")
Back to Journals » International Medical Case Reports Journal » Volume 17
c.1103T>C (p.Ile368Th) de novo Variant in Synaptotagmin 1 (SYT1) Gene is Pathogenic, Leading to an Ultra-Rare Neurodevelopmental Disorder: The Baker-Gordon Syndrome
Authors Porto MB , Castro GDME, Pereira SSS, Uchoa EMGS , Zatarin R, Minasi LB, da Cruz AD
Received 29 November 2023
Accepted for publication 17 January 2024
Published 24 January 2024 Volume 2024:17 Pages 63—70
DOI https://doi.org/10.2147/IMCRJ.S448555
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Milena Barbosa Porto,1 Geovanna da Mata e Castro,1 Samara Socorro Silva Pereira,2 Elza Maria Gonçalves Santos Uchoa,2 Raffael Zatarin,3 Lysa Bernardes Minasi,1 Aparecido D da Cruz1– 3
1Graduate Program in Genetics, School of Medical and Life Sciences, Pontifical Catholic University of Goiás, Goiânia, GO, Brazil; 2Federal University of Goiás, Graduate Program in Genetics and Molecular Biology, Goiânia, GO, Brazil; 3Clinical Genetics Service, Center for Rehabilitation and Readaptation Dr. Henrique Santillo, State Health Secretary of Goiás, Goiânia, GO, Brazil
Correspondence: Aparecido D da Cruz, Email [email protected]
Abstract: Baker-Gordon Syndrome (BAGOS) is a genetically determined 4 (NDD), represented by a phenotypic spectrum of moderate to severe intellectual disability, resulting from mutations in the synaptotagmin 1 (SYT1) gene. Its prevalence is estimated at 1:1,000,000 and the known gene variants have indicated complete penetrance with variable expressivity. SYT1 is a membrane trafficking protein in presynaptic vesicles, which exerts a complex influence on synaptic transmission, with fundamental roles in the release of neurotransmitters and facilitators of endocytosis, impacting both neurotransmission and neuron plasticity. The current case report describes the first Brazilian male patient diagnosed at 17-year-old, and the 39th reported case globally using whole-exome sequencing. A de novo heterozygous missense mutation at chr12q:79448958 (NM_005639.2; c.1103T>C; p.Ile368Thr) in the SYT1 was found and classified as a pathogenic variant. The proband’s clinical phenotype was compatible with BAGOS, involving behavioral changes such as irritability and severe intellectual disability. Knowledge about the mechanism of action and the extent of the genotypic and phenotypic presentations of the mutations in the SYT1 is still unfolding. Thus, we aimed to describe additional genotype–phenotype correlation for BAGOS, contributing to the expansion of the existing knowledge of such a heterogeneous ultra-rare syndrome, and, therefore, improve its diagnostic yield, case management, and therapeutic journey for future patients.
Keywords: SYT1, hypotonia, congenital malformations, NDD, WES
Graphical Abstract:
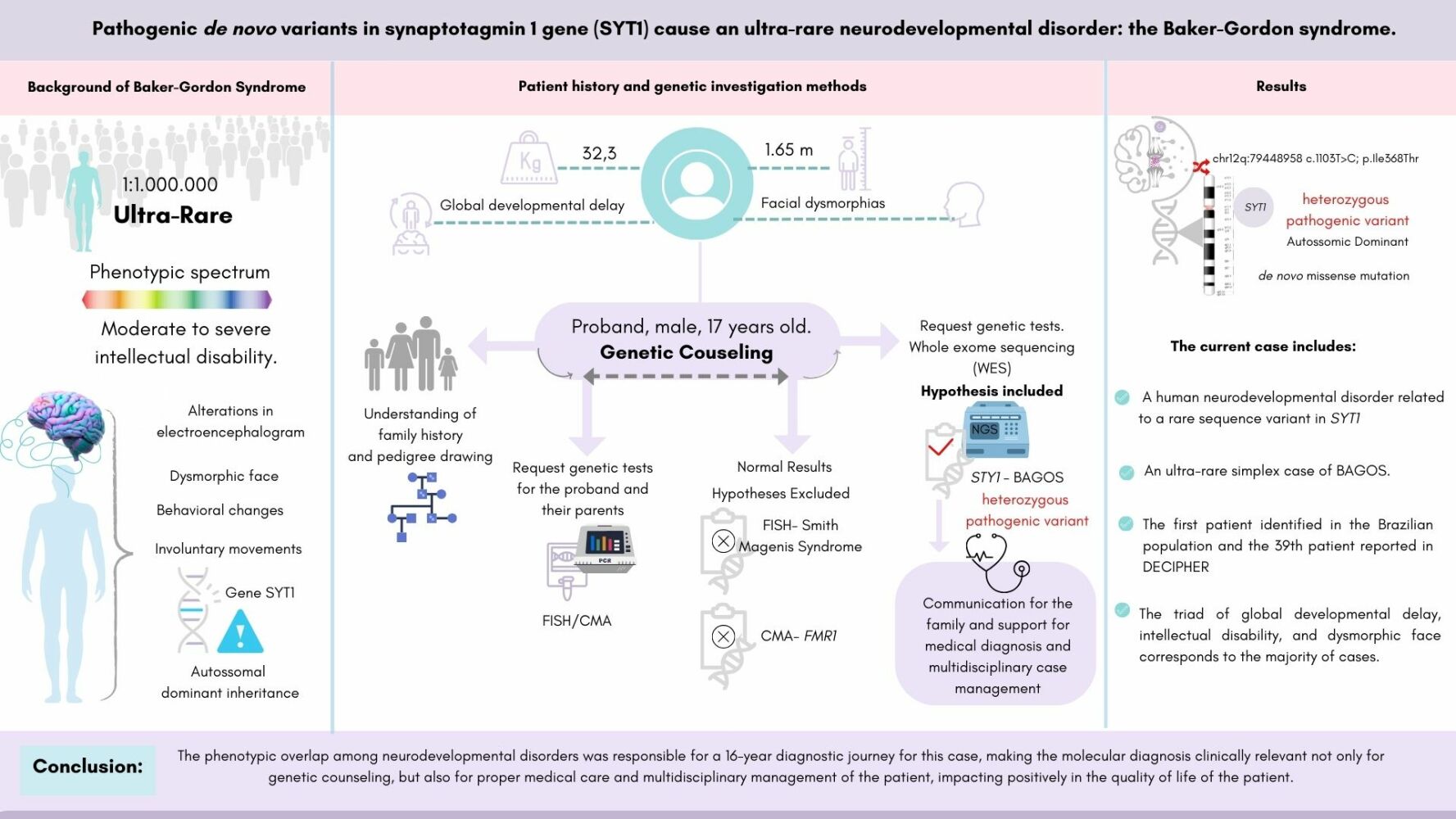
Introduction
