")
Back to Journals » Risk Management and Healthcare Policy » Volume 14
Distribution of β-Thalassemia and Other Hemoglobinopathies in Bangladeshi University Students and Ready-Made Garment Workers
Authors Islam MM, Hossain F, Sakib N , Zeba Z, Bhuiyan AI, Mamun MA , Kaggwa MM , Yoshimura K, Afrin S, Selim S, Hossain M
Received 3 May 2021
Accepted for publication 2 June 2021
Published 24 June 2021 Volume 2021:14 Pages 2707—2714
DOI https://doi.org/10.2147/RMHP.S317852
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Marco Carotenuto
Mohammad Manirul Islam,1,2,* Fahima Hossain,2,3 Najmuj Sakib,4,* Zebunnesa Zeba,5 AKM Israfil Bhuiyan,6 Mohammed A Mamun,5– 7 Mark Mohan Kaggwa,8 Ken Yoshimura,2,7,9 Sadia Afrin,2,7 Sheikh Selim,10 Moazzem Hossain2,7
1BMT Unit, Department of Hematology, Dhaka Medical College, Dhaka, Bangladesh; 2Thalassemia Hospital and Institute, Savar, Dhaka, Bangladesh; 3International Center for Diarrheal Diseases and Research, Bangladesh, Dhaka, Bangladesh; 4Department of Microbiology, Jashore University of Science and Technology, Jashore, Bangladesh; 5Department of Public Health and Informatics, Jahangirnagar University, Savar, Dhaka, Bangladesh; 6CHINTA Research Bangladesh, Savar, Dhaka, Bangladesh; 7Institute of Allergy and Clinical Immunology of Bangladesh, Savar, Dhaka, Bangladesh; 8Department of Psychiatry, Faculty of Medicine, Mbarara University of Science and Technology, Mbarara, Uganda; 9Japan Snake Institute, Ota Gunma, Japan; 10Humanity Beyond Barriers, Davis, CA, USA
*These authors contributed equally to this work
Correspondence: Mohammed A Mamun; Mark Mohan Kaggwa Email [email protected]; [email protected]
Background: The inheritable thalassemia and other structural hemoglobinopathies are prevalent globally. In Bangladesh, there is a scarcity of studies concerning thalassemia and other structural hemoglobinopathies. Hence, the present study investigated the prevalence and distribution of β-thalassemia and other hemoglobinopathies among the university students and ready-made garment (RMG) workers.
Methods: A cross-sectional study was conducted, whereas a total of 989 subjects were selected randomly from 10 garments industries (n=495) and five universities (n=494) for diagnosis and counseling from April 2018 to August 2018. Basic information related to the study and venous blood was collected and subjected to capillary Hb-electrophoresis for each participant.
Results: The present study identified thalassemia carriers and other hemoglobinopathies among 13.6% of the participants (14.1% university students and 13.2% RMG workers). The most common form of hemoglobin formation disorder was the Hb-E trait (10.5%), considering both participants’ groups. Other forms of hemoglobin formation disorders observed were a β-thalassemia trait, Hb-D trait, Hb-E disease, and β-thalassemia major. The hemoglobinopathies were significantly associated with the individuals who had a family history of thalassemia and were not vaccinated against hepatitis B in the total sample. Although not statistically suggestive, the most considerable portion (87.3%) of the carriers was unmarried in the present findings.
Conclusion: The present study findings are anticipated to help the public health authorities by providing novel data (of a neglected cohort, ie, RGM workers), facilitating the upcoming comprehensive national thalassemia prevention programs.
