")
Back to Journals » OncoTargets and Therapy » Volume 7
Doxorubicin-conjugated bacteriophages carrying anti-MHC class I chain-related A for targeted cancer therapy in vitro
Authors Phumyen A, Jantasorn S, Jumnainsong A, Leelayuwat C
Received 12 June 2014
Accepted for publication 30 August 2014
Published 28 November 2014 Volume 2014:7 Pages 2183—2195
DOI https://doi.org/10.2147/OTT.S69315
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
Achara Phumyen,1–3 Siriporn Jantasorn,1 Amonrat Jumnainsong,1 Chanvit Leelayuwat1–4
1The Centre for Research and Development of Medical Diagnostic Laboratories (CMDL), Faculty of Associated Medical Sciences, 2The Liver Fluke and Cholangiocarcinoma Research Center, Faculty of Medicine, 3Research Cluster: Specific Health Problem of Grater Maekong Subregion (SHeP-GMS), 4Department of Clinical Immunology and Transfusion Sciences, Faculty of Associated Medical Sciences, Khon Kaen University, Khon Kaen, Thailand
Background: Cancer therapy by systemic administration of anticancer drugs, besides the effectiveness shown on cancer cells, demonstrated the side effects and cytotoxicity on normal cells. The targeted drug-carrying nanoparticles may decrease the required drug concentration at the site and the distribution of drugs to normal tissues. Overexpression of major histocompatibility complex class I chain–related A (MICA) in cancer is useful as a targeted molecule for the delivery of doxorubicin to MICA-expressing cell lines.
Methods: The application of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC) chemistry was employed to conjugate the major coat protein of bacteriophages carrying anti-MICA and doxorubicin in a mildly acid condition. Doxorubicin (Dox) on phages was determined by double fluorescence of phage particles stained by M13-fluorescein isothiocyanate (FITC) and drug autofluorescence by flow cytometry. The ability of anti-MICA on phages to bind MICA after doxorubicin conjugation was evaluated by indirect enzyme-linked immunosorbent assay. One cervical cancer and four cholangiocarcinoma cell lines expressing MICA were used as models to evaluate targeting activity by cell cytotoxicity test.
Results: Flow cytometry and indirect enzyme-linked immunosorbent assay demonstrated that most of the phages (82%) could be conjugated with doxorubicin, and the Dox-carrying phage-displaying anti-MICA (Dox-phage) remained the binding activity against MICA. Dox-phage was more efficient than free drugs in killing all the cell lines tested. The half maximal inhibitory concentration (IC50) values of Dox-phage were lower than those of free drugs at approximately 1.6–6 times depending on MICA expressions and the cell lines tested.
Conclusion: Evidently, the application of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide chemistry is effective to conjugate doxorubicin and major coat protein of bacteriophages without destroying binding activity of MICA antibodies. Dox-carrying bacteriophages targeting MICA have been successfully developed and may enable a broad range of applications in cancer-targeting chemotherapy.
Keywords: MHC class I chain–related A (MICA), phage display, doxorubicin, targeted therapy
Introduction
Over the past two decades, the therapeutic efficiency of chemotherapeutic drugs or toxins targeting tumors have been improving, which relies on the specific binding and internalization of these conjugates into the target cells. Immunoconjugates–monoclonal antibodies (mAbs) or immunoconjugates–nanoparticles coupled to highly toxic agents are becoming a significant component in anticancer treatments.1 By combining the exquisite targeting specificity of mAbs with enhanced tumor-killing power of toxic effector molecules, immunoconjugates permit sensitive discrimination between targets and normal tissues, resulting in reduced administration of drug concentration and fewer toxic side effects than most conventional chemotherapeutic drugs.2 Recently, a new platform of targeted anticancer therapy has been generated in the form of drug-carrying bacteriophages.3,4 This approach is based on genetically modified and chemically manipulated filamentous bacteriophages. The genetic manipulation endows the phages with the ability to display host specificity–conferring ligands. The phages are loaded with a large payload to a cytotoxic drug by chemical conjugation. Targeting of phage nanomedicines results in endocytosis, intracellular degradation, and drug release, leading to growth inhibition of the target cells in vitro with potentiation factor of more than 1,000 over the corresponding free drug.4
Natural killer group 2, member D (NKG2D) ligands show restricted expression in normal tissues, but they are frequently overexpressed in cancer and viral-infected cells.5 Many tumor cell lines and primary tumors from diverse tissue origins express NKG2D ligands such as carcinoma (lung, breast, kidney, prostate, ovary, colon, liver, and bile duct), melanoma, and some primary leukemia.5–12 The binding of NKG2D ligands to receptor activates natural killer (NK) cells and T cells and promotes cytotoxic lysis of the cells that express these molecules.13 Therefore, the expression of major histocompatibility complex (MHC) class I chain-related A (MICA) on tumor cells provide us opportunities for the use of immunoconjugate drug therapy for cancer. Immunotherapy via NKG2D recognition has been exploited using chimeric antitumor mAb/NKG2D ligand reagents, which specifically bind to human tumor cells and enhance the lysis by NK cells in an NKG2D-dependent manner as well as elicit adaptive immune responses against cancer cells independent of NKG2D ligand expression.14 We have previously produced bacteriophages carrying mutated anti-MICA antibodies with high binding activities.15 In order to generate an effective platform for killing cancer cells, the application of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC) chemistry was employed to conjugate anticancer drug (doxorubicin) to major coat g8p protein of M13 filamentous phages that carry anti-MICA antibodies. These drug-carrying phages specific to MICA antigens are more effective than free doxorubicin in killing cell lines expressing MICA.