Baker-Gordon Syndrome (BAGOS, OMIM #618218) is a genetically determined neurodevelopmental disorder (NDD), encompassing a phenotypic spectrum of moderate to severe intellectual disability, caused by pathogenic mutations in the synaptotagmin 1 gene (SYT1, OMIM *185605) at 12q21.1 BAGOS is characterized by involuntary movements, facial dysmorphology, behavioral abnormalities, and alterations in electroencephalogram (EEG) patterns.1,2 It is a rare genetic syndrome, with an estimated prevalence of 1:1,000,000 and early childhood onset.3 The syndrome is characterized by global developmental delay since infancy, including hypotonia, delayed or inability to walk, poor or absent speech, and moderate to profound impaired intellectual development.2,4–6 Other common characteristics include feeding difficulties, gastroesophageal reflux, sleep disorders, self-mutilation, outbursts of anger, episodic agitation, and stereotypical and/or unpredictable behaviors that may resemble autism spectrum disorder. Additionally, there are congenital ophthalmological abnormalities such as strabismus, esotropia, nystagmus, hyperopia, lack of visual attention, and central visual impairment. Less frequent features include skeletal disorders such as joint laxity, foot deformities, scoliosis, central sleep apnea, and nonspecific abnormalities of the white matter.1–3,6
Many genes and postsynaptic processes have been associated with neurodevelopmental disorders. It is becoming increasingly evident that presynaptic dysfunction plays an important role in the pathophysiology of neurodevelopmental disorders.6,7 Healthy brain function depends on the precise regulation of the probability, timing, and bioavailability of neurotransmitter release in the synaptic cleft. In this context, a fundamental step involves calcium-dependent triggering of fusion between the synaptic vesicle and plasma membranes to allow rapid and coordinated neurotransmitter release.6,8
SYT1 is a membrane trafficking protein present in presynaptic vesicles, which requires calcium binding for exocytosis and endocytosis of the neurotransmitter in synaptic clefts. The primary brain isoform expressed throughout the neocortex and subcortical structures in postnatal life, functions as a calcium sensor for rapid synchronous neurotransmitter release. This mechanism has been molecularly confirmed, as SYT1 deletion results in specific ablation of the mechanism in model organisms. Thus, SYT1 exerts a complex influence on neurotransmission and neuron plasticity, acting as a cargo molecule for extracellular vesicles.5,7 In the aforementioned context, de novo pathogenic or likely pathogenic mutations in SYT1 manifest in a dominant manner. Variants located in the C2B domain have been considered phenotypically more severe as they are more efficient to disrupt endocytosis, leading to a defect in the dynamics of presynaptic vesicle release and disrupting neurotransmission.6,9–12 Contemporary findings suggest a genotype–phenotype relationship where variation in patient phenotype reflects the specific impact of the mutation, affecting neurotransmitter function in a genotype-dependent manner in the synaptic vesicle.5,6 Therefore, knowledge about the mechanism of action and the extent of the genotype–phenotype relationship of observed mutations in the SYT1 are still limited. Thus, it is necessary to assess the extent of clinical manifestations and the variability of phenotypes in relation to the identified genotypes.5–8
