")
Back to Journals » ImmunoTargets and Therapy » Volume 13
Cold Tumour Phenotype Explained Through Whole Genome Sequencing in Clinical Nasopharyngeal Cancer: A Preliminary Study
Authors Handoko, Adham M, Rachmadi L , Wibowo H , Gondhowiardjo SA
Received 12 December 2023
Accepted for publication 23 February 2024
Published 18 March 2024 Volume 2024:13 Pages 173—182
DOI https://doi.org/10.2147/ITT.S452117
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Michael Shurin
Handoko,1– 3,* Marlinda Adham,2,4,* Lisnawati Rachmadi,2,5,* Heri Wibowo,6,* Soehartati A Gondhowiardjo1,2,*
1Department of Radiation Oncology, Cipto Mangunkusumo National General Hospital, Jakarta, Indonesia; 2Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia; 3Doctoral Program in Biomedical Sciences, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia; 4Department of Otorhinolaryngology - Head and Neck Surgery Department, Cipto Mangunkusumo National General Hospital, Jakarta, Indonesia; 5Department of Anatomical Pathology, Cipto Mangunkusumo National General Hospital, Jakarta, Indonesia; 6Integrated Laboratory, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
*These authors contributed equally to this work
Correspondence: Soehartati A Gondhowiardjo, Faculty of Medicine, Universitas Indonesia, Jl. Salemba Raya No. 6, Jakarta, 10430, Indonesia, Email [email protected]; [email protected]
Introduction: Nasopharyngeal cancer (NPC) is a complex cancer due to its unique genomic features and association with the Epstein–Barr virus (EBV). Despite therapeutic advancements, NPC prognosis remains poor, necessitating a deeper understanding of its genomics. Here, we present a comprehensive whole genome sequencing (WGS) view of NPC genomics and its correlation with the phenotype.
Methods: This study involved WGS of a clinical NPC biopsy specimen. Sequencing was carried out using a long read sequencer from Oxford Nanopore. Analysis of the variants involved correlation with the phenotype of NPC.
Results: A loss of genes within chromosome 6 from copy number variation (CNV) was found. The lost genes included HLA-A, HLA-B, and HLA-C, which work in the antigen presentation process. This loss of the major histocompatibility complex (MHC) apparatus resulted in the tumour’s ability to evade immune recognition. The tumour exhibited an immunologically “cold” phenotype, with mild tumour-infiltrating lymphocytes, supporting the possible etiology of loss of antigen presentation capability. Furthermore, the driver mutation PIK3CA gene was identified along with various other gene variants affecting numerous signaling pathways.
Discussion: Comprehensive WGS was able to detect various mutations and genomic losses, which could explain tumour progression and immune evasion ability. Furthermore, the study identified the loss of other genes related to cancer and immune pathways, emphasizing the complexity of NPC genomics. In conclusion, this study underscores the significance of MHC class I gene loss and its probable correlation with the cold tumour phenotype observed in NPC.
Keywords: nasopharyngeal cancer, genomic, MHC class I, antigen processing and presentation, copy number variation, deletion
Introduction
Nasopharyngeal cancer (NPC), characterized by its distinct genomic and biological features, remains a formidable challenge in the realm of oncology, owing to its intricate association with the Epstein–Barr virus (EBV) and sophisticated immune evasion mechanisms.1 Evading immune surveillance is characteristic of cancer and phenotypically can be seen as a “cold” tumour, which has minimal tumour infiltrating lymphocytes. This NPC’s unique genetic and virological characteristics has long perplexed the scientific community and posed significant clinical challenges. Despite advancements in therapeutic modalities, the prognosis for NPC remains suboptimal, underscoring the pressing need for a comprehensive understanding of its genomic underpinnings.
Whole genome sequencing (WGS) has emerged as an indispensable tool, offering unprecedented insights into the genomic landscape of NPC.2 It has potential implications for assessing how the tumourgenomics translate into its aggressive phenotype. WGS, a cutting-edge technology, allows for in-depth exploration of the entire genomic repertoire of NPC tumours, enabling the identification of molecular aberrations that may contribute to the development of various cancer hallmarks, including immune evasion.3,4
Here, we report on the captivating crossroads of nasopharyngeal cancer genomics, the association with immune evasion, and the NPC phenotype, through comprehensive WGS. We assessed the NPC’s intricate genomics, seeking a relationship between and an association with aberrant NPC genomics, and correlated them with the relevant tumour phenotype. We aim to cast new light on molecular and genetic alterations in clinical NPC specimens that potentially drive cancer progression and render the cancer unseen by our immune system.
