")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 15
Antibacterial and Antioxidant Activities, in silico Molecular Docking, ADMET and DFT Analysis of Compounds from Roots of Cyphostemma cyphopetalum
Authors Degfie T, Ombito JO, Demissie TB, Eswaramoorthy R , Dekebo A, Endale M
Received 13 June 2022
Accepted for publication 28 September 2022
Published 26 October 2022 Volume 2022:15 Pages 79—97
DOI https://doi.org/10.2147/AABC.S377336
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr James Briggs
Teshome Degfie,1 Japheth O Ombito,2 Taye B Demissie,2 Rajalakshmanan Eswaramoorthy,3 Aman Dekebo,1,4 Milkyas Endale1
1Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, Ethiopia; 2Department of Chemistry, University of Botswana, Gaborone, Botswana; 3Department of Biomaterials, Saveetha Dental College and Hospitals, Saveetha Institute of Medical and Technical Sciences, Saveetha University, Chennai, India; 4Institute of Pharmaceutical Sciences, Adama Science and Technology University, Adama, Ethiopia
Correspondence: Milkyas Endale; Teshome Degfie, Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, P.O. Box 1888, Adama, Ethiopia, Email [email protected]; [email protected]
Background: Cyphostemma cyphopetalum is a medicinal plant traditionally used to treat various ailments. Limited studies on C. cyphopetalum inspired us to investigate the chemical nature and therapeutic potential of the plant.
Methods: Silica gel column chromatographic separation was used for isolation. 1D and 2D NMR spectroscopic analysis and literature data were used for structural elucidation. Agar well diffusion assay was used for evaluation of antibacterial activity against E. coli, P. aeruginosa, and S. aureus. DPPH assay was used to evaluate radical scavenging activities. Molecular docking was done by AutoDock Vina 4.2 open-source program. DFT calculations were performed using the Gaussian 16 program package.
Results: Dichloromethane/methanol (1:1) roots extract afforded a new hydroxyl-spongiane diterpenoid lactone derivative, 3-hydroxyisoagatholactone ( 1), along with β-sitosterol ( 2) and ϵ-viniferin ( 3) whereas methanol extract afforded trans-resveratrol ( 4), gnetin H ( 5), tricuspidatol A ( 6), ϵ-viniferin-diol ( 7) and parthenostilbenin B ( 8). At 50 μg/mL, compound 3 recorded the highest inhibition against E. coli (8.55 ± 0.45 mm) and S. aureus (9.30 ± 1.39 mm). Against P. aeruginosa, compound 5 consistently outperformed chloramphenicol (11.76 ± 0.77 mm, at 30 g/mL). Maximum binding affinity were observed by compound 3 against DNA gyrase B (-7.6 kcal/mol) where as compound 5 displayed maximum binding against PqsA (-8.8 kcal/mol) and S. aureus PK (-5.8 kcal/mol). Compounds 1, 3 and 4 satisfy Lipinski’s rule of five. Trans-resveratrol ( 4) demonstrated strong DPPH scavenging activity at 12.5 g/mL, with IC50 values of 0.052 μg/mL, compared to ascorbic acid (IC50 value of 0.0012 μg/mL).
Conclusion: In this work, eight compounds were identified from the roots extracts of C. cyphopetalum including a new hydroxyl-spongiane diterpenoid lactone, 3-hydroxyisoagatholactone ( 1). Compounds 3 and 5 exhibited good antibacterial activity and binding affinities. The docking result is in agreement with the in vitro antibacterial study. Overall, the study result suggests that the isolated compounds have the potential to be used as therapeutic agents, which supports the traditional uses of C. cyphpetalum roots.
