletions (<1 exon) and point mutations, accounted for 27.5% of patients (Table 1). This finding is similar to the data reported in the global TREAT-NMD-DMD database (a 20% is reported).8 Mutations leading to the generation of premature termination codons (PTC) were the genetic alterations in 11.6% of our patients. PTC mutations are associated with the reduction or complete absence of DMD mRNA. The complete absence is related to mRNA degradation by the Nonsense Mediated Decay system (NMD), which is a cellular regulatory control mechanism that prevents the expression of truncated proteins.20 For this reason, a drastic effect of these mutations on the protein is to be expected and therefore, a severe phenotype is estimated.19,20
Regarding mutations in splice sites, we identified three variants, which according to the in-silico prediction, affect the donor site and therefore, the normal splicing of the messenger RNA.
It is estimated that three possible molecular mechanisms can generate abnormal transcripts: intron retention, activation of non-canonical splicing sites and exon skipping. In any case, the protein is functional. Therefore, transcriptomic analysis of the patient’s muscle biopsy or functional studies (Minigenes), are required to confirm this prediction.29
We did not identify missense mutations in our sample of 69 previously clinically diagnosed patients. According to the literature, this type of mutation is the less frequent within DMD/BMD patients, which was also supported by our results.30–33 Missense mutations are rare and are located mainly in the DMD protein–protein interaction domains.30,33,34 In the largest cohort of Latin American patients with DMD analyzed to date, only one pathogenic missense variant was identified,17 probably with a larger sample size, we could have identified missense mutations that would contribute to the DMD mutational spectrum of Colombian patients. The molecular strategy used in our study (sanger and NGS) allows to identify 100% of the missense variants and we can conclude that for the population analyzed the frequency of this type of variant is 0%.
Taking together the findings obtained with the molecular strategies used (MLPA, Sanger, and NGS), 72.4% of CNVs and 27.5% of small pathogenic sequence variants were detected in the Colombian patients. These results are in concordance with data from Latin American populations such as Brazil,35 Argentina,17 Puerto Rico36 and Mexico37 where CNVs frequencies between 64.8 and 75% were reported. Interestingly, nonsense mutations throughout most Latin American countries represent 10.1 to 12.4% and, as our findings (11.6%), are the predominant molecular cause within sequence variants. The impact of this type of mutation lies in their potential relationship with mutation-directed therapy. Additionally, it supports the importance of using molecular diagnostic algorithms that, in addition to MLPA, include sanger sequencing or NGS. To date only four Latin American countries, use these combined strategies, therefore our results contribute to molecular characterization in Latin American populations.
Other populations such as Peru and Venezuela have reported significantly lower frequencies of large rearrangements (52.5 and 37% respectively), which is mainly explained by the methodology used (multiplex PCR).38,39
Assessment of autosomal and sexual markers in Latin American populations has revealed a high heterogeneity in the ethnic structure of these populations,40 and for Colombia, a higher inter-population variability has been described.41 Most of the patients were born in Bogotá (72.5%), a city with an estimated population structure with predominance of native ancestry (52%), followed by European and African (45% and 3% respectively). None of the participants were asked for their self-reported ethnicity, and ancestry was assumed as indicated in previous studies based on the analysis of Ancestry informative markers (AIMs) in individuals from this same population.41,42 Interestingly, despite the intra- and inter-population ethnic variability, our results and those of the Latin American populations are concordant and reflect a mutational profile in patients with DMD that highlights a high proportion of nonsense mutations,17 which is in agreement with our results (Table 1). For Latin America, only 7 studies have been published (about 725 patients) and potential differences derived from specific ancestry may be lost. Large cohorts of patients analyzed in multiple European populations have shown a marked heterogeneity in the frequency of large deletions versus sequence variants, indicating the importance of ancestry in the dystrophin mutation profile.43
Since 27.5% of all identified mutations were small deletions and point mutations, we support the use of both tools (sequencing and MLPA) for the molecular diagnosis of DMD/BMD. The MLPA has a sensitivity over 88%,24 however, this tool has some limitations such as non-detection of changes that lie outside the target sequences analyzed by the probes or non-delimitation of deleted regions. In our study, nine mutations were not clearly identified using MLPA (Supplemental Table 1, represented with b) because the deleted regions could not be precisely defined. In these cases, the large rearrangements included both introns and exons, causing alterations that could not be predicted by in silico analysis, however, we conducted an analysis based only on the exons deleted to predict the protein consequence.