Keywords: thalassemia, hemoglobinopathies, premarital carrier screening, RMG workers, thalassemia education and awareness, Bangladesh
Introduction
Thalassemia is one of the most familiar forms of autosomal recessive disorders, characterized by the reduction or absent production of one or more globin chains that make up the hemoglobin (Hb) tetramers.1 There are two fundamental forms of thalassemia: α-thalassemia and β-thalassemia. Impaired synthesis of α and β-globin chain is diagnosed as α-thalassemia, and β-thalassemia, respectively, and the latter is the most prevalent.2 An estimated 330,000 babies having clinically significant hemoglobin disorders are born every year,3 whereas about 60,000–70,000 children are born each year with severe forms of thalassemia.4 Notably, most of the affected children are born in low-resource areas like Bangladesh.5
Mass people’s movement and intermarriage between ethnic groups have paved the way to thalassemia in most countries globally, including the regions where the disease was previously oblivious.6 As estimated, over 360 million, at least 5.2% of the world population, carry a range of hemoglobin variants.3 More than 100 million β-thalassemia carriers are found with a global frequency of 1.5%,6 while South Asia belonging to 23% of the world population,7 is a hotspot of hemoglobinopathies.3 Most information on thalassemia in South Asia has mainly originated from the studies based in India, where the overall prevalence of β-thalassemia carriers has been estimated within 2.78 to 4%.8 However, the non-representative Bangladeshi studies suggested heterogeneous prevalence rates in terms of hemoglobinopathies. Khan et al estimated that Hb-E trait carriers to be 6.1% among general children and 41.1% in tribal school children in Bangladesh,9 whereas Farhana et al found 17.39% Hb-E trait and 13.04% Hb-E disease.10 Bangladesh is one of the densely populated countries globally, where over 70% of the population lives in highly resource-limited rural areas.5 However, Bangladesh has achieved a remarkable reduction in infant and under-five mortality rates by combating malnutrition, infectious diseases and improving the health system. On the contrary, thalassemia, like inherited diseases, receives little attention. Though many countries have set up comprehensive national thalassemia prevention programs, the Bangladeshi government still has less attention to combat the issue. Even mentionable public awareness was not achieved in the country.11
Bangladesh is a developing country with culturally sensitive people who lack literacy related to the consequences of thalassemia and its potential prevention.11 However, its prevention can largely be achieved through building awareness before marital engagement. Thalassemia transmission reduction is somewhat unimaginable in Bangladesh because less studies or initiatives are conducted to screen thalassemia or build awareness of its consequences.9,15 Hence, this study was carried out to screen the thalassemia carriers among the university and Ready-Made Garment (RMG) workers considering their marital status (ie, most of them unmarried). The study paves the way to fulfill the national knowledge gap for the cohorts and will facilitate the adoption of comprehensive national integrated prevention programs that includes public awareness and education, carrier screening, genetic counseling, premarital screening, and prenatal diagnosis.
Methods
Study Site and Participants
The present cross-sectional study was carried out to implement the “Thalassemia Screening and Awareness Program” by the Institute of Allergy and Clinical Immunology of Bangladesh (IACIB), Dhaka, Bangladesh. Although the prior Bangladeshi studies were conducted covering the screening process of the thalassemia status among students,5,9 Ready Made Garment (RMG) workers were not studied ever. The RMG workers are approximately 4.6 million in population, less facilitated regarding inaccessibility to health literacy and awareness,16 which instigated the research team to include them in this screening test and students. Student participants from five universities (ie, Jahangirnagar University, Daffodil International University, City University, Manarat International University, and Eastern University) and workers from ten garment factories (ie, Chantik, Crystal Garments, Elaine, Eve Dress-Shirts Ltd., Mehnaj Fashions, Metro Knitting & Dyeing Mills Ltd, Murad Apparels Ltd, New Asia Fashions Ltd., Radiance Fashions and Rizvi Fashion) located in the capital (Dhaka, Bangladesh) were selected based on simple random sampling technique. A total of 989 participants (ie, 494 university students and 495 garment factory workers) were engaged in the survey who also partook in the awareness program. The study was conducted based on voluntary participation within the program implementation time (ie, April 2018 to August 2018).
Data Collection
A pre-tested semi-structured questionnaire having three sections (ie, socio-demographics, blood and family history-related variables, and laboratory test profile) was used for data collection. Where, information related to sociodemographic and blood and family history were self-reported. In each university and garment, an awareness program consisting of a PowerPoint presentation and video documentation was projected; besides, leaflets and posters were also distributed for further informational access. After this session, they partook in the survey. Lastly, blood samples were collected by three skilled nurses and then transferred to the EDTA tube on the spot. Finally, the tubes with samples were carried out to the Thalassemia Hospital and Institute, Savar, Dhaka, Bangladesh, for the laboratory confirmation test. Blood samples were marked distinctively to avoid mistakes and biases.