Materials and Methods
A freshly frozen nasopharyngeal cancer biopsy specimen was obtained. Informed consent had already been obtained from the patient for further genetic analysis. Ethical approval for collection, storage, and further molecular or genetic testing was received from the Faculty of Medicine, Universitas Indonesia’s ethics committee (approval number: KET-149/UN2.F1/ETIK/PPM.00.02/2023). The specimen was collected from a patient whose nasopharyngeal cancer was clinically evident but not yet pathologically confirmed. A biopsy from the nasopharynx mass was taken as part of a routine diagnostic procedure. A small piece of biopsied tissue, around 50 mg, was concurrently collected and stored in a liquid nitrogen tank in −80°C for further molecular and genetic testing.
Pathological confirmation was obtained through examination by a senior pathologist. Information on the tumour immune microenvironment, assessed through histopathological slides, was also collected. The immune microenvironment on the histopathological slide was identified as mild, moderate or heavy tumour lymphocyte infiltration by assessing the overall region of the slides.5 Relevant tumour phenotype and WGS finding were correlated.
DNA Extraction
Upon pathological confirmation of nasopharyngeal cancer, 30 mg of freshly frozen specimen was retrieved. It was thawed and mechanically dissected on 100 µL of phosphate-buffered saline (PBS) with a sterile surgical scalpel. Further, the sample underwent mechanical shearing with another 250 µL of PBS using an ultrasonic sonicator for 10–15 seconds, repeated until no solid granular mass was visible. The sample DNA was then extracted using the QIAamp DNA Mini kit (QIAGEN) following the manufacturer’s instruction. Extracted DNA underwent quality control for assessment of its purity and quantity. DNA purity was assessed using a NanoDrop™ Spectrophotometer; DNA quantification was assessed using a Qubit™ Fluorometer (both Thermo Fisher).
DNA Library Preparation and DNA Sequencing
Extracted DNA was further processed through a library preparation step, following the protocol of the Oxford Nanopore Ligation sequencing DNA V14 (SQK-LSK114) instructions. No PCR step was involved. Sequencing of the whole genome was done using a PromethION 2 Sequencing Unit Solo from Oxford Nanopore with PromethION flow cell R10.4.1 and kit 14 chemistry. The library DNA concentration loaded was around 100–200 fmol, as suggested by the manufacturer. Sequencing was carried out for 71 hours until all pores were run out to achieve maximal data. Reloading of library DNA samples to sequencing flow cells was done twice during the whole sequencing process (when the pore occupancy dropped to around 25%), as recommended by the manufacturer’s manual.
Bioinformatics
The raw sequencing data was in POD5 format.6 The raw data was further basecalled using EPI2ME wf-basecalling (available from https://github.com/epi2me-labs/wf-basecalling) and the output was FASTQ data. The FASTQ data was assessed for quality using NanoPlot (available from https://github.com/wdecoster/NanoPlot).7 Furthermore, alignment was carried out using EPI2ME wf-alignment (available from https://github.com/epi2me-labs/wf-alignment). An alignment reference file was downloaded from UCSC reference human genome GRCh38 and NCBI reference genome for EBV. Alignment to the EBV reference genome was just to ensure that EBV DNA was present (commonly present concurrently with nasopharyngeal cancer).
The alignment BAM file, which was mapped to the human reference genome, then underwent copy number variation (CNV) calling using EPI2ME wf-human-variation (available from https://github.com/epi2me-labs/wf-human-variation). Single nucleotide variation (SNV) calling was done from a BAM file using an EPI2ME wf-human variation SNV calling pipeline. Annotation was carried out with SnpEff. The SNV variants were classified based on their genes’ function associated with specific pathways according to those available in the Kyoto Encyclopaedia of Genes and Genomes (KEGG database). A graphical summary according to SNV types of mutation by relevant cancer-related KEGG pathways was presented. Possible driver mutation based on SNV data was screened for manually by analysing the biological function of those genes.
Visualization of aligned data was carried out using Integrative Genomics Viewer (IGV) Desktop Application.8–10 Genes’ location, names and other details within the specific human chromosomal region were obtained from ENSEMBLE database. Reported CNV copy number gains or losses were summarized. The loss or gain genes were checked to see if they contained any genes related to the human cancer pathway or immune system-related pathway based on the KEGG database. The biological explanation for loss or gain genes was analysed to find an association between cancer phenotype and genomic findings.