Materials and methods
Materials
Phage-displaying anti-MICA antibody
Phage-displaying mutant Fab WW9B8.21 displays high-affinity anti-MICA derived from monoclonal WW9B8.15,16 These phages were used for conjugation with the anticancer drug doxorubicin · HCl (Enzo Life Sciences, Inc., Matford Court, Exeter, UK). All phages were amplified by infected bacteria, Escherichia coli XL1 blue (Stratagene, La Jolla, CA, USA) and rescued by VSCM13 (New England Biolabs, Inc., Ipswich, MA, USA), the helper phages, for phage stock. The stocked phages were counted and calculated as plaque-forming unit (PFU) before performing conjugation and then were stored in Tris-buffered saline (USB; Affymetrix, Inc., High Wycombe, UK)/1% bovine serum albumin (Sigma-Aldrich, St Louis, MO, USA) at 4°C.
Soluble MICA antigen
Soluble MICA (sMICA) antigen was produced from a eukaryotic expression clone carrying a full-length coding sequence of the MICA*009 gene with inserted stop codons before the transmembrane portion to produce a soluble form of MICA according to a previous report.16
Cell lines
Cell lines used were a cervical cancer cell line, Hela (CCL-2; American Type Culture Collection, Manassas, VA, USA), and the cholangiocarcinoma cell lines, KKU-M055, KKU-M213, KKU-M214, KKU-M13, and KKU-MMNK1 provided by The Liver Fluke and Cholangiocarcinoma Research Center, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand. Cells have an epithelial-like morphology growing as monolayer in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) and HAM’s F12 (Thermo Fisher Scientific) with 10% (v/v) heat inactive fetal calf serum (PAA GmbH, Cölbe, Germany) and 100 μg/mL of penicillin/streptomycin sulfate (Sigma-Aldrich).
Methods
Chemical conjugation of doxorubicin to phage-displaying anti-MHC class I chain-related A antibodies
The conjugation of doxorubicin to M13 bacteriophages was performed according to the previous studies.4,17,18 The phage major coat protein g8p contains three carboxylic amino acids (glu2, asp4, and asp5), which can be conjugated directly with the primary amine of doxorubicin by the application of EDC chemistry (Thermo Fisher Scientific, Waltham, MA, USA), a rapid reaction performed at mild pH 4–6.19 All conjugations were performed within a 1.5 mL tube. The reagents for conjugation which were the EDC solution at pH 4-6 were used in the reaction as the condition below. The concentration and ratio of carboxylate groups on phage particles, amine groups on doxorubicin molecules, and concentration of EDC were calculated at a ratio of 1 mol COOH:1 mol amine:4 mol EDC.
In the experiment, doxorubicin, phages, 0.1 M sodium citrate buffer pH 4, 5, or 6, and 0.75 NaCl (Vivantis, Inc., Oceanside, CA, USA) were added into a 1.5 mL tube. The conjugation reaction was initiated by the addition of EDC solution, which was repeated four times at time intervals of 30 minutes (0, 30, 60, and 90 min). Each reaction was carried out at room temperature with gentle stirring (10 revolutions per minute [rpm]) for a total of 2 hours. The doxorubicin-conjugated phages were separated from the reaction by two dialysis steps against 1,000 mL of sterile 0.3 M NaCl each for 16 hours. Then, the doxorubicin-conjugated phages were precipitated using 4% polyethylene glycol (PEG) (Bio Basic, Inc., Markham, ON, Canada)/3% NaCl on ice for 1 hour and were centrifuged at 9,000 rpm, 4°C, for 20 minutes. The supernatant was discarded and the pellet was resuspended with 1,000 μL of phosphate Buffered Saline (PBS) (USB), pH 7.4.
Evaluation of binding specificity of doxorubicin-carrying phages to soluble MICA antigen by enzyme-linked immunosorbent assay
To evaluate the specific binding of doxorubicin-conjugated phages carrying anti-MICA, the reactions were tested as follows. After conjugation and dialysis, the solution from each pH (4, 5, and 6) was precipitated with 4% PEG/3%NaCl (v/v) on ice for 30 minutes and then centrifuged at 9,000 rpm, 4°C for 20 minutes. The supernatants and pellets of each were collected and tested for specific binding to sMICA antigen. A 96-well microtiter plate (Corning Incorporated Life Sciences, Tewksbury MA, USA) was treated with 300 μL of carbonate buffer pH 9.6 for 2 hours at room temperature. After discarding coating buffer, the sMICA antigens were coated on each well plate at 5 and 1 μg in PBS (pH 7.4). The plate was wrapped with parafilm tape and incubated at 4°C overnight. The solution was discarded, and the plate was then washed with phosphate buffer saline tween-20 (PBST) three times, followed by the addition of blocking solution, 300 μL of 5% skim milk (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) PBST and incubated at room temperature for 2 hours. The blocking solution was discarded, and the plate was washed three times with PBST followed by the addition of 1011 PFU of doxorubicin-conjugated phages in PBS (pH 7.4, 100 μL) and incubated at room temperature for 1 hour and the solution was then discarded. PBS (pH 7.4) was used as a negative control. After washing three times with PBST, 100 μL of anti-M13-conjugated horseradish peroxidase (HRP) (diluted at 1:15,000 in blocking solution; GE Healthcare Europe GmbH, Freiburg, Germany) was added to each well and incubated at room temperature for 1 hour. The antibody solution was discarded, and the plate was washed three times with PBST followed by the addition of 100 μL of TMB substrate per well containing substrate A and substrate B at 1:1 (v/v) ratio (Kirkegaard and Perry Laboratories, Inc., Gaithersburg, MD, USA) and then incubated for 20 minutes in the dark at room temperature. After the blue color was developed, 50 μL of stop reaction solution (2 N H2PO4; BDH Laboratories Supplies, Poole, UK) was added to each well and yellow color was developed. The absorbance (optical density [OD]) at 450 nm was measured by the Sunrise enzyme-linked immunosorbent assay (ELISA) plate reader (Tecan, Grödig, Austria) and analyzed by the Magellen7 software (Tecan).