Ethical Considerations
The present study was conducted in accordance with the Declaration of Helsinki. The local ethical committee of the Institute of Allergy and Clinical Immunology of Bangladesh (Ref: IRBIACIB/CEC/032018012) approved the research protocol. Additionally, informed written consent (or verbal consent for the illiterate garment workers) was taken from each participant beforehand. Anonymity and confidentiality of the personal data were ensured for all participants during the entire study. Furthermore, no financial or other gain was offered to the participants in the study.
Study Variables
This study assessed several sociodemographic variables, including gender, marital status, home district (being inside the capital, Dhaka, or not), and ethnicity for both cohorts. For students, study discipline and academic year were also assessed. Besides, self-rated information of blood group status and vaccination status for Hepatitis B of the participants were collected. Lastly, whether the participants had consanguineous parents and their family history of thalassemia were also assessed.
HB-Electrophoresis
Capillary electrophoresis (gold standard for thalassemia carrier detection) was performed by using MINICAPSebia flex piercing (fully automated; Sebia, France) to allow separation of the hemoglobin variants (Hb-D, Hb-E) associated with the study and quantification of the Hb-A, Hb-A2, and Hb-F.12 HbA2 level of >3.5% was used as a cutoff to screen β-thalassemia carriers.13,14 Two medical technologists performed the tests, and the test results (eg, β-thalassemia trait) were confirmed by a physician (specialist in Hematology).
Statistical Analysis
Statistical Package for Social Science (SPSS) version 20 was used for data analysis. Descriptive statistics (eg, frequencies, percentages) and first-order analysis (chi-squares, Fisher’s exact tests) were carried out by the SPSS. A p-value of <0.05 was considered significant in the present study.
Results
Characteristics of the Participants
Of the 989 participants (including both the university students and the RMG workers), the male was almost double of the female with a combined mean age of 22.7 ± 3.9 years. A large portion (89.6%) of the participants were unmarried, while about 15% of the RMG workers were married. Most of the participants (88.3%) were from outside of the capital, whereas a few individuals (0.9%, n = 9) were originated in tribal regions (Table 1).
 |
Table 1 Sociodemographic, Blood and Family History-Related Variables of the Participants |
About 25.7% reported having no knowledge about their respective blood groups in the total sample, whereas the rate was 47.2% and 4.3% within the RMG workers and students, respectively (χ2 = 518.40, p <0.001). About 85.1% of the participants were not vaccinated with Hepatitis B, and the non-vaccination status was more severe in the RMG workers than university students (94.9% vs 75.5%; χ2 = 70.09, p <0.001). Only 3.7% (n = 37) of the participants had consanguineous parents in the total sample. Besides, nearly all of the RMG workers were not informed about their family history of thalassemia (99.2%), which was 85.9% for the university students (χ2 = 58.023, p<0.001) (Table 1).
Prevalence of the Thalassemia and Other Hemoglobinopathies
Since Sebia capillary electrophoresis was able to separate Hb-A2 distinctly from Hb-E and other Hb- variants, the presence of Hb-E fractions or other hemoglobin variants could indicate the carriers as well. Out of the total participants, 134 (13.6%) had one or other forms of hemoglobinopathies, and the rest were found normal. Hb-E trait was found to be the most prevalent trait with a frequency of 10.4% (103/989) followed by 2.4% β-thalassemia trait (24/989), 0.4% Hb-D trait (4/989), 0.2% Hb-E disease (Hb-E beta-thalassemia disease; 2/989) and 0.1% β-thalassemia Major (1/989). The ratio was almost equal for the number of Hb-E traits among university students and RMG workers (Table 2).
 |
Table 2 Types of Thalassemia Carriers and Other Hb Variants Across Participants |
Distribution of the Thalassemia and Other Hemoglobinopathies
shown in Table 3. Thalassemia carrier and/or other hemoglobinopathies were significantly associated with those individuals having a family history of thalassemia (χ2 = 37.65, p<0.001); alarmingly, the most significant portion of them did not have any idea regarding their family history. About 12.2% of the participants who did not know their blood group carried thalassemia and/or other hemoglobinopathies, while the result was not statistically significant. Individuals not vaccinated with hepatitis B in the total sample (12.5%) were found to be one of the significant carriers (χ2 = 5.63, p <0.05) (Table 3).