In other reports, about 2% of DMD/BMD patients, mutations were not identified by MLPA and sequencing. This is potentially related to deep intronic changes produced by modifications located more than 100 bp upstream or downstream of the exon and the presence of complex rearrangements.44 In these cases, the analysis of the RNA from the muscle biopsy, by transcriptome and/or RNAseq analysis is necessary to verify the genetic cause of the disease.13
Adequate DNA diagnostic analysis is relevant since it provides information on eligibility for therapeutic strategies in DMD treatment.45,46 The FDA or MDA has approved molecular therapies based on specific mutations such as nonsense and out-of-frame mutations. To date, Ataluren is the bioavailable treatment for patients affected with PTC-related mutations. Recent reports indicated that in approximately 10–15% of boys with DMD, the disorder is due to a nonsense mutation, data similar to our findings (11.6% of cases).47 Among the 7 new mutations identified in our study, one of them is a nonsense mutation, a candidate for pharmacological treatment, which further highlights the importance of a comprehensive molecular diagnosis. In this context, a percentage of our population is expected to benefit from Ataluren treatment, which has been designed to generate ribosomal readthrough and enable the production of the full-length protein.48
One promising strategy for out-of-frame mutations is exon skipping, which restored the translational reading frame using synthetic analog nucleic acids analogs called antisense oligonucleotides (AOs).49 At the time, two drugs have received the US FDA approval for the treatment of DMD in patients with mutation amenable to exon skipping of the 51 (Eteplirsen) and 53 (Golodirsen) exons. Those AOs bind on pre-mRNA and influence the splicing machinery to exclude specific exon from the final transcript. The molecular therapies achieve the production of shortened functional dystrophin proteins.49,50 It has been estimated that in about 14% of all DMD patient’s single exon skipping is applicable.51 However, in our study, we identified deletions amenable to exon 51 and 53 skipping in 7.2% (5/69) of the cases (Table 3). Taken together, our results suggest that 19% of DMD patients analyzed would potentially benefit from nonsense readthrough or exon skipping therapy.
Recently other strategies have been developed for DMD therapy, evidence demonstrates that ncRNAs play a role in dystrophin regulation and represent a strategy with potential efficacy when combined with oligonucleotides/gene therapy approaches.52 Additionally, CRISPR/Cas9 is a potential strategy to correct muscular dystrophies by editing disease-causing mutations at the genomic level.53
Despite the potential application of therapies based on specific mutations, the lack of clinical information does not allow us to know whether our patients meet the inclusion criteria required for the benefit of these therapies.
Our results allowed us to establish a mutation profile for the dystrophin gene in a cohort of Colombian patients affected with DMB/BMD. Understanding the molecular variability and specific mutations in DMD/BMD patients will potentially lead to personalized therapies, genetic counseling, reproductive options and carrier detection.
Limitations of Study
We consider some limitations in our study: a) Lacks an analysis of correlation genotype-phenotype, b) For MLPA, the lack of exact delimitation of deleted regions in some cases and c) Clinical information limited to the treating physician d) the small sample size.
Conclusion
Our results describe the genomic profile of mutations for the Dystrophin gene in a cohort of DMB/BMD Colombian patients. These findings, including the discovery of new mutations, are relevant due to the possibility of including patients with mutations amenable to molecular therapies such as nonsense read-through or exon skipping.
Finally, this work contributes with the efforts to characterize the frequencies and molecular variants in Latin America populations affected with DMD/BMD.