Results
The WGS yielded a total 53,141,702 reads and 64,805,867,700 bases; its N50 read length was 1798 bases and its median read quality was 17. The average sequencing depth was 18.7x. The reads were successfully mapped against both EBV and the human GrCh38 reference genome. It showed EBV DNA was present as it is commonly infected and remains latent in nasopharyngeal cancer.11,12
From the CNV calling result, it was found that there was a total loss on chromosome 6 base position 28,500,000 until 33,500,000, and a total loss on chromosome X base position 1 until 2,000,000. There was also a single copy loss on chromosome 11 base position 1 until 3,500,000, and on chromosome 15 base position 23,500,000 until 31,500,000. An overview visualization of the CNV result is shown in Figure 1.
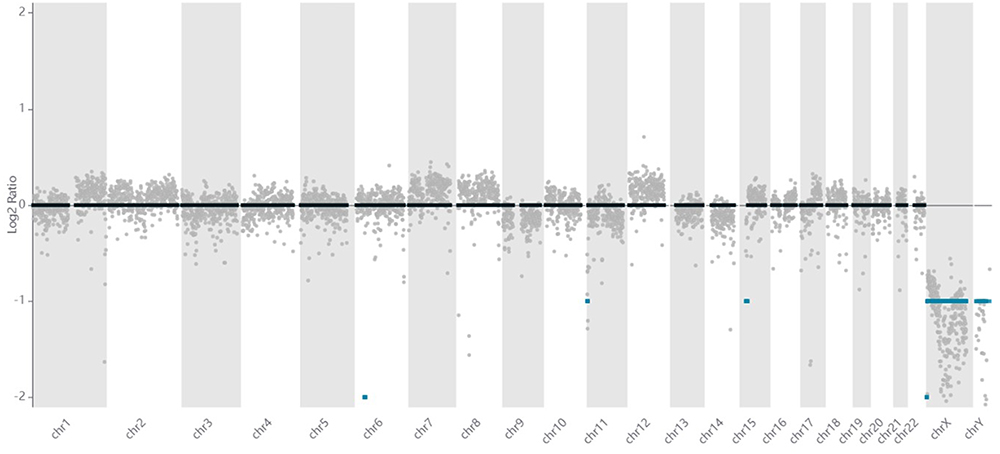 |
Figure 1 Overview of CNV result from WGS NPC data. |
There were 425 genes with a total loss within chromosome 6 base position 28,500,000 until 33,500,000. From those 425 genes, 22 genes are protein-coding genes and have a function related to antigen processing and presentation based on the KEGG database immune system pathway. There were 4 protein coding gene losses related to complement and coagulation cascades. One gene loss related to Fc gamma R-mediated phagocytosis. One gene loss related to NOD-like receptor. One gene loss related to NOTCH signalling. One gene loss related to both complement and coagulation cascades, and apoptosis signalling. One gene loss was related to both ERK signalling and chemokine signalling. These genes and their related roles are summarised in Supplementary Table 1.
There were 30 genes with total losses within chromosome X base position 1 until 2,000,000. Out of 61 genes, three were protein-coding and play a role in B cell receptor signalling, T cell receptor signalling, or JAK-STAT signalling. These genes and their related roles are summarised in Supplementary Table 2. Genes with a single copy loss are summarised in Supplementary Table 3 and Supplementary Table 4. There was no significant copy number gain from this WGS sample.
The dominant gene loss within this WGS NPC sample was related to antigen processing and presentation. All MHC class I molecules, HLA-A, HLA-B, and HLA-C, were completely lost. The MHC class I molecules are necessary for normal cells to present antigens, including tumour antigens, to CD8 T cells and other immune cells. Thus, it can be inferred that tumours escape recognition because they have lost that important and necessary repertoire to tell immune cells that there is something wrong within the cells.
Gene loss was confirmed using an integrative genomics viewer (IGV; as shown in Supplementary Figure 1a). An example of loss of the HLA-A gene is shown in Supplementary Figure 1b. Identifying the loss of other genes, such as TNF, NOTCH4, IL3RA, etc., is less straightforward. Those genes are more context dependent, though they might be related to a cancer-related pathway or an immune-related pathway. The effects of loss of those genes remain uncertain.