In the context present above, in the future, the diversity of molecular mechanisms associated with STY1 sequencing variants will lead to the understanding of the complex genotype–phenotype associations found in BAGOS and may even shed light into the severity of symptoms and prognosis, facilitating precision medical care. Prospectively, it is also very likely large-scale genome sequencing analysis, combined with experimental neuroscience, investigating presynaptic mechanisms will lead to the discovery of other clinically relevant rare phenotypes of neurodevelopmental disorders.
The present report aims to describe another genotype–phenotype relationship for BAGOS, including additional information to contribute to the underlying mechanisms of this disorder. The description of rare cases is important for better understanding patient prognosis, inheritance, appropriate genetic counseling, treatment, case management and follow-up, and quality of life improvement.
Case Report
A 17-year-old male patient from Goiânia, Central Brazil, was referred to the State Rehabilitation and Readaptation Center for medical assistance and genetic counseling due to a rare undiagnosed syndrome with congenital malformations predominantly affecting the facial aspects. The father was healthy, and the mother reported a thyroid cancer treated before conception with surgery and radioactive iodine therapy. The parent’s age at conception were 29 (mother) and 32 (father) years old. The parents did not have difficulty getting pregnant and received regular prenatal care. The pregnant mother was prescribed the standard prenatal vitamin supplements. During pregnancy, she denied infections, bleeding, amniotic fluid loss, rashes, fever, hypertension, or edema. However, she had gestational diabetes controlled without medication.
During pregnancy, the fetus started moving around the 18th week and remained active until delivery. Delivery was performed by cesarean section due to gestational diabetes as a medical decision. No unusual placenta nor umbilical cord presentations were reported. The newborn weighed 3370kg, measured 54cm in length, and had Apgar scores of 8, 9, and 10 at 1, 5, and 10 minutes, respectively. The newborn required no respiratory assistance, did not show the crying reflex and was unresponsive to stimuli, causing delayed suction and latching. As the newborn did not adapt to breastfeeding, he was transitioned to formula with the help of a speech therapist. The family stayed in the maternity ward for 2 days.