Detection of doxorubicin molecules on phage particles
Detection of doxorubicin molecules on phage coat protein by X-ray diffraction assay
For the X-ray diffraction (XRD) assay, the samples were crystallized by lyophilizer (Martin Christ, Osterode am Harz, Germany) before analysis (provided by the Faculty of Dentistry, Khon Kaen University, Thailand). The samples, including doxorubicin 770 μg in 1,000 μL of distilled water, doxorubicin-conjugated phage 1012 PFU, doxorubicin and phage 1012 PFU mixture in the solution of conjugation reaction with replaced EDC solution with distilled water and the naked phages 1012 PFU in the same solution of conjugation reaction, were frozen at −80°C overnight followed by frozen evaporation for 24 hours. All samples in crystallized form were analyzed by Philips X’Pert MPD X-ray diffractometer (Philips, Almelo, the Netherlands) using scanning angle 2θ (theta) ranging from 10¸ to 70θ, which were performed at and supported by the Department of Physics, Faculty of Sciences, Ubon Ratchathani University, Thailand. The spectrums from XRD were analyzed using the X’Pert data organizer software (Philips).
Detection of doxorubicin molecules on phage coat protein by flow cytometry
Doxorubicin is an autofluorescent agent, which can be excited and the emission detected by a fluorescent detector.20 The samples used in this experiment included two tubes of phages and doxorubicin-conjugated phages with 107 phage particles. All samples were precipitated with 4% PEG/3% NaCl on ice for 30 minutes and then centrifuged at 9,000 rpm, 4°C for 20 minutes. The supernatant was discarded, and the pellet was resuspended with 100 μL of PBS pH 7.4. The naked phages and doxorubicin-conjugated phages were stained with 2 μg/mL of anti-M13-conjugated FITC antibodies (Antibodies-online Inc, Atlanta, USA) and incubated on ice for 1 hour. Simultaneously, detection of doxorubicin molecules on phage coat proteins by the excitation at 488 nm with emission integrated above 530 nm and fluorescent signal of conjugated FITC anti-M13 was performed using the BD FACSCanto II Flow cytometer (Becton, Dickinson and Company).21–23 The raw data from all samples were analyzed by the BD FACSDiva software.
Calculation of amount of doxorubicin on conjugated phages
The standard curve of different concentrations of doxorubicin was performed to calculate the amount of doxorubicin on conjugated phages. The fluorescent intensity of doxorubicin was measured by Spectramax Gemini XPS fluorescence microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA), using excitation at 485 nm. The amount of doxorubicin tested was 20 μg, 10 μg, 5 μg, 2.5 μg, 1.25 μg, 0.625 μg, 0.312 μg, 0.156 μg, 0.078 μg, 0.039 μg, and 0.019 μg. This standard curve was used to estimate the amount of doxorubicin on conjugated phages.
Evaluation of cytotoxicity of doxorubicin-conjugated phages by sulforhodamine B assay
The evaluation of cytotoxicity of doxorubicin-conjugated phages by SRB assay was modified from the protocol reported by Vichai and Kirtikara.24 In vitro cytotoxicity of doxorubicin-conjugated phages was evaluated on Hela, KKU-M055, KKU-M213, KKU-M214, KKU-M139, and KKU-MMNK1, which are cancer cell lines expressing MICA antigens on their cell surface. Cells were harvested from an exponential phase culture by trypsinization with 1× trypsin/EDTA (Thermo Fisher Scientific, Waltham, MA, USA), counted and plated in 96-well plates in a triplicate manner. Optimal seeding densities for each cell line were determined to ensure exponential growth during 64 hours. Seeding densities were 4,500 cells per well. Thereafter, cells were incubated for 16–18 hours before exposure to testing samples.