From the SNV calling result, multiple variants were detected. These variants are summarized in Figure 2. The majority were G to A and missense variants. Among the detected variants were some that are also involved in various signalling pathways. Those pathways are known to be important and, when deranged, to be associated with cancer (Figure 3).
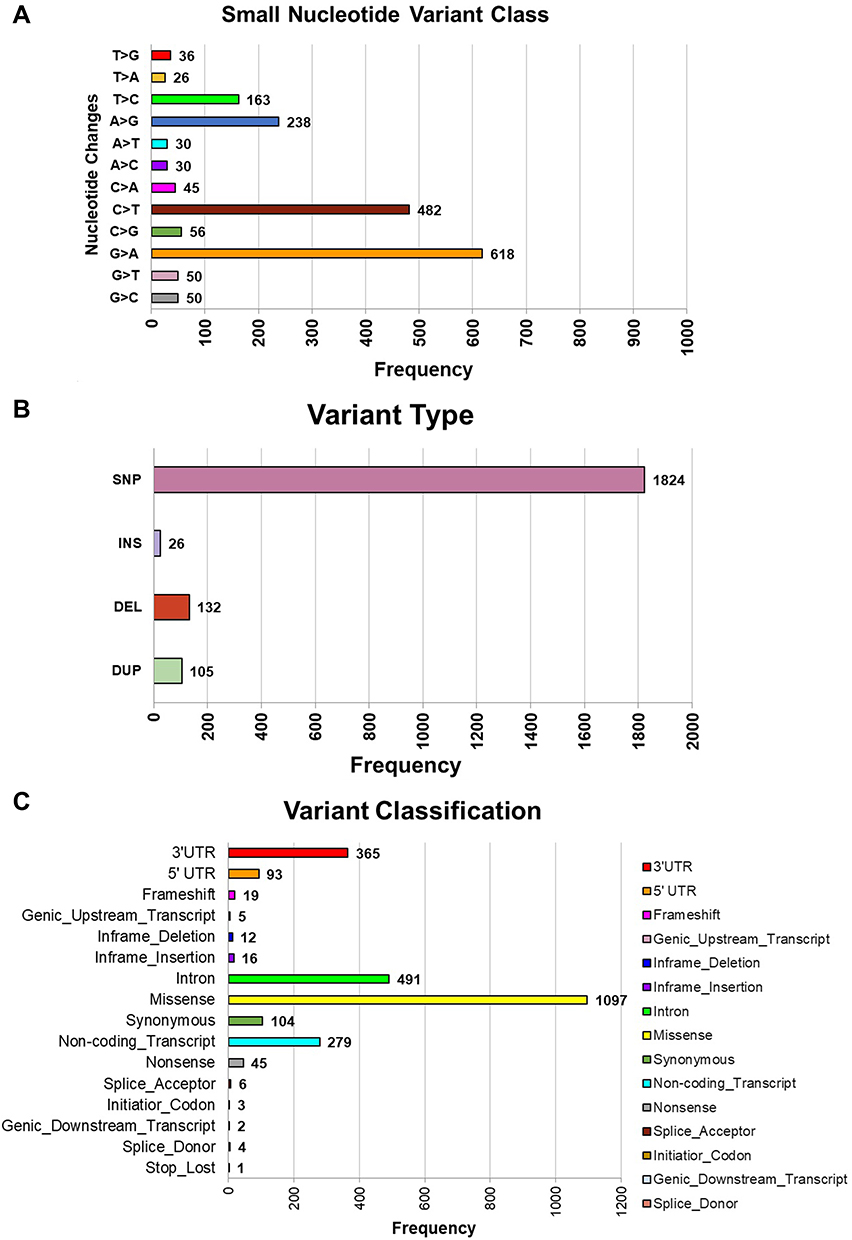 |
Figure 2 Summary of small variant from a comprehensive WGS of a clinical NPC. (A) Summary of the frequency of single base changes; (B) summary of variant type; (C) summary of variant classification. |
 |
Figure 3 Important cellular pathways with the listed genes associated with those pathways. Colour coding indicates variant detected in that gene. More than 1 colour coding indicates more than 1 variant classification detected. |
Among all variants, one notable driver mutation was identified. It was a mutation in the PIK3CA gene. It was a missense mutation: Human Genome Variation Society (HGVS) NM_006218.4:c.1634A>C. The change happened in position 1634 of the PIK3CA gene from A to C nucleotide, which initially coded for amino acid glutamic acid, but changed to alanine in amino acid sequence 545 (rs ID: rs121913274 SNP). There were 43 reads within that position, with 22 reads showing C instead of A, confirming the presence of the variant (see Supplementary Figure 2). There is a clinically approved targetable drug, alpelisib, for that specific mutation.13 It works by selectively inhibiting class I PI3K p110α, which is the catalytic subunit of PI3K.14
Clinically, this patient had an aggressive nasopharyngeal cancer, initially diagnosed at stage IV. The tumour was localised within the nasopharynx, neck nodes, and very small early lung metastasis. Neoadjuvant chemotherapy consisting of cisplatin and 5-fluorouracil (5-FU) was administered for 3 cycles, then followed by chemoradiation to the nasopharynx and neck nodes. The local nasopharynx and regional node masses shrank, but 3 months later the lung had multiple lung metastases. The patient underwent further chemotherapy but the cancer continued to progress. The histopathological specimen from the initial biopsy revealed mild tumour infiltrating lymphocytes, which sort of excluded lymphocytes from the bulk of cancer cells. This was considered an immunologically cold tumour, which generally has a poorer prognosis. The representative histopathological finding is shown in Figure 4.