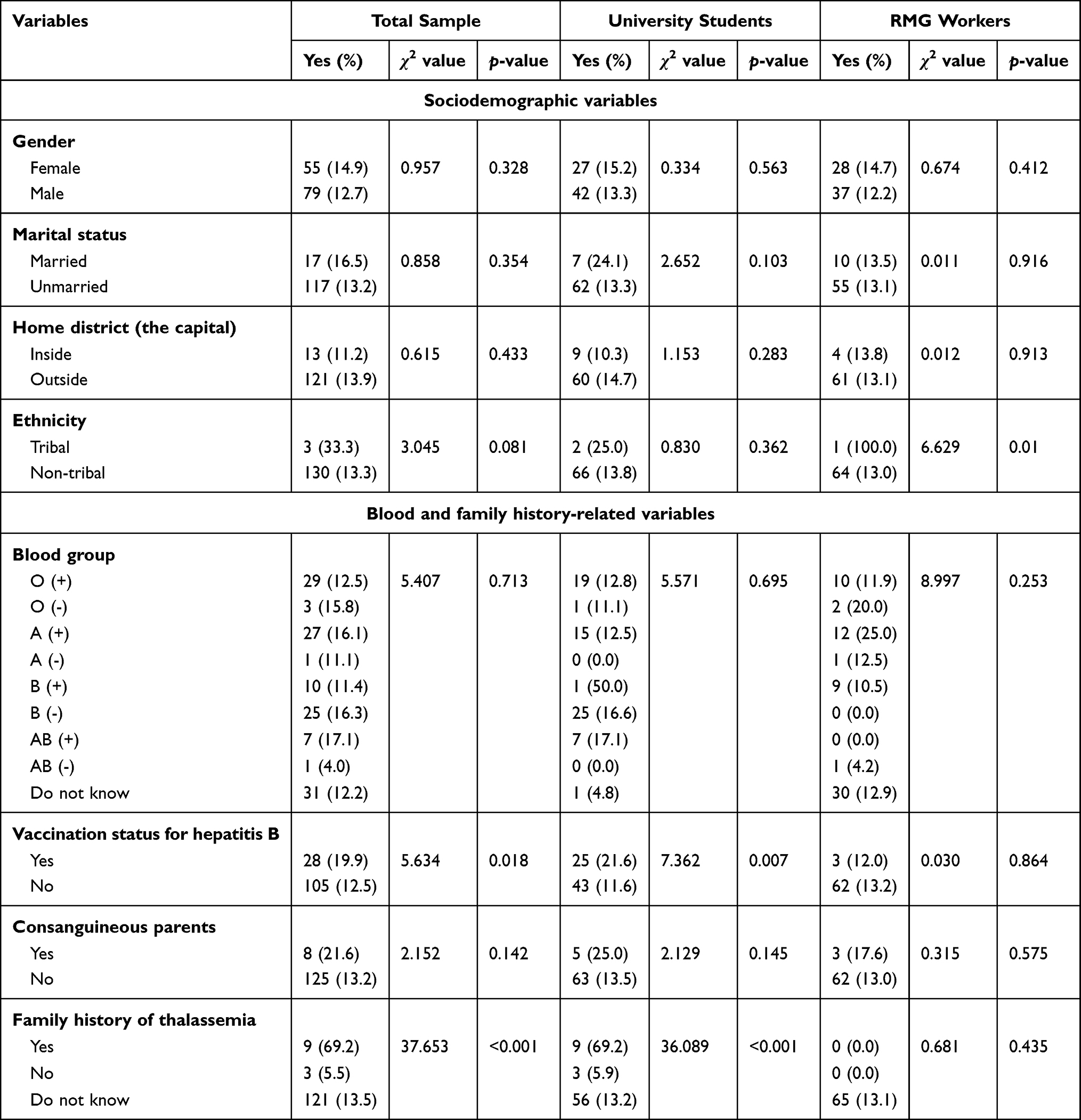 |
Table 3 Distribution of the Variables Among the Participants Carrying Thalassemia and/or Other Hemoglobinopathies |
Discussion
The present study is the first-ever study identifying thalassemia status among the Bangladeshi Ready-Made Garment (RMG) workers to the authors’ best of knowledge. As aforementioned, since about 4.6 million RMG workers in the country and a major portion reside in a poor living environment, they eventually have low health awareness, leading them to less conversant of health-related fatal issues thalassemia.16 Thus, thalassemia carrier detection and rising awareness before marriage can be stated as actionable protective initiatives to suppress its prevalence across generations as no effective treatment is available.17,20 Therefore, this study provides novel data on implementing national thalassemia programs and strategies among this neglected cohort.