Cell lines were treated in three conditions in triplicates. First, cell lines were treated with dilutions of free doxorubicin (1 μg, 0.5 μg, 0.1 μg, 0.05 μg, 0.01 μg, 0.005 μg, and 0.001 μg). Second, cell lines were treated with dilutions of each conjugated phages ranging from 107 to 1012 particles as well as unconjugated or naked phages of 1012 particles. Third, cell lines were blocked with 1012 particles of anti-MICA-carrying phages with no drug conjugation 2 hours before Doxorubicin (Dox)-carrying phage-displaying anti-MICA (Dox-phage) treatment. The amount of doxorubicin on phage particles are shown in Table 1. All testing samples were diluted in 100 μL of complete Dulbecco’s Modified Eagle’s Medium and HAM’s F12 medium. The treated cells were incubated in humidified incubator with 5% CO2 at 37°C for 48 hours followed by the fixing step. The aside plate containing only cell suspension in three columns for no growth (day 0) was set up, and the plate was incubated at 37°C in a humidified incubator with 5% CO2 until cell attachment completed and it was moved to the fixing step. In the fixing step, 100 μL of cold 10% (w/v) tricholo acetic acid (Sigma-Aldrich) was added to each well, and the plates were incubated at 4°C for 1 hour and then washed four times with slow tap water and the excess water was removed, followed by air-drying at room temperature. The cells were then stained with 100 μL of 0.4% (w/v) SRB (Sigma-Aldrich, Munich, Germany) dissolved in 1% (v/v) acetic acid (Merck, Germany) for 30 minutes and subsequently washed five times with 1% acetic acid to remove unbound stain. The plate was left to dry at room temperature, and bound protein stain was solubilized with 200 μL of 10 mM Tris base (pH 10.5) per well and then shaken in the gyrator for 1 hour. The amount of SRB was measured for the absorbance at 492 nm using the ELISA plate reader (Tecan). The percentage of cell death was calculated using the formulae: (% of cell death =100 – ([mean ODsample − mean ODday 0]/[mean ODcontrol − mean ODday 0]) × 100).17 For IC50 determination, a dose–response curve was plotted between the sample concentrations and percent growth inhibition, which was derived from the IC50 calculation software (the CalcuSyn software; Biosoft Great Shelford, Cambridge, UK).
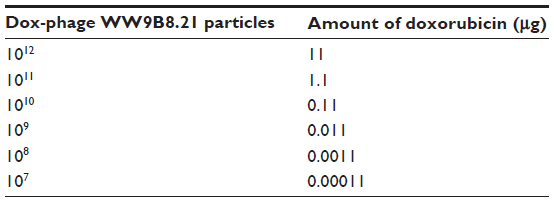 | Table 1 The amount of doxorubicin on each dilution of Dox-phage WW9B8.21 |
Results
Specific binding of doxorubicin- carrying phages to sMICA antigens
The specific binding of doxorubicin-conjugated phages displaying mutated anti-MICA antibodies (WW9B8.21) was evaluated by ELISA. The doxorubicin-conjugated phages at pH 5 showed the highest OD when compared to those derived from pH 4 and 6 using the same EDC concentration (Figure 1). The results indicate that the process of doxorubicin conjugation did not destroy the antibody activities and that the activities against MICA were preserved. Most of conjugated phages were in the pellets after precipitation.
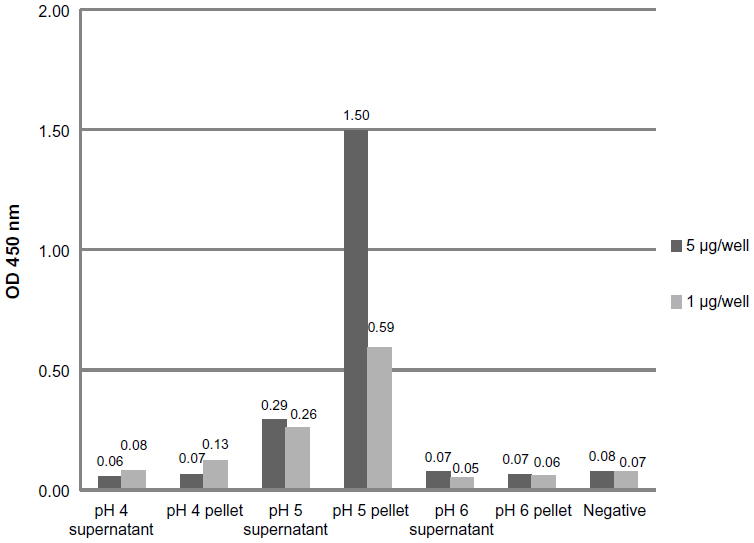 | Figure 1 Determination of specific binding doxorubicin-conjugated phages at different pH conditions. |
Detection of doxorubicin molecules on phages by XRD assay
The existence of doxorubicin molecules on phage particles was confirmed by two methods: the XRD assay and flow cytometry analysis. The spectrum patterns of free doxorubicin, naked phages, doxorubicin and phage mixture, and doxorubicin-conjugated phages are shown in Figure 2. Doxorubicin revealed an amorphous spectrum, while the spectra of phages, doxorubicin-conjugated phages, and doxorubicin and phage mixture were shaped peaks. These spectra were similar; however, an additional peak was only found in the spectrum of doxorubicin-conjugated phages (Figure 2), indicating that the spectrum of phage particles changed after doxorubicin conjugation.