The child’s medical history included recurrent urinary tract infections, strong urine odor since birth, idiopathic high fever at 12 months, a medication-induced seizure episode at 4 years, gastroesophageal reflux, and infantile hypotonia. The proband showed global neurodevelopmental delay, not having achieved most developmental milestones and lacked independence in all his daily activities. Some milestones were reached with delay, such as closure of the wide fontanelle at 1 year, eruption of the first teeth at 1 year, and sitting with assistance at 2 years old. He showed behavioral changes such as irritability and had severe intellectual disability making social interactions ineffective. The proband has had an updated vaccination schedule. Upon his last physical examination at 18 years old, he weighed 32.3 kg (P1%; −6.61 SD), had a height of 165 cm (P4%; −1.72 SD), and a head circumference of 57.1 cm (P46%; −0.09 SD). The weight deficit is likely due to dysphagia and difficulty ingesting liquid and solid foods, accepting only pureed or small pieces of solid foods.
The complementary tests showed variable results. The electrocardiogram, ophthalmological evaluation, and brain magnetic resonance imaging were unremarkable. However, electroneuromyography indicated chronic motor neuropathy in all four limbs and a slight increase of glycine in the urine was found. For follow-ups, the patient has been monitored by a multidisciplinary team including nutrition, speech, occupational, and physio therapies, besides genetic counseling.
Based on the family history and phenotypic synopsis, diagnostic hypotheses were formulated, which guided the genetic tests. Initially, fluorescent in situ hybridization (FISH) was performed for Smith-Magenis syndrome, genotyping of the FMR1 gene expansion by polymerase-chain reaction (PCR), and chromosomal microarray analysis (CMA) of the proband and his parents. All tests showed normal results. Subsequently, whole-exome sequencing (WES) using next-generation sequencing (NGS) was performed. Preparation and enrichment of genomic DNA were carried out using Agilent SureSelectXT Human All Exon 50Mb kit. Exome sequencing was performed on the HiSeq 2500 System platform (Illumina, USA). Quality parameters for WES included average number of reads of 175x, 20x coverage of >94% of targeted bases. A de novo heterozygous pathogenic variant at chr12q:79448958 (NM_005639.2; c.1103T>C; p.Ile368Thr) in the SYT1 (GRCh38) was found. The variant was classified as pathogenic according to the criteria of the American College of Medical Genetics (ACMG) and described in patients with intellectual disability and facial dysmorphisms, associated with the phenotype of BAGOS (OMIM #618218). Only the reported variant was found.
During genetic counseling, the family history revealed a simplex case, consistent with autosomal dominant inheritance (ADI). The proband was the second child of a non-consanguineous, healthy couple with no future reproductive interest. Although the penetrance of the variant is unknown, in the present case, it behaved with complete penetrance with onset at birth, consistent with all previously reported cases. In the literature, there are records of variable expressivity for this phenotype. The prevalence of BAGOS is estimated at 1:1,000,000.3 So far, the described cases of genomic variants in SYT1 suggest complete penetrance with variable expressivity. However, the limited number of cases is not sufficient to confirm this evidence. Therefore, for genetic counseling in the present case, although healthy, the proband’s sisters were considered individuals at risk. Consequently, the recurrence risk in the sibling of an isolated case of ADI with high penetrance (>90%) and low fitness (<30%) with two healthy sisters was estimated to be <0.021. Additionally, predictive testing recommendations for asymptomatic children and adolescents were discussed with the family, which should be done only for genes that determine medically actionable conditions. Therefore, in the present case, there was no indication for genetic testing of the siblings. As it is a variant with dominant manifestation, with healthy parents without phenotypic manifestation, genetic testing for the couple was not offered, following national guidelines. Subsequently, the parents participated voluntarily in a larger study regarding global developmental delay and had their genotyped confirmed with no variants found. Thus, the family history included a de novo missense mutation following dominant manifestation.