Abbreviations
DMD/BMD, Duchenne and Becker muscular dystrophies; NGS, next-generation sequencing; MLPA, multiplex ligation dependent-probes amplification; NHEJ, the non-homologous end joining mechanism; NAHR, the non-allelic homologous recombination; ORF, open reading frame; ASD, α-syntrophin; DBD, dystrobrevin; PTC, premature termination codons; NMD, Nonsense Mediated Decay system; AIMs Ancestry informative markers.
Ethics Approval and Consent to Participate
All experimental procedures were approved by the Ethics Committee of the Universidad del Rosario. The study followed the guidelines of the Declaration of Helsinki (Approved CEI-AMH002-000185). All participants provided informed consent to take part in this study and for clinical attention in the private laboratory Genética Molecular de Colombia.
Acknowledgments
We thank Genética Molecular de Colombia and Universidad del Rosario for providing all the data and infrastructure necessary for carrying out this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Funding was not applicable. Data was obtained for molecular diagnosis of analyzed patients. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Silva CT, Mateus H, Contreras N. Restrepo CM Distrofia muscular de Duchenne y Becker: una visión molecular. Acta Med Colomb. 2005;3(30):112–116.
2. Bernal J, Obando F. La carga de la enfermedad genética en Colombia, 1996-2025. Universitas Médica. 2007;49:12–28.
3. Mercuri E, Bonnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019;394(10213):2025–2038.
4. Waldrop MA, Flanigan KM. Update in Duchenne and Becker muscular dystrophy. Curr Opin Neurol. 2019;32(5):722–727. doi:10.1097/WCO.0000000000000739
5. Gao QQ, McNally EM. The Dystrophin complex: structure, function, and implications for therapy. Compr Physiol. 2015;5(3):1223–1239. doi:10.1002/cphy.c140048
6. Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355(6362):696–702. doi:10.1038/355696a355690
7. Le Rumeur E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosnian J Basic Med Sci. 2015;15(3):14–20. doi:10.17305/bjbms.12015.17636
8. Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395–402. doi:10.1002/humu.22758
9. Mah JK, Selby K, Campbell C, et al. A population-based study of dystrophin mutations in Canada. Can J Neurol Sci. 2011;38(3):465–474. doi:10.1017/S0317167100011896
10. Aravind S, Ashley B, Mannan A, et al. Targeted sequencing of the DMD locus: a comprehensive diagnostic tool for all mutations. Indian J Med Res. 2019;150(3):282–289. doi:10.4103/ijmr.IJMR_4290_4118
11. Polavarapu K, Preethish-Kumar V, Sekar D, et al. Mutation pattern in 606 Duchenne muscular dystrophy children with a comparison between familial and non-familial forms: a study in an Indian large single-center cohort. J Neurol. 2019;266(9):2177–2185. doi:10.1007/s00415-00019-09380-00413
12. Tian PC, Wang Y, Shi DD, Chen Z, Luo Q, Wang HL. [Application of next-generation sequencing in the molecular diagnosis of Duchenne muscular dystrophy]. Zhongguo Dang Dai Er Ke Za Zhi/Chinese J Contemporary Pediatrics. 2019;21(3):244–248. Chinese.
13. Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PLoS One. 2015;10(8):e0135189. doi:10.0131371/journal.pone.0135189
14. Deburgrave N, Daoud F, Llense S, et al. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum Mutat. 2007;28(2):183–195. doi:10.1002/humu.20422
15. Muntoni F, Desguerre I, Guglieri M, et al. Ataluren use in patients with nonsense mutation Duchenne muscular dystrophy: patient demographics and characteristics from the STRIDE Registry. J Comp Eff Res. 2019;8(14):1187–1200. doi:10.2217/cer-2019-0086
16. Nakamura A. Mutation-based therapeutic strategies for Duchenne Muscular Dystrophy: from genetic diagnosis to therapy. J Personalized Med. 2019;9(1):16. doi:10.3390/jpm9010016
17. Luce L, Carcione M, Mazzanti C, et al. Theragnosis for Duchenne muscular dystrophy. Front Pharmacol. 2021;12:648390. doi:10.3389/fphar.2021.648390
18. Elhawary NA, Jiffri EH, Jambi S, et al. Molecular characterization of exonic rearrangements and frame shifts in the dystrophin gene in Duchenne muscular dystrophy patients in a Saudi community. Hum Genomics. 2018;12(1):18. doi:10.1186/s40246-40018-40152-40248
19. Vieitez I, Gallano P, Gonzalez-Quereda L, et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: study of 284 cases. Neurol. 2017;32(6):377–385. doi:10.1016/j.nrl.2015.1012.1009
20. Magri F, Del Bo R, D’Angelo MG, et al. Clinical and molecular characterization of a cohort of patients with novel nucleotide alterations of the Dystrophin gene detected by direct sequencing. BMC Med Genet. 2011;12:37. doi:10.1186/1471-2350-1112-1137
21. Silva CT, Fonseca DJ, Mateus H, Contreras N, Restrepo C. Deleciones en el gen de la distrofina en 62 familias colombianas: correlación genotipo-fenotipo para la distrofia muscular de Duchenne y Becker. Colomb Med. 2009;35:191–198.
22. Vieitez I, Gallano P, Gonzalez-Quereda L, et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: study of 284 cases. Neurol. 2017;32(6):377–385. doi:10.1016/j.nrl.2015.12.009
23. Chen C, Ma H, Zhang F, et al. Screening of Duchenne muscular dystrophy (DMD) mutations and investigating its mutational mechanism in Chinese patients. PLoS One. 2014;9(9):e108038. doi:10.1371/journal.pone.0108038
24. Manjunath M, Kiran P, Preethish-Kumar V, Nalini A, Singh RJ, Gayathri N. A comparative study of mPCR, MLPA, and muscle biopsy results in a cohort of children with Duchenne muscular dystrophy: a first study. Neurol India. 2015;63(1):58–62. doi:10.4103/0028-3886.152635
25. Okubo M, Minami N, Goto K, et al. Genetic diagnosis of Duchenne/Becker muscular dystrophy using next-generation sequencing: validation analysis of DMD mutations. J Human Genetics. 2016;61(6):483. doi:10.1038/jhg.2016.1037
26. Zhou J, Xin J, Niu Y, Wu S. DMDtoolkit: a tool for visualizing the mutated dystrophin protein and predicting the clinical severity in DMD. BMC Bioinform. 2017;18(1):87. doi:10.1186/s12859-017-1504-4
27. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–95. doi:10.1016/0888-7543(1088)90113-90119
28. Gurvich OL, Maiti B, Weiss RB, Aggarwal G, Howard MT, Flanigan KM. DMD exon 1 truncating point mutations: amelioration of phenotype by alternative translation initiation in exon 6. Hum Mutat. 2009;30(4):633–640. doi:10.1002/humu.20913
29. Wang Z, Lin Y, Qiu L, et al. Hybrid minigene splicing assay verified the pathogenicity of a novel splice site variant in the dystrophin gene of a Chinese patient with typical Duchenne muscular dystrophy phenotype. Clin Chem Lab Med. 2016;54(9):1435–1440. doi:10.1515/cclm-2015-1042
30. Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30(12):1657–1666. doi:10.1002/humu.21114
31. Mohammed F, Elshafey A, Al-Balool H, et al. Mutation spectrum analysis of Duchenne/Becker muscular dystrophy in 68 families in Kuwait: the era of personalized medicine. PLoS One. 2018;13(5):e0197205. doi:10.1371/journal.pone.0197205
32. Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395–402. doi:10.1002/humu.22758
33. Tuffery-Giraud S, Béroud C, Leturcq F, et al. Genotype-phenotype analysis in 2405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30(6):934–945. doi:10.1002/humu.20976
34. Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PLoS One. 2015;10(8):e0135189. doi:10.1371/journal.pone.0135189
35. de Almeida PAD, Machado-Costa MC, Manzoli GN, et al. Genetic profile of Brazilian patients with dystrophinopathies. Clin Genet. 2017;92(2):199–203. doi:10.1111/cge.12975
36. Ramos E, Conde JG, Berrios RA, Pardo S, Gómez O, Mas Rodríguez MF. Prevalence and genetic profile of Duchene and Becker muscular dystrophy in Puerto Rico. J Neuromusc Dis. 2016;3(2):261–266. doi:10.3233/JND-160147
37. López-Hernández LB, Gómez-Díaz B, Luna-Angulo AB, et al. Comparison of mutation profiles in the Duchenne muscular dystrophy gene among populations: implications for potential molecular therapies. Int J Mol Sci. 2015;16(3):5334–5346. doi:10.3390/ijms16035334
38. Huamán-Dianderas FD, Guevara-Fujita ML, Málaga DR, Estrada-Cuzcano A, Fujita R. [Detection of mutations causing Duchenne and Becker muscular dystrophies: multiplex polymerase chain reaction vs. Multiplex ligation dependent probe amplification]. Rev Peru Med Exp Salud Publica. 2019;36(3):475–480. Catalan.