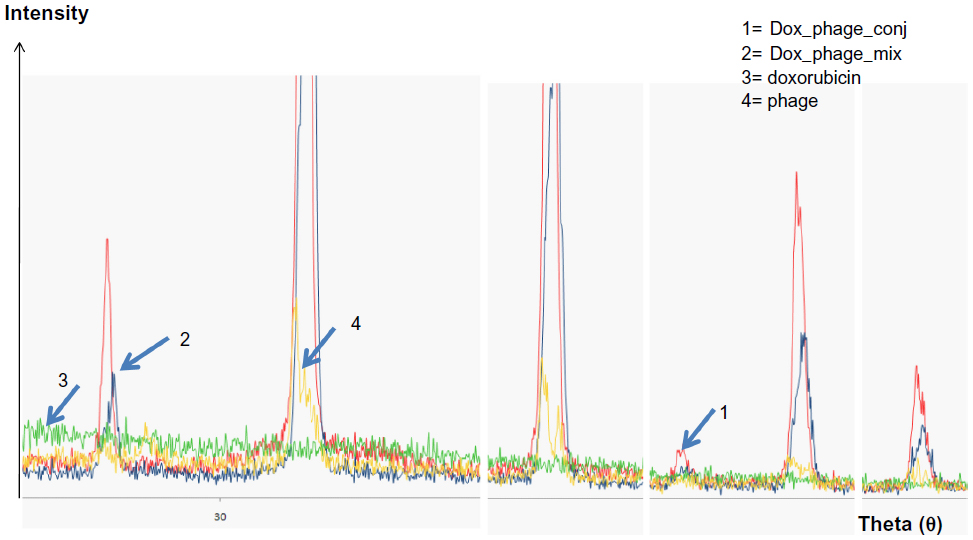 | Figure 2 Magnified spectra of X-ray diffraction from crystallized samples of doxorubicin, phages, and conjugated phages. |
Detection of doxorubicin molecules on phage particles by flow cytometry
The doxorubicin molecules are autofluorescent and can be detected by any fluorometer. Thus doxorubicin on phage particles could be excited and detected by the channel of peridinin–chlorophyll–protein complex by flow cytometer. With anti-M13 FITC staining, doxorubicin conjugated to phage particles could be indicated by double-fluorescent particles of peridinin–chlorophyll–protein complex and FITC. As shown in Figure 3, 98.8% of phage particles were double fluorescent, indicating the existence of doxorubicin in phage particles. Using a doxorubicin standard curve (Figure 4), doxorubicin carried by phage solution used in the experiment could be calculated (Table 1).
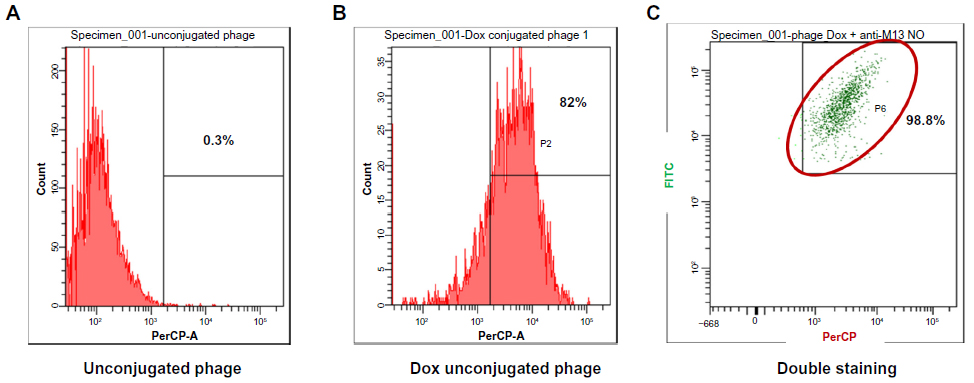 | Figure 3 Detection of doxorubicin molecules on phage particles by flow fluorometry. |
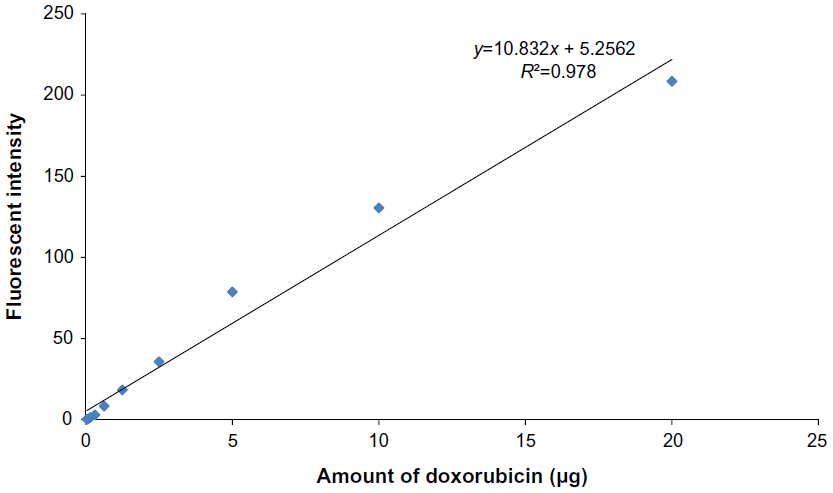 | Figure 4 The standard curve of doxorubicin. |
Amount of doxorubicin on conjugated phages
The standard curve of different concentrations of doxorubicin and the equation for calculating the amount of doxorubicin on conjugated phages are shown in Figure 4. The fluorescence intensity of doxorubicin-conjugated phages at 1012 PFU was 124.38. This standard curve was used to estimate the amount of doxorubicin on conjugated phages. The amount of doxorubicin was calculated using the formulae as follows:
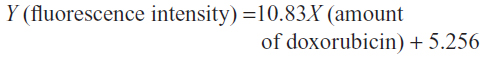
Cytotoxicity of doxorubicin-conjugated phages on MICA-expressing cell lines
The cytotoxic effect of doxorubicin-conjugated phages on MICA-expressing cell lines KKU-M214, KKU-M213, KKU-M055, KKU-M139, KKU-MMNK1, and Hela was determined by adding conjugated phages at different dilutions into the cells and comparing with doxorubicin-free drugs (Figure 5). The IC50 values were evaluated using the CalcuSyn demo version software and shown in Table 2 with the intensity of MICA expression on each cell line analyzed by flow cytometry. The doxorubicin-conjugated phages could kill the cell lines more effectively than free drug with a ratio of 0.8 to 6 (Table 2). In order to show the targeting effect, the MICA-blocking experiments were performed. Obviously, after blocking MICA by mAb, the IC50 value of each cell line was increased and was comparable to that of free drug as shown in Table 2 and Figure 5 with the exception of KKU-M055, which is a doxorubicin-resistant cell line.