Discussion
Neurotransmission requires a balance between exocytosis and endocytosis of synaptic vesicles for effective and healthy neuronal communication. This process is calcium-dependent, as calcium triggers the release of neurotransmitter to initiate signal transmission.2,6,10 SYT1 is considered the primary calcium sensor for vesicular trafficking. Considering the relevance of SYT1 in synaptic neurotransmission physiology, it is essential to fully understand the role of the protein as a relevant biomarker for synaptic dysregulation, as its dysfunction can result in a wide range of deleterious effects in the central nervous system (CNS), leading to brain and neurodevelopmental disorders.5–7
The current case reported a rare missense sequence variant in SYT1, the mutation replaced isoleucine, a nonpolar amino acid, with threonine, a neutral polar amino acid. The difference in hydropathy indices between these two amino acids alters the protein’s spatial structure, as the more hydrophilic threonine tends to occupy more external regions in the protein’s spatial organization, leading to loss of function and haploinsufficiency of SYT1. Therefore, there is a significant reduction in the availability of functional calcium sensors involved in triggering neurotransmitter release in the synaptic cleft. Consequently, it causes intellectual disability and facial dysmorphisms associated with the phenotype of BAGOS (OMIM #618218).1
The proband’s clinical presentation was consistent with the cardinal phenotypes known for BAGOS, including the age of onset in childhood.2,3,6 On the other hand, electromyography indicated motor neuropathy in all four limbs with signs of chronicity. These findings had not been previously reported in association with BAGOS. The current case supported that neurodevelopmental disorders associated with SYT1 have a wide phenotypic variability, modulated by the genotype.5,6 Thus, the advancement of genomic technologies and the increase in casuistic SYT1 sequence variants will contribute to further understanding the role of the genotypes in BAGOS outcomes.11 Elucidating the genotype–phenotype relationship through case reports in the literature would, for example, confirm how mutations in SYT1 behave in terms of penetrance and expressivity, providing pieces of information crucial to case management and genetic counseling.