 |
Figure 4 Histopathological examination with region of dominant cancer cells (marked in red oval circle) and lymphocytes marked by yellow arrows. There was only mild true tumour infiltrating lymphocyte. Examination was done in 100x magnification. |
Discussion
Chromosomal instability in cancer that leads to chromosomal aberration is actually well documented.15 Various reasons can lead to chromosomal aberration. One very plausible mechanism is a defect in mitotic DNA repair capability.16 In normal conditions, an effective DNA sensing mechanism keeps track and activates a DNA repair response if an error occurs.17 This is a protective mechanism. A DNA repair mechanism fixes all errors inherently occurring during the cell life cycle or cell mitosis until an exogenous assault occus that may also cause DNA error.18
Not all DNA repair mechanisms are error-free, however. For instance, in a double-strand DNA break (DSB), there are generally 2 main DNA repair mechanisms: non-homologous end joining (NHEJ) and homologous recombination (HR).19 NHEJ is considered error prone, while HR is an error-free repair mechanism.19 NHEJ is active in all cell cycles. HR is a DNA repair mechanism that typically operates during the S and G2 phases of the cell cycle, when a sister chromatid is available as a template for repair.20 This ensures accurate repair of double-strand DNA breaks by using the intact sister chromatid as a template.20 Impairment of HR by any means, such as due to EBV in NPC cases, can result in reliance on an error-prone DNA repair mechanism, thus accumulating DNA mutations.21
Activation of homologous recombination (HR) during mitosis is generally suppressed and considered unfavourable. When HR is activated during mitosis, several problems can occur. During mitosis, the chromosomes condense and are not in a state in which HR can readily occur. Attempting HR during mitosis can result in a loss of genetic material, as there is no homologous chromatid available for repair.16 Furthermore, activating HR during mitosis can disrupt the normal process of chromosome segregation, potentially leading to aneuploidy (abnormal chromosome number) and mitotic errors.22 Aneuploidy can be a hallmark of cancer and is associated with genomic instability.3
In certain conditions or due to specific factors, HR can be mistakenly activated during mitosis. During the G2 phase, cells have mechanisms to check for DNA damage and ensure proper repair before proceeding to mitosis.23 If DNA damage is detected but not adequately repaired, cells may enter mitosis with unresolved DNA lesions.23 This can potentially trigger HR during mitosis. Furthermore, mutations or deficiencies in other DNA repair pathways, such as non-homologous end joining (NHEJ),24 can lead to HR being used as a backup mechanism during mitosis when other pathways fail. This can also result in the inappropriate activation of HR.
Conditions that cause replication stress, such as stalled replication forks or excessive DNA damage, can lead to HR activation during mitosis as a means of repairing the DNA damage.25 Some viruses can also interfere with cells’ DNA repair mechanisms, potentially leading to the inappropriate activation of HR.26 Some carcinogens, such as benzene, can disrupt the regulation of DNA repair pathways and lead to genomic instability.27 This instability can manifest as the activation of HR during mitosis.27
The activation of HR during mitosis is usually considered a last-resort repair mechanism and is associated with the potential for adverse consequences, including genomic instability and the generation of aneuploid cells. Cells have evolved mechanisms to tightly control the timing of HR to ensure accurate repair, primarily during the S and G2 phases, when a sister chromatid is available as a template. Inappropriate HR activation during mitosis can result in genetic abnormalities, for example loss of certain genomic regions, and can be associated with cancer development and progression.