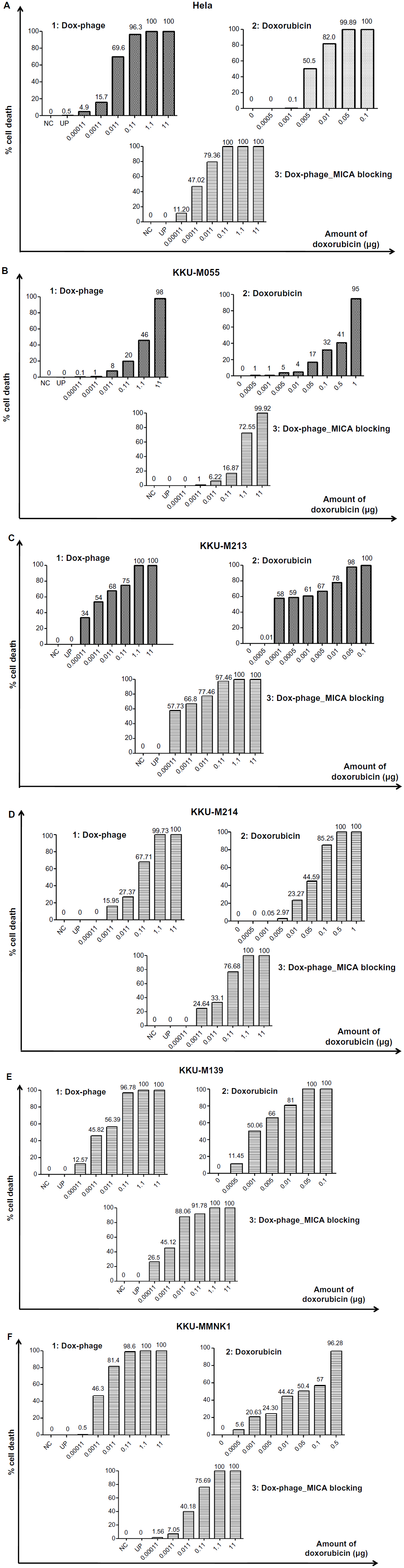 | Figure 5 The toxicity effect of doxorubicin and Dox-phage mutant WW9B8.21. |
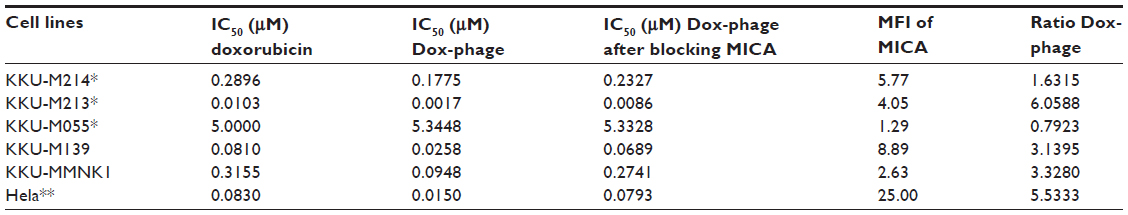 | Table 2 IC50 values of doxorubicin and doxorubicin-conjugated phages on MICA-expressing cell lines |
Discussion
Conventional chemotherapeutic agents are distributed nonspecifically in the body where they affect both cancer cells and normal cells. Molecularly targeted therapy has emerged as one approach to overcome the lack of specificity of conventional chemotherapeutic drugs. The mAbs that bind to specific markers on the surface of tumor cells offer an alternative therapy that is tumor specific and thus less toxic. The antibody–drug conjugates have been developed to increase the specific cytotoxicity on targeted tumor cells due to specific moieties and higher accumulated dose of anticancer drugs at targeted sites.25,26 However, this approach is limited by the amount of drug molecules that can be conjugated to antibody molecules. To overcome suboptimal dose, several nanoparticles are used as drug carriers using both passive and active targeting strategies that can enhance the accumulation of drug concentration at the tumor site while avoiding toxicity in normal tissues.1 Viral nanoparticles are virus-based nanoparticle formations27 that can be used for novel materials with a variety of properties including dynamics and self assembling systems. They can be produced in large quantities in a short time with programmable scaffolds. In addition, they are biocompatible and biodegradable with ability to display moiety for targeting.