Currently, DECIPHER database contains 39 cases of genomic variants in SYT1.13 Out of this, 9 patients had sequence variants like the patient in this report (Table 1; Figure 1). Despite the small sample size, among reported cases the sex ratio was 2:1, suggesting BAGOS appears twice as common among boys. The missense mutation described here corresponded to the most reported variant among patients with sequence variants (3/9), having a predicted molecular impact of inhibited membrane penetration.6 Additionally, a total of 28 patients in DECIPHER had copy number variations (CNVs) involving the SYT1, distributed in 14% gains and 86% losses.13 Regarding CNVs, about 14% of them were inherited, and half of them were inherited from parents with phenotypes like the proband’s. The quantitative sex ratio also revealed the predominance of males (2:1). On the other hand, aneuploidy was important for the manifestation of the phenotype in two girls, one with trisomy and the other with uniparental disomy of chromosome 12, likely resulting from a trisomy rescue. Although the casuistic data is still extremely rare, the observed genotypic diversity among patients with BAGOS supports the observation that multiple genomic mechanisms may be involved in the pathophysiology and dominant manifestation of the phenotype in this ultra-rare disease. Figure 2 illustrates the synopsis of the phenotypes of cases with variants in the sequence of the SYT1, including the present case. Moreover, 22 patients, which included some previously deposited in DECIPHER database, were reported with missense variants in SYT1.6 The paper thoroughly described the phenotype heterogeneity in BAGOS and presented a rich approach detailing the potential molecular mechanisms underlying the pathogenic potential of STY1 variants worth reading by all clinicians.
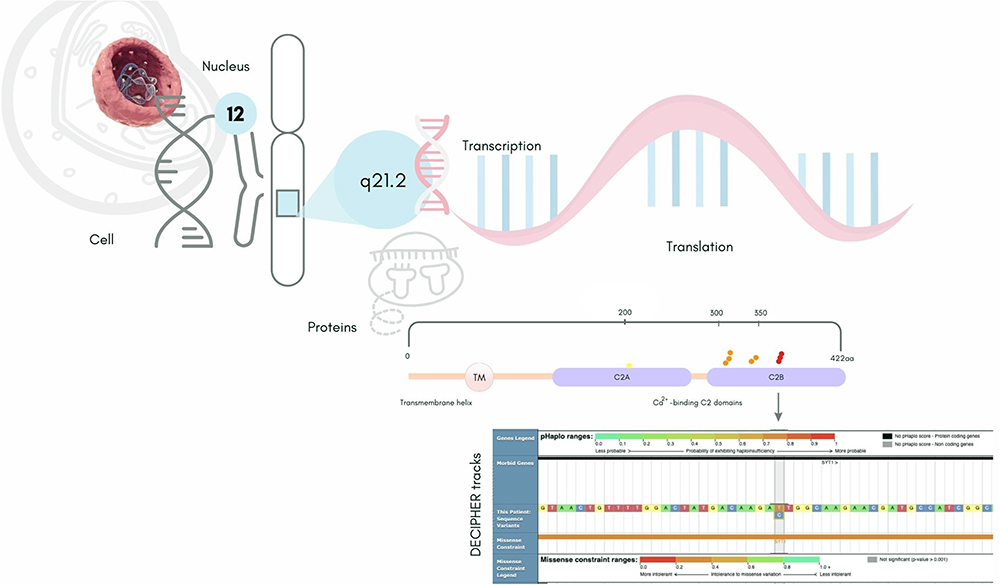 |
Figure 1 Missense sequence variants of SYT1 deposited in DECIPHER13 indicating their location on the protein domains by yellow, Orange, and red dots classified as variant of uncertain significance, Likely pathogenic, and pathogenic respectively. DECIPHER severity tracks related to variant identified in a patient with Baker-Gordon Syndrome from Central Brazil is also included. Note: This figure makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from [email protected]. DECIPHER is hosted by EMBL-EBI and funding for the DECIPHER project was provided by the Wellcome Trust [grant number WT223718/Z/21/Z]. |