In our WGS study of a clinical NPC sample specimen, it was revealed that there was a portion of the genome with total loss. Genomic aberration in our sample can be caused by multiple factors, including accumulated pre-existing mutations caused by many factors, including aberrant mitotic repair ability. The chronic process of genomic aberration resulted in constant selection of clones of cells that harbour important pro-survival cancer genes and deletion of anti-survival cancer genes.28
Our study showed that significant total losses were observed on chromosome 6, where multiple genes related to antigen processing and presentation are located. This loss encompassed essential genes like HLA-A, HLA-B, and HLA-C, which play a pivotal role in enabling normal cells to present antigens, including tumor antigens, to CD8 T cells and other immune cells.29 The absence of these MHC-I molecules suggests that tumor cells within the NPC specimen may have acquired the ability to evade immune recognition by lacking the necessary molecular machinery to communicate with immune cells.29
We also confirmed that the tumour phenotype from this histopathological specimen was an immunologically cold tumour, as evidenced by the excluded tumour-infiltrating lymphocytes. There were only mild tumour-infiltrating lymphocytes, which is in line with the WGS finding on the loss of genes necessary to conduct proper antigen processing and presentation. This resulted in the absence of immune recognition of tumour cells.30
Various mechanisms can account for how the loss occurred, but the fact that there was a loss on those antigen processing and presentation genes can have important clinical implications. For instance, adoptive T cell transfer or CAR T cells might not work in this scenario. T cell therapy requires the presence of tumour antigen on the tumour cell surface.31 This was abolished in our specimen, rendering this kind of therapy ineffective. Another example, radiotherapy with a hypofractionated dosing regimen generally of around 6–20 Gy per fraction, has been known to induce upregulation of MHC class I molecules to improve the tumour neoantigen presentation to CD8 T cells.1,30,32
However, in the case of total loss of MHC class I related genes, we also suspected that no such upregulation can be observed. Therefore, the treatment strategy of using hypofractionated radiotherapy to act as a tumour in-situ vaccine and combined with checkpoint inhibitors might not work in this scenario. Furthermore, our study identified the loss of other genes through CNVs, including TNF, NOTCH4, and IL3RA, etc. While these genes are associated with cancer and immune-related pathways, their precise functions and impacts on NPC remain context-dependent and warrant further investigation.33,34
In our SNV calling, we found small variants in multiple genes that were involved in important cellular signalling. Not all those variants might have important clinical consequences. In general, mutation in genes directly regulating signalling pathways might drive the development, aggressiveness, and survival of these genes until ability to metastasise of malignant cancer.35,36 The one that was very likely to have an important clinical relevance in our comprehensive sequencing was the mutation in the PIK3CA gene. The mutation found in PIK3CA was a missense mutation in amino acid position 545, which has been reported to be oncogenic in multiple cancer sites.37–39 The PIK3CA inhibitors have also been shown to produce meaningful clinical tumour regression in multiple solid tumours with PIK3CA mutation.14,40 However, clinical trial results have shown that the PIK3CA inhibitor only confers the benefit of additional cancer progression-free survival by a median of 5.3 months compared to placebo in PIK3CA mutated breast cancer.41 Though great, this is not extraordinary; eventually, the cancer still grows.
It is very likely that cancer cells are progressively multiclonal.42 Continuous mutation and natural clonal selection result in the most aggressive and fittest cancer stem cells surviving and proliferating. Those different clones might have completely different genomic landscapes within an individual, which drive progression and complicate treatment strategy.43 Discovering the genomic profile of an initial site is the first step in devising an effective treatment strategy. Eventually, if the cancer continues to progress, another profiling is needed to alter the treatment strategy based on the current and main pattern of the cancer molecular landscape.
Conclusion
In conclusion, our study provides valuable insights into the genomic landscape of NPC with its driver mutation and its potential links to immune evasion, with the loss of MHC-I genes standing out as a key mechanism by which NPC tumours may escape immune recognition. The findings underscore the necessity for further research on such cancer subsets. Various mechanisms might lead to the observed phenotype of this immunologically cold tumour, such as, in our case, total loss of the MHC class I repertoire.44,45 We need to devise a new strategy for dealing with such a tumour subset to improve therapeutic outcomes in the future.