Previously, the immunotherapy via NKG2D recognition has been established using chimeric antitumor mAb/NKG2D ligand complex,28 which specifically bind to tumor cells and enhance the in vitro lysis by NK cells in an NKG2D-dependent manner as well as elicit adaptive immune responses against cancer cells independent of NKG2D ligand expression.14 Recently, a new form of targeted anticancer therapy has been generated in the form of a drug-carrying phage nanoparticles approach, which is based on genetically modified and chemically manipulated filamentous bacteriophages by Bar et al.3,4,17 The genetic manipulation endows the phages with the ability to display host specificity-conferring ligands. The phages are loaded with a large payload to a cytotoxic drug by chemical conjugation, which show that targeting of phage nanomedicines results in endocytosis, intracellular degradation, and drug release, resulting in growth inhibition of the target cells in vitro.4
In our study, MICA was used as a targeted moiety for the construction of anticancer-carrying filamentous phage nanoparticles. Doxorubicin was used as a therapeutic agent because it is widely used for treatment of many cancers.29 Conjugation of doxorubicin anticancer drug to phage g8p coat proteins was performed using the EDC chemistry at mild acidic pH condition. The ratio of all reagents was optimized by calculation to gain the maximal drug conjugation. Previously, EDC, EDC-NHS, and EDC-sulfoNHS were employed to crosslink anti-HFA antibodies on APTES-functionalized platforms for immunodiagnostic applications. The results demonstrated more efficient antibody crosslinking by EDC in comparison to EDC-NHS and EDC-sulfoNHS at a normal pH of about 7.4, which increased the analytical performance and cost-effectiveness of immunoassays.30 In addition, following NHS drug conjugation, the major coat g8p protein of phage particles was no longer recognized in ELISA by the commercial antiphage antibody.15
After conjugation reaction, detection of doxorubicin molecules on phage particles was performed by XRD assay and flow cytometry. The XRD assay is scanning of crystallized forms of particles by X-ray beam that is reflected by the crystallized particles. The reflected pattern can be observed as a spectrum. Particles with more crystallized forms show more shaped peak, while particles that are not crystallized or amorphous show a random spectrum. When particles have changed in structural conformation, different patterns can be observed when compared to those of original particles. The results indicated that doxorubicin-conjugated phages have structurally conformational changes after conjugation reaction. Additional shaped peak of doxorubicin-conjugated phages could be observed, suggesting the existence of doxorubicin molecules that were linked to phage amino acids. Higher intensity of doxorubicin-conjugated phages was observed indicating higher amount of conjugated doxorubicin (Figure 2). To ensure the drug conjugation on phages, flow cytometry was employed to confirm the Dox-phage conjugation. Flow cytometry was able to detect the signal of doxorubicin molecules due to the autofluorescent property, which can be excited and emitted. This has been successfully detected by flow cytometer in previous studies.21,22,31 The doxorubicin molecules can be triggered at 488 nm with emission integrated above 530 nm and can be detected by flow cytometer using the peridinin-chlorophyll-protein complex channel. To ensure that signal from doxorubicin was emitted from phage particles, second dye staining was required. The additional dye is anti-M13-conjugated FITC with emission at 530 nm. This antibody is specific to g8p coat proteins of phages. The samples of naked phages and doxorubicin-conjugated phages stained with anti-M13-conjugated FITC were analyzed by flow cytometer. Conjugated doxorubicin can be simultaneously detected with the signal from FITC indicating the co-existence of doxorubicin and g8p. The percentage of phage particles conjugated with doxorubicin was about 82% with 98.8% of phages being double fluorescent (Figure 3). Detection of doxorubicin on conjugated phages that were stored at 4°C in PBS (pH 7.4) for 1 month showed that the signal from doxorubicin was decreased by 10%. This was presumed to be phage particles that had lost doxorubicin molecules during storage (data not show).
SRB assay was used to evaluate the cytotoxicity of doxorubicin-conjugated phages, Dox-phage WW9B8.21. Hela, KKU-M055, KKU-M213, KKU-M214, KKU-M139, and KKU-MMNK1 were treated in this experiment. These cells expressed MICA antigens at different levels indicated by mean fluorescence intensity. These cells were sensitive to doxorubicin-free drug with various IC50 values as shown in Table 2. The IC50 values of Dox-phages were two to six times lower than those of free drug, except for KKU-M055, which is more resistant to doxorubicin. Thus, Dox-phages have more killing efficiency than free drugs. In the case of KKU-M055, the use of free drug was not different from Dox-phages because of decreased MICA expression. However, the efficiency of killing is not only based on MICA expressions but also the drug-resistant factors carried by each cell line. The targeting effect of Dox-phages was indicated by the MICA-blocking experiments. The IC50 values of Dox-phages when they were preblocked with anti-MICA were close to those of free drugs (Table 2). These results indicate that we have successfully developed the Dox-phages that are targeting MICA.
Conjugation of doxorubicin to phage g8p coat proteins using EDC chemistry is one approach to construct the targeted therapeutic nanoparticles with no effect on binding moiety of antibodies on phage particles. The process of conjugation is easy and is done by adding EDC to the reaction at mild acidic pH. However, amide bond between drugs and phage coat proteins is also sensitive to mild acidic pH as seen in the releasing experiment (data not shown). This is an advantage of these particles, which when reaching the tumor site with acidic pH environment were internalized into cells by endocytosis. Regarding immunogenicity of phage particles, antibacterial drug-carrying phages were evaluated in vivo, were shown to be nontoxic to mice and greatly reduced in immunogenicity and quickly cleared from the bloodstream.18 However, this has not been studied in humans. Thus, to avoid immunogenicity and other complications in humans, other nanoparticles such as dendrimer or liposome should be considered as a new platform of drug delivery system.