In this study, the carrier frequency of the Hb-E trait and β-thalassemia trait was 12.9% combinedly, where the Hb-E trait has counted the most (10.5%). An earlier study conducted by Khan et al showed the prevalence of the Hb-E trait was 6.1%, and β-thalassemia trait was 4.1% among children of grades nine and ten in Bangladesh.9 Our result coincided with a very recent work in which the combined carrier frequency was 10.92%, with the Hb-E trait the highest.15 Besides, Noor et al used both the conventional hematological approaches and the capillary electrophoresis method, although the current study only utilized the latter one (while the false positives and false negatives were undoubtedly reduced to some extent).15 In addition, Hb-E disease and E-trait are homozygotic and heterozygotic conditions of a β-chain variant where glutamic acid substitutes a lysine residue.17 The thalassemia also has a broad dispersal throughout the Mediterranean, the Middle East, and the Indian subcontinent.18,19 While the present study could only detect a single individual with β-thalassemia major and a couple of individuals with Hb-E disease, in another study in Chittagong, Hb-E thalassemia was the most common one among all types of thalassemia (38.65%), followed by thalassemia trait (30.43%), Hb-E trait (22.71%).21
Patients with thalassemia major generally need to regularly receive a blood transfusion to maintain optimum hemoglobin level.22 Such transfusion may increase the exposure to hepatitis viruses. It was reported that transmission of hepatitis infections is not unusual; also, hepatitis B virus (HBV) infection is more prevalent than others.23 In the present study, many HBV non-vaccinated individuals (12.5%) were found with any of the thalassemia traits, although 19.9% of the total population carrying the traits was vaccinated. This concern coincides within the RMG workers as well (ie, 13.2% of the cases). The situation demands many HBV vaccination among the population, while studies showed that the HBV vaccine is immunogenic and safe in multi-transfused thalassemia patients.24 In individuals with thalassemia carriers and/or other hemoglobinopathies, as expected, there was a significant association with those who have a previous family history of thalassemia. This finding is consistent with the inherited nature of beta-thalassemia, including sickle cell anemia and Hb-E disorders reported globally.3 Consanguinity did not carry that much significance in the current study, although previous reports showed the relation of this factor,15 which is subjected to further studies.
Bangladesh is a resource-limited, developing country where most people are culturally sensitive, and most of them are unaware of thalassemia due to a lack of knowledge about this hereditary disease.11 Besides, according to a recent study, most of the thalassemia patients admitted in a hospital of Chittagong, Bangladesh belonged to the lower-middle class, and a good number of them did not have any educational background.25 However, the supports are not up to the mark at present. The failure to render sufficient support for thalassemia will result in excessive burdens of overall public health, especially for the vulnerable cluster such as RMG workers. Being members of the low-income groups, RMG workers cannot afford appropriate foods that lead to anemia with the iron imbalance in many cases.26 On top of that, thalassemia like genetic disorders can aggravate the situation.27 Other developing countries, regardless of religion, ethnicity, or race, have similar problems concerning thalassemia; but the situation has been improved dramatically by Pre-Marital Screening and Genetic Counseling for thalassemia.28,29 Therefore, the free screening and counseling program for thalassemia should be introduced in Bangladesh throughout that cohort to recognize the carrier and create awareness. That is why the present findings are expected to be helpful and effective. Considering the situation, the results of screening tests performed in this study were provided to all participants free of cost. Thalassemia carriers were appropriately informed that they should not marry another carrier. Moreover, the carriers from both groups were enlisted in a database so that their future action might be followed up. To sum up, the present study also suggests that the Bangladeshi people (especially the illiterate cohorts like RMG workers) need to be concerned about the importance of blood-related knowledge essential for life emergencies.