Ethics Statement
This study has received ethical clearance from the ethics committee of the Faculty of Medicine, Universitas Indonesia (KET-149/UN2.F1/ETIK/PPM.00.02/2023). All informed consent was obtained prior to inclusion of the subject in this study.
Acknowledgments
The authors thank Joshua Garin Dwicahyo Listianto Linggo for his help in formatting the figures.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Handoko, Louisa M, Permata M, Gondhowiardjo S. Deciphering driver of nasopharyngeal cancer development. Oncol Rev. 2022;16. doi:10.3389/or.2022.10654
2. De S, Ganesan S. Looking beyond drivers and passengers in cancer genome sequencing data. Ann Oncol off J Eur Soc Med Oncol. 2017;28(5):938–945. doi:10.1093/annonc/mdw677
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
4. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. doi:10.1158/2159-8290.CD-21-1059
5. Gondhowiardjo SA, Handoko, Adham M, Rachmadi L, Kodrat H, Tobing DL, Haryoga IM, Dwiyono AG, Kristian YA, Mayang Permata TB. Tumor microenvironment predicts local tumor extensiveness in PD-L1 positive nasopharyngeal cancer. PLoS One. 2020;15(3):e0230449.
6. Samarakoon H, Ferguson JM, Jenner SP, et al. Flexible and efficient handling of nanopore sequencing signal data with slow5tools. Genome Biol. 2023;24(1):69. doi:10.1186/s13059-023-02910-3
7. De Coster W, Rademakers R. NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics. 2023;39(5):btad311. doi:10.1093/bioinformatics/btad311
8. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative Genomics Viewer. Vol. 29. Nature biotechnology. United States; 2011:24–26
9. Robinson JT, Thorvaldsdottir H, Turner D, Mesirov JP. igv.js: an embeddable javascript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics. 2023;39(1):btac830. doi:10.1093/bioinformatics/btac830
10. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi:10.1093/bib/bbs017
11. Gondhowiardjo SA, Adham M. Epstein–Barr Virus (EBV) viral load in tumor cells did not predict tumor extensiveness in nasopharyngeal cancer. Microbiol Res. 2021;12(1):150–156. doi:10.3390/microbiolres12010011
12. Gondhowiardjo S. Epstein-Barr virus latent membrane protein 1 (EBV-LMP1) and tumor proliferation rate as predictive factors of nasopharyngeal cancer (NPC) radiation response. Gan To Kagaku Ryoho. 2000;27 Suppl 2:323–331.
13. Narayan P, Prowell TM, Gao JJ, et al. FDA approval summary: alpelisib plus fulvestrant for patients with HR-positive, HER2-negative, PIK3CA-mutated, advanced or metastatic breast cancer. Clin Cancer Res an off J Am Assoc Cancer Res. 2021;27(7):1842–1849. doi:10.1158/1078-0432.CCR-20-3652
14. Wilhoit T, Patrick JM, May MB. Alpelisib: a Novel Therapy for Patients With PIK3CA-Mutated Metastatic Breast Cancer. J Adv Pract Oncol. 2020;11(7):768–775. doi:10.6004/jadpro.2020.11.7.9
15. Bakhoum SF, Cantley LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell. 2018;174(6):1347–1360. doi:10.1016/j.cell.2018.08.027
16. Bakhoum SF, Kabeche L, Compton DA, Powell SN, Bastians H. Mitotic DNA damage response: at the crossroads of structural and numerical cancer chromosome instabilities. Trends Cancer. 2017;3(3):225–234. doi:10.1016/j.trecan.2017.02.001
17. Chen C, Xu P. Cellular functions of cGAS-STING signaling. Trends Cell Biol. 2023;33(8):630–648. doi:10.1016/j.tcb.2022.11.001
18. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–263. doi:10.1002/em.22087
19. Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7(18):2902–2906. doi:10.4161/cc.7.18.6679
20. Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7(10):739–750. doi:10.1038/nrm2008
21. McIntosh MT, Koganti S, Boatwright JL, et al. STAT3 imparts BRCAness by impairing homologous recombination repair in Epstein-Barr virus-transformed B lymphocytes. PLOS Pathog. 2020;16(10):e1008849. doi:10.1371/journal.ppat.1008849
22. Epum EA, Haber JE. DNA replication: the recombination connection. Trends Cell Biol. 2022;32(1):45–57. doi:10.1016/j.tcb.2021.07.005