Conclusion
This study demonstrated the condition for conjugation of doxorubicin on phage particles displaying high-affinity anti-MICA without affecting their MICA-binding activities. After conjugation, the doxorubicin molecules could be detected as conformational changes by XRD assay and double-fluorescent detection by flow cytometry. The linkages between conjugated doxorubicin molecules and phage proteins were sensitive to acidic pH and the affinity of targeted moiety also affected the cytotoxic activities on targeted cells. For further study, liposomes or dendrimers will be employed as new drug delivery platforms to avoid the problem of immunogenicity and safety in humans. The soluble Fab of anti-MICA antibodies will be produced and tagged on the surface of liposome/dendrimer that contains the anticancer drug. The new delivery system may be able to target selectively and show specificity to cancer cells overexpressing MICA.
Acknowledgments
This work was supported by the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education, through the Center of Excellence in Specific Health Problems in Greater Mekong Subregion Cluster (SheP-GMS), Project no NRU 542022, and the Centre for Research and Development of Medical Diagnostic Laboratories, Khon Kaen University.
Disclosure
All authors have read and approved the manuscript and there are no conflicts of interest.
References
Sharkey RM, Goldenberg DM. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J Clin. 2006;56:226–243. | |
Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. | |
Yacoby I, Benhar I. Targeted filamentous bacteriophages as therapeutic agents. Expert Opin Drug Deliv. 2008;5:321–329. | |
Bar H, Yacoby I, Benhar I. Killing cancer cells by targeted drug-carrying phage nanomedicines. BMC Biotechnol. 2008;8:37. | |
Pende D, Rivera P, Marcenaro S, et al. Major histocompatibility complex class I-related chain a and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002;62:6178–6186. | |
Friese MA, Platten M, Lutz SZ, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003;63:8996–9006. | |
Salih HR, Antropius H, Gieseke F, et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 2003;102:1389–1396. | |
Watson NF, Spendlove I, Madjd Z, et al. Expression of the stress-related MHC class I chain-related protein MICA is an indicator of good prognosis in colorectal cancer patients. Int J Cancer. 2006;118:1445–1452. | |
Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A. 1999; 96(12):6879–6884. | |
Vetter CS, Groh V, Thor SP, et al. Expression of stress-induced MHC class I related chain molecules on human melanoma. J Invest Dermatol. 2002;118:600–605. | |
Melum E, Karlsen TH, Schrumpf E, et al. Cholangiocarcinoma in primary sclerosing cholangitis is associated with NKG2D polymorphisms. Hepatology. 2008;47(1):90–96. | |
Jinushi M, Takehara T, Tatsumi T, et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int J Cancer. 2003;104:354–361. | |
Das H, Groh V, Kuijl C, et al. MICA engagement by human Vgamma2Vdelta2 T cells enhances their antigen-dependent effector function. Immunity. 2001;15(1):83–93. | |
Wodnar-Filipowicz A, Kalberer CP. Function of natural killer cells in immune defence against human leukaemia. Swiss Med Wkly. 2006; 136(23–24):359–364. | |
Phumyen A, Jumnainsong A, Leelayuwat C. Improved binding activity of antibodies against major histocompatibility complex class I chain-related gene A by phage display technology for cancer-targeted therapy. J Biomed Biotechnol. 2012;2012:597647. | |
Wongsena W, Sconocchia G, Cho HS, et al. Production and characterization of monoclonal antibodies against major histocompatibility complex class I chain-related gene A. Tissue Antigens. 2008; 72(5):431–440. | |
Yacoby I, Shamis M, Bar H, Shabat D, Benhar I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob Agents Chemother. 2006;50(6):2087–2097. | |
Yacoby I, Bar H, Benhar I. Targeted drug-carrying bacteriophages as antibacterial nanomedicines. Antimicrob Agents Chemother. 2007;51(6):2156–2163. | |
Nakajima N, Ikada Y. Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media. Bioconjug Chem. 1995;6(1):123–130. | |
Karukstis KK, Thompson EH, Whiles JA, Rosenfeld RJ. Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys Chem. 1998;73(3):249–263. | |
Krishan A. Monitoring of cellular resistance to cancer chemotherapy: drug retention and efflux. Methods Cell Biol. 2001;64:193–209. | |
Krishan A, Sauerteig A, Wellham LL. Flow cytometric studies on modulation of cellular adriamycin retention by phenothiazines. Cancer Res. 1985;45(3):1046–1051. | |
Krishan A. Flow cytometric monitoring of drug resistance in human tumor cells. Methods Cell Sci. 2002;24(1–3):55–60. | |
Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. | |
Ricart AD, Tolcher AW. Technology insight: cytotoxic drug immunoconjugates for cancer therapy. Nat Rev Clin Oncol. 2007;4:245–255. | |
Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147–159. | |
Yildiz I, Shukla S, Steinmetz NF. Applications of viral nanoparticles in medicine. Curr Opin Biotechnol. 2011;22:901–908. | |
Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66(11):5927–5933. | |
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. | |
Sandeep KV. Comparison of 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide based strategies to crosslink antibodies on amine-functionalized platforms for immunodiagnostic applications. Diagnostics. 2012;2(3):23–33. | |
Krishna R, St-Louis M, Mayer LD. Increased intracellular drug accumulation and complete chemosensitization achieved in multidrug-resistant solid tumors by co-administering valspodar (PSC 833) with sterically stabilized liposomal doxorubicin. Int J Cancer. 2000;85(1):131–141. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.