The present findings are limited due to the cross-sectional nature of the study. Furthermore, hematological parameters (eg, OF/DCIP or MCV, MCH/DCIP etc.) were not used for initial screening as the Hemoglobin electrophoresis was performed as a gold standard,30 which can be an arguable limitation. Also, the molecular characterization of the beta-globin gene mutations is necessary for identifying the type of beta-thalassemia mutation. Aiming only at screening the most frequently thalassemia carriers, this study did not identify the alpha thalassemia. There is no data for alpha thalassemia trait, Hb-H disease. In addition, this study can also be limited because of issues related to self-reporting data, lack of control groups (eg, married individuals). Despite these limitations, the present study is the first approach for providing baseline data concerning β-thalassemia and other hemoglobinopathies among the RMG workers in Bangladesh.
Conclusions
This study shows that the Hb-E trait was the most common hemoglobinopathies pattern between the study groups (ie, RMG workers and university students). Other disorders like β-thalassemia trait, Hb-D trait, Hb-E disease, and β-thalassemia major were also identified. However, no case with alpha thalassemia was detected as it remains undetected in most cases.31 Further analysis should be directed towards the correlation of genetic mutation with clinical diversity. Finally, although thalassemia is prevalent throughout Bangladesh, the services providing facilities to combat thalassemia are limited to the urban region of the country.15 Therefore, thalassemia prevention programs are suggested and should be nurtured and supported according to the government policy by coordinating the country’s local structures, social values, religious laws, and cultural traditions. However, the present findings are anticipated to be helpful for public health authorities in modifying existing facilities to reduce the thalassemia carriers in Bangladesh.
Acknowledgments
We are grateful to Humanity Beyond Barriers USA for providing support for this study. Besides, Mr Mamun likes to acknowledge their affiliation, the CHINTA Research Bangladesh, formerly known as the Undergraduate Research Organization.
Funding
The present study did not get any financial supports. Besides, the authors involved in this research communication do not have any relationships with other people or organizations that could inappropriately influence (bias) the findings.
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi:10.1182/blood-2010-01-251348
2. Safizadeh H, Farahmandinia Z, Nejad SS, Pourdamghan N, Araste M. Quality of life in patients with thalassemia major and intermedia in Kerman-Iran (I.R.). Mediterr J Hematol Infect Dis. 2012;4(1):e2012058. doi:10.4084/mjhid.2012.058
3. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;2008:480–487. doi:10.2471/BLT.06.036673
4. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712.
5. Weatherall DJ. The challenge of haemoglobinopathies in resource-poor countries. Br J Haematol. 2011;154:736–744. doi:10.1111/j.1365-2141.2011.08742.x
6. Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of β-thalassemias and hemoglobin e disorders. Expert Rev Hematol. 2010;3:103–117. doi:10.1586/ehm.09.74
7. Central Intelligence Agency. The World Factbook 2016. Washington, DC: Central Intelligence Agency; 2016.
8. World health organization. Management of haemoglobin disorders: report of a joint WHO-TIF meeting, Nicosia, Cyprus, 16–18 November 2007. 2008.
9. Khan WA, Banu B, Amin SK, et al. Prevalence of Beta thalassemia trait and Hb E trait in Bangladeshi school children and health burden of thalassemia in our population. DS (Child) H J. 2005;21:1–7.