39. Delgado Luengo W, Pineda-Del Villar L, Borjas L, et al. Molecular diagnosis of Duchenne/Becker muscular dystrophy in Venezuelan patients with the polymerase chain reaction. Invest Clin. 1994;35(4):195–207.
40. Adhikari K, Mendoza-Revilla J, Chacón-Duque JC, Fuentes-Guajardo M, Ruiz-Linares A. Admixture in Latin America. Curr Opin Genet Dev. 2016;41:106–114. doi:10.1016/j.gde.2016.09.003
41. Salzano FM, Sans M. Interethnic admixture and the evolution of Latin American populations. Genet Mol Biol. 2014;37(1 Suppl):151–170. doi:10.1590/S1415-47572014000200003
42. De Castro M, Restrepo CM. Genetics and genomic medicine in Colombia. Mol Genetics Genomic Med. 2015;3(2):84–91. doi:10.1002/mgg3.139
43. Selvatici R, Rossi R, Fortunato F, Trabanelli C, Sifi Y. Ethnicity-related DMD genotype landscapes in European and non-European countries. Neurol Genetics. 2021;7(1):e536.
44. Laing NG, Davis MR, Bayley K, Fletcher S, Wilton SD. Molecular diagnosis of Duchenne muscular dystrophy: past, present and future in relation to implementing therapies. Clin Biochem Rev. 2011;32(3):129–134.
45. Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53(3):145–151. doi:10.1136/jmedgenet-2015-103387
46. Sun C, Shen L, Zhang Z, Xie X. Therapeutic strategies for Duchenne muscular dystrophy: an update. Genes. 2020;11:8. doi:10.3390/genes11080837
47. Muntoni F, Desguerre I, Guglieri M, et al. Ataluren use in patients with nonsense mutation Duchenne muscular dystrophy: patient demographics and characteristics from the STRIDE Registry. J Comp Eff Res. 2019;8(14):1187–1200. doi:10.2217/cer-2019-0086
48. Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med. 2013;64:407–425. doi:10.1146/annurev-med-120611-144851
49. Stein CA, Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol Ther. 2017;25(5):1069–1075. doi:10.1016/j.ymthe.2017.03.023
50. Heo YA. Golodirsen: first approval. Drugs. 2020;80(3):329–333. doi:10.1007/s40265-020-01267-2
51. Charleston JS, Schnell FJ, Dworzak J, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology. 2018;90(24):2146–2154. doi:10.1212/WNL.0000000000005680
52. Brusa R, Magri F, Bresolin N, Comi GP, Corti S. Noncoding RNAs in Duchenne and Becker muscular dystrophies: role in pathogenesis and future prognostic and therapeutic perspectives. Cell Mol Life Sci. 2020;77(21):4299–4313. doi:10.1007/s00018-020-03537-4
53. Chemello F, Bassel-Duby R, Olson EN. Correction of muscular dystrophies by CRISPR gene editing. J Clin Invest. 2020;130(6):2766–2776. doi:10.1172/JCI136873
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.