23. Krenning L, van den Berg J, Medema RH. Life or death after a break: what determines the choice? Mol Cell. 2019;76(2):346–358. doi:10.1016/j.molcel.2019.08.023
24. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495–506. doi:10.1038/nrm.2017.48
25. Wu X, Xu S, Wang P, et al. ASPM promotes ATR-CHK1 activation and stabilizes stalled replication forks in response to replication stress. Proc Natl Acad Sci U S A. 2022;119(40):e2203783119. doi:10.1073/pnas.2203783119
26. Hu C, Bugbee T, Dacus D, Palinski R, Wallace N. Beta human papillomavirus 8 E6 allows colocalization of non-homologous end joining and homologous recombination repair factors. PLoS Pathog. 2022;18(2):e1010275. doi:10.1371/journal.ppat.1010275
27. Yang X, Lu Y, He F, et al. Benzene metabolite hydroquinone promotes DNA homologous recombination repair via the NF-κB pathway. Carcinogenesis. 2019;40(8):1021–1030. doi:10.1093/carcin/bgy157
28. Dhital B, Rodriguez-Bravo V. Mechanisms of chromosomal instability (CIN) tolerance in aggressive tumors: surviving the genomic chaos. Chromosom Res. 2023;31(2):15. doi:10.1007/s10577-023-09724-w
29. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of mhc class i antigen presentation. Front Immunol. 2021;12:636568. doi:10.3389/fimmu.2021.636568
30. Gondhowiardjo SA, Handoko, Jayalie VF, et al. Tackling resistance to cancer immunotherapy: what do we know? Molecules. 2020;25(18):4096.
31. Adu-Berchie K, Brockman JM, Liu Y, et al. Adoptive T cell transfer and host antigen-presenting cell recruitment with cryogel scaffolds promotes long-term protection against solid tumors. Nat Commun. 2023;14(1):3546. doi:10.1038/s41467-023-39330-7
32. Laurent PA, Morel D, Meziani L, Depil S, Deutsch E. Radiotherapy as a means to increase the efficacy of T-cell therapy in solid tumors. Oncoimmunology. 2023;12(1):2158013. doi:10.1080/2162402X.2022.2158013
33. Zhou B, Lin W, Long Y, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther. 2022;7(1):95. doi:10.1038/s41392-022-00934-y
34. Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, Ségui B. The TNF paradox in cancer progression and immunotherapy. Front Immunol. 2019;10:1818. doi:10.3389/fimmu.2019.01818
35. Yip HYK, Papa A. Signaling pathways in cancer: therapeutic targets, combinatorial treatments, and new developments. Cells. 2021;10(3):659. doi:10.3390/cells10030659
36. He H, Shao X, Li Y, et al. Targeting signaling pathway networks in several malignant tumors: progresses and challenges. Front Pharmacol. 2021;12. doi:10.3389/fphar.2021.675675
37. MacConaill LE, Garcia E, Shivdasani P, et al. Prospective enterprise-level molecular genotyping of a cohort of cancer patients. J Mol Diagn. 2014;16(6):660–672. doi:10.1016/j.jmoldx.2014.06.004
38. Chang MT, Asthana S, Gao SP, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34(2):155–163. doi:10.1038/nbt.3391
39. Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. doi:10.1158/2159-8290.CD-13-0642
40. Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K inhibitors in cancer: clinical implications and adverse effects. Int J Mol Sci. 2021;22(7):3464. doi:10.3390/ijms22073464
41. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–1940. doi:10.1056/NEJMoa1813904
42. Erickson A, He M, Berglund E, et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature. 2022;608(7922):360–367. doi:10.1038/s41586-022-05023-2
43. Martínez-Jiménez F, Movasati A, Brunner SR, et al. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature. 2023;618(7964):333–341. doi:10.1038/s41586-023-06054-z
44. Benoit A, Vogin G, Duhem C, Berchem G, Janji B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Vol. 12. Cells; 2023.
45. Wang MM, Coupland SE, Aittokallio T, Figueiredo CR. Resistance to immune checkpoint therapies by tumour-induced T-cell desertification and exclusion: key mechanisms, prognostication and new therapeutic opportunities. Br J Cancer. 2023;129(8):1212–1224. doi:10.1038/s41416-023-02361-4
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.