10. Farhana DT, Nahar Q, Choudhury S. Pattern of haematological disorders in a tertiary diabetic hospital: a pilot study. J Bangladesh Coll Phys Surg. 1970;27:148–154. doi:10.3329/jbcps.v27i3.4292
11. Tahura S, Selimuzzaman M, Khan WA. Thalassaemia prevention: Bangladesh perspective - a current update. Bangl J Child Health. 2017;40:31–38. doi:10.3329/bjch.v40i1.31553
12. Brancaleoni V, Di Pierro E, Motta I, Cappellini MD. Laboratory diagnosis of thalassemia. Int J Lab Hematol. 2016;38:32–40. doi:10.1111/ijlh.12527
13. Giordano PC. Strategies for basic laboratory diagnostics of the hemoglobinopathies in multi-ethnic societies: interpretation of results and pitfalls. Int J Lab Hematol. 2013;35:465–479. doi:10.1111/ijlh.12037
14. Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of Haemoglobin A 2. Int J Lab Hematol. 2012;34:1–13. doi:10.1111/j.1751-553X.2011.01368.x
15. Noor FA, Sultana N, Bhuyan GS, et al. Nationwide carrier detection and molecular characterization of β-Thalassemia and Hemoglobin E variants in Bangladeshi population. Orphanet J Rare Dis. 2020;15:15. doi:10.1186/s13023-020-1294-z
16. Bhuiyan MZA. Present status of garment workers in Bangladesh: an analysis. IOSR J Bus Manag. 2012;3:38–44. doi:10.9790/487X-0353844
17. Uddin MM, Akteruzzaman S, Rahman T, Hasan AKMM, Shekhar HU. Pattern of β-Thalassemia and other Haemoglobinopathies: a cross-sectional study in Bangladesh. ISRN Hematol. 2012;2012:1–6. doi:10.5402/2012/659191
18. Harano T, Harano K, Okada S, Shimono K. A wider molecular spectrum of β-Thalassaemia in Myanmar. Br J Haematol. 2002;117:988–992. doi:10.1046/j.1365-2141.2002.03539.x
19. Hill AVS. Molecular epidemiology of the Thalassaemias (including Haemoglobin E). Baillières Clin Haematol. 1992;5:209–238. doi:10.1016/S0950-3536(11)80042-9
20. Palit S, Bhuiyan RH, Aklima J, Emran TB, Dash R. A study of the prevalence of thalassemia and its correlation with liver function test in different age and sex group in the Chittagong district of Bangladesh. J Basic Clin Pharm. 2012;3:352. doi:10.4103/0976-0105.105339
21. Mannan A, Kawser J, Ahmed AMA, Faruk Sikd MO, Islam MJ, Chowdhury MA. A demographic approach for understanding the prevalence of β Thalassemia patterns and other Hemoglobinopathies: selective study in Chittagong city perspective. Asian J Biol Sci. 2013;6:124–130. doi:10.3923/ajbs.2013.124.130
22. Prati D. Benefits and complications of regular blood transfusion in patients with Beta-Thalassaemia major. Vox Sang. 2000;79:129–137. doi:10.1046/j.1423-0410.2000.7930129.x
23. Schreiber GB, Busch MP, Kleinman SH, Korelitz JJ. The risk of transfusion-transmitted viral infections. N Engl J Med. 1996;334:1685–1690. doi:10.1056/NEJM199606273342601
24. Sharifi Z, Milani S, Shooshtari MM. Study on efficacy of hepatitis B immunization in vaccinated beta-Thalassemia children in Tehran. Iran J Pediatr. 2010;20:211–215.
25. Barua T, Das AK, Sultana R, Das D, Arju MA. Socio-demographic profile of patients admitted in Thalassemia care center of Chattogram Maa Shishu-O-General Hospital. Chattagram Maa Shishu-O-Gen Hosp Med Coll J. 2020;19(1):33–37. doi:10.3329/cmoshmcj.v19i1.48800
26. Hossain M, Islam Z, Sultana S, et al. Effectiveness of workplace nutrition programs on anemia status among female Redaymade Garment Workers in Bangladesh: a program evaluation. Nutrients. 2019;11(6):1259. doi:10.3390/nu11061259
27. Saliba AN, Atoui A, Labban M, et al. Thalassemia in the emergency department: special considerations for a rare disease. Ann Hematol. 2020;99:1967–1977. doi:10.1007/s00277-020-04164-6
28. Saffi M, Howard N. Exploring the effectiveness of mandatory premarital screening and genetic counselling programmes for β-Thalassaemia in the Middle East: a scoping review. Public Health Genom. 2015;18:193–203. doi:10.1159/000430837
29. Mudiyanse RM, Senanayake MP, Rathnayake RMS. “Safe Marriages” for Thalassaemia prevention: a KAP survey in SriLanka. Transl Biomed. 2015;6:3.
30. Oyaert M, Van Laer C, Claerhout H, et al. Evaluation of the Sebia Minicap Flex Piercing capillary electrophoresis for hemoglobinopathy testing. Int J Lab Hematol. 2015;37(3):420–425. doi:10.1111/ijlh.12305
31. Panja A, Ghosh TK, Basu A. Genetics of thalassemia in Indian population. J Commun Nutr Health. 2012;1(1):39–46.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.