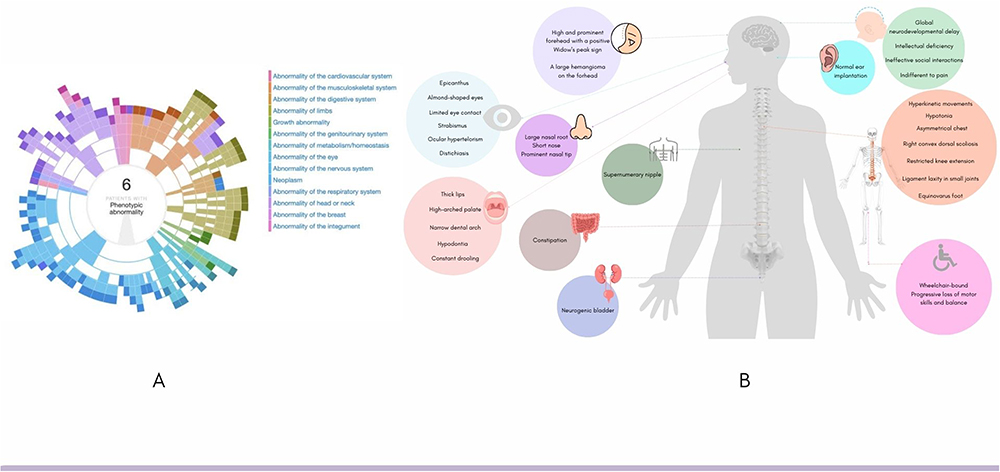 |
Figure 2 Phenogram of patients harboring sequence variants in SYT1 (12q21.2). (A) abnormalities by ontological clustering in open access DECIPHER patients;13 (B) Synopsis of phenotype of a male patient diagnosed with Baker-Gordon Syndrome from Central Brazil. |
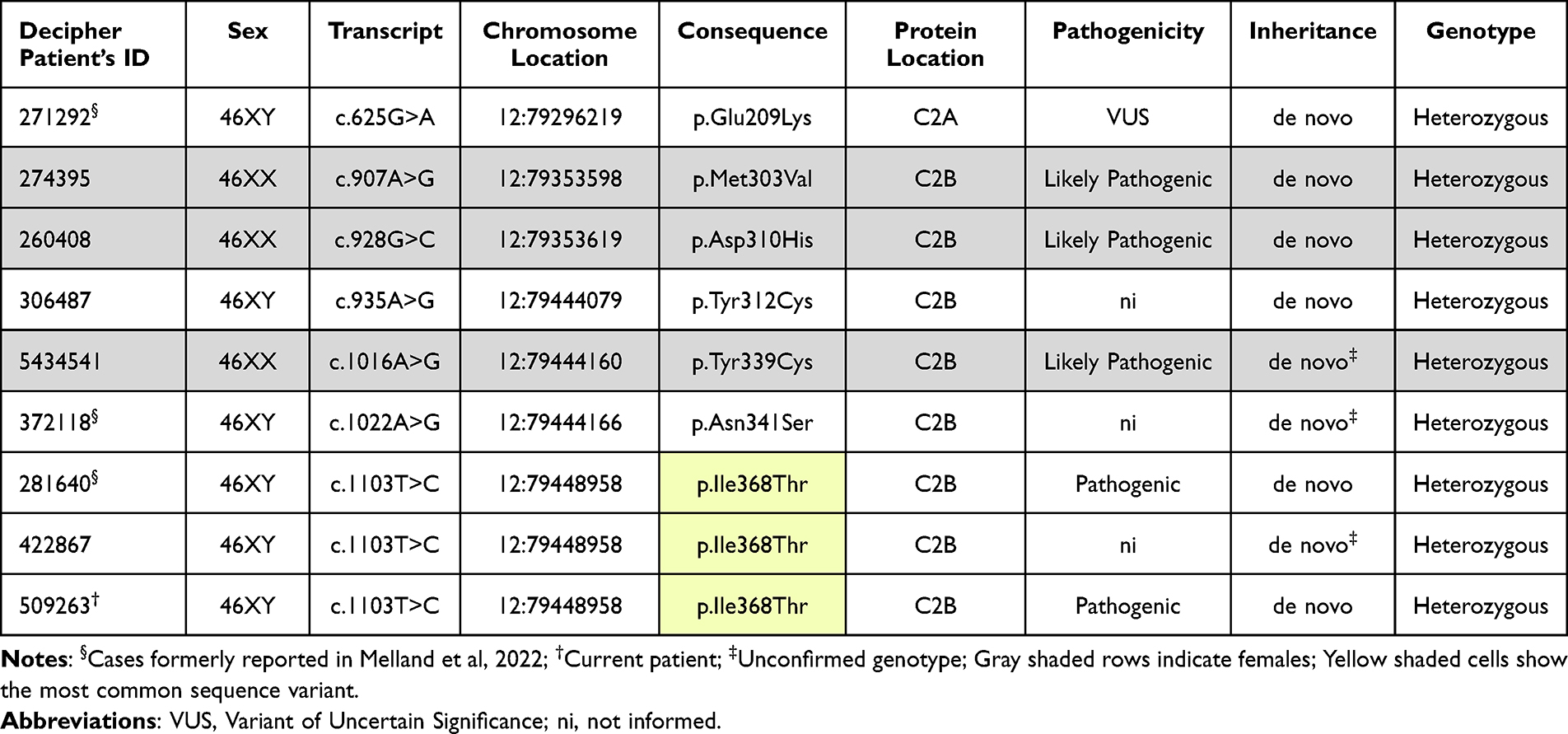 |
Table 1 Patients with BAGOS Phenotype (OMIM #618218), Included in DECIPHER13 Until October 2023, Who Exhibited the Syndrome Due to Missense Sequence Variants |
Conclusion
This case describes an ultra-rare case of BAGOS, corresponding to the first patient identified in the Brazilian population with a de novo heterozygous pathogenic variant at chr12q:79448958 (NM_005639.2; c.1103T>C; p.Ile368Thr) in the SYT1 (GRCh38). Regarding the phenotypic synopsis of BAGOS patients included in DECIPHER, the triad of global developmental delay, intellectual disability, and facial dysmorphisms corresponds to most cases. The phenotypic overlap among neurodevelopmental disorders was responsible for a 16-year diagnostic journey for our case, making the molecular diagnosis clinically relevant not only for genetic counseling but also for proper medical care and multidisciplinary management of patients. The data described here and made public in DECIPHER contributed to expand the knowledge about the phenotypic and genotypic presentations of this syndrome. However, most single variant cases from DECIPHER include patients with missense mutations in the C2B domain and some do not include phenotype information. For BAGOS, phenotypes vary according to variants’ location and genotypes, therefore, the community dealing with the spectrum of SYT1-associated neurodevelopmental disorders should consider updating the information in DECIPHER and those with new patients should also include their information in the databank. That approach would increase the likelihood of finding patients for future functional studies, reducing the chances of biases due to uncontrolled variables.
Data Sharing Statement
All data relevant to the current study were included in this manuscript and in DECIPHER database. This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of Pontifical Catholic University of Goiás under CAAE # 67838022.9.0000.0037. Written informed consent was obtained from the proband’s parents which included consent to publish.
Acknowledgments
The authors are grateful for the important and generous contributions of each family to DECIPHER. We also appreciate the support of the multidisciplinary teams involved in the care of our patient, from clinicians to laboratory scientists involved in ending his diagnostic journey and assisting him and his family through this challenging health experience. We are also in debt to DECIPHER and its community for allowing the inclusion of information regarding individual patients in this report. Finally, we are grateful to Mr. Sean Quail for proofreading the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) of Brazil. This work has also been supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) of Brazil as the corresponding author holds a fellowship 1D. This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from [email protected]. DECIPHER is hosted by EMBL-EBI and funding for the DECIPHER project was provided by the Wellcome Trust [grant number WT223718/Z/21/Z].
Disclosure
Dr Raffael Zatarin reports non-financial support from BioMarin Pharmaceutical, non-financial support from PTC therapeutics, non-financial support from Chiesi Pharmaceuticals, non-financial support from Sanofi, non-financial support from Novartis, outside the submitted work. The authors have no other relevant financial or non-financial interests to disclose.
References
1. Baker-Gordon Syndrome (BAGOS). An Online Catalog of Human Genes and Genetic Disorders (OMIM); 2018 [updated December 29, 2021]. Available from: https://omim.org/entry/618218.
2. Melland H, Arvell EH, Gordon SL. Disorders of synaptic vesicle fusion machinery. J Neurochem. 2021;157(2):130–164. doi:10.1111/jnc.15181
3. Baker-Gordon Syndrome (BAGOS). MalaCards Human Disease Database; [updated September 29, 2023]. Available from: https://www.malacards.org/card/baker_gordon_syndrome.
4. Baker K, Gordon SL, Grozeva D, et al. Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J Clin Invest. 2015;125(4):1670–1678. doi:10.1172/JCI79765
5. Baker K, Gordon SL, Melland H, et al. SYT1-associated neurodevelopmental disorder: a case series. Brain. 2018;141(9):2576–2591. doi:10.1093/brain/awy209
6. Melland H, Bumbak F, Kolesnik-Taylor A, et al. Expanding the genotype and phenotype spectrum of SYT1-associated neurodevelopmental disorder. Genet Med. 2022;24(4):880–889. doi:10.1016/j.gim.2021.12.002
7. Riggs E, Shakkour Z, Anderson CL, Carney PR. SYT1-associated neurodevelopmental disorder: a narrative review. Children. 2022;9(10):1439–1449. doi:10.3390/children9101439
8. Bradberry MM, Courtney NA, Dominguez MJ, et al. Molecular basis for synaptotagmin-1-associated neurodevelopmental disorder. Neuron. 2020;107(1):52–54. doi:10.1016/j.neuron.2020.04.003
9. Paddock BE, Wang Z, Biela LM, et al. Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo. J Neurosci. 2011;31(6):2248–2257. doi:10.1523/jneurosci.3153-09.2011
10. Bello OD, Jouannot O, Chaudhuri A, et al. Synaptotagmin oligomerization is essential for calcium control of regulated exocytosis. Proc Natl Acad Sci USA. 2018;115(32):E7624–E7631. doi:10.1073/pnas.1808792115
11. Moutton S. L’approche basée sur le génotype déterminé par séquençage haut-débit en première intention et le partage international des données pour identifier de nouveaux gènes et nouveaux syndromes responsables d’anomalies du développement [The approach based on the genotype determined by first-line high-throughput sequencing and international data sharing to identify new genes and new syndromes responsible for developmental anomalies]. Médecine humaine et pathologie. Université Bourgogne Franche-Comté; 2019. Français. Français: https://theses.hal.science/tel-02477164.
12. Tagliatti E, Bello OD, Mendonça PRF, et al. Synaptotagmin 1 oligomers clamp and regulate different modes of neurotransmitter release. Proc Natl Acad Sci USA. 2020;117(7):3819–3827. doi:10.1073/pnas.1920403117
13. Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84(4):524–533. doi:10.1016/j.ajhg.2009.03.010
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.