Keywords: Cyphostemma cyphopetalum, 3-hydroxyisoagatolactone, antibacterial, docking, drug-likeness, antioxidant studies
Introduction
The genus Cyphostemma (Planch.) Alston, belonging to the Vitaceae family, includes approximately 259 taxonomically accepted species of erect, prostrate, or climbing perennial herbs or shrubs, with or without tendrils, distributed throughout the tropics and subtropics of the world.1 Members of this genus have long been used in folk medicine to treat various ailments, such as malaria, inflammation, kwashiorkor, and marasmus in children, snake bite, abortifacient, rabies, tooth ache, tumor.2–5 The phytochemical constituents of the genus Cyphostemma include flavonoids, alkaloids, coumarins, saponins, terpenoids, stilbenoids, and tannins.6–10 These compounds exhibit diverse bioactivities, including antioxidant, antibacterial, antifungal, hepatoprotective, antiploriferation, and chemopreventive.11–16 C. cyphopetalum (Fresen.) is a medicinal plant up to 4–6 m having scrambling succulent leaves and creamy yellow flowers distributed in tropical and subtropical African countries, including Ethiopia, Cameroon, Sudan, Uganda, Kenya, Tanzania, Rwanda, Congo, and Zambia.1,17 In Ethiopia, it is locally called as ‘Milas Golgul’ (Amharic),18,19 and Gindosh (Amharic)3 and available in different parts of the country, including Harerge, Afar, Ilubabor, Bale, Arsi, Welo, Shewa, Tigray, Gonder, Gojam, Welega, Sidamo, and Gamogofa.17 Various traditional uses of the plant have been reported in Ethiopia, including treating tumor,9 snake bite, rabies,3 eczema (Atopic dermatitis).20 In Kenya, the plant has various traditional uses, including eradicating insects.21
The lack of report on the phytochemical and pharmacological studies on C. cyphopetalum, despite the reported ethnomedicinal uses, inspired us to investigate and explore the chemical nature and therapeutic potential of the plant. Reported herein are the isolation, structure elucidation, antibacterial and antioxidant activity evaluation of eight compounds from the dichloromethane/methanol (1:1) and methanol root extracts of which 3-hydroxyisoagatholactone (1) is a novel compound.
Experimental Part
General Experimental Procedure
IR spectra were recorded using KBr pellets on PerkElmer BX Infrared Spectrometer in the range 4000 to 400 cm−1. The NMR spectra were recorded on Bruker Avance 400 MHz spectrometer with deuterated chloroform and methanol using TMS as internal standard. Analytical TLC plates with silica gel 60 F254 TLC (Merck, Germany) were used to determine TLC profiles. The spots on TLC plates were visualized using a UV lamp (254 and 365 nm). Silica gel column chromatography was performed at silica gel (60–120 mesh). All chemicals, solvents and reagents were used to analytical grade.
Plant Material Collection and Identification
The roots of C. cyphopetalum were collected from the district of Huruta town, Lode Hetosa Woreda, Arsi Zone, Oromia, Ethiopia, in September 2019. The plant material was authenticated by Melaku Wendafrash (Chief Technician, Botanist), and voucher specimen (TCY09) was deposited at the National Herbarium, Department of Biology, Addis Ababa University, Ethiopia.
Extraction and Isolation of Compounds from the Roots of C. cyphopetalum
The powdered roots of C. cyphopetalum (500 g) were extracted sequentially with n-hexane, dichloromethane/methanol (1:1), and methanol (100%) by maceration, each for 72 h, thrice at room temperature. The filtrates were concentrated in vacuo on a rotary evaporator at 40 ℃ to obtain 31 g, and 24 g of crude extracts, respectively.
The dichloromethane/methanol (1:1) roots extract (10 g) was subjected to silica gel column chromatography (silica gel 180 g) and eluted with increasing gradient of n-hexane/EtOAc followed by dichloromethane/methanol mixtures. A total of 145 fractions were collected (each 50 mL). Fractions 37–44 obtained with n-hexane/EtOAc (1:1) were subjected to silica gel column chromatography (20 g) eluting with DCM/MeOH (9.5:0.5) isocratic mode to afford the 3-hydroxyisoagatholactone (1, 53 mg). Compound 2 (45 mg) was obtained from fractions 17–36 using n-hexane/EtOAc (3:1) as eluent. Fractions 45–52 obtained from n-hexane/EtOAc (2:3) were subjected to silica gel column chromatography (1–5% EtOAc in n-hexane as eluent) to afford compound 3 (41 mg). The methanol roots extract (10 g) was subjected to gravity column chromatography and eluted with increasing gradient of n-hexane/EtOAc and DCM/MeOH mixtures to afford 98 fractions. Fractions 27–29 eluted with n-hexane/EtOAc (5:2) were purified further using isocratic elution of 5% MeOH in DCM to afford compounds 4 (11 mg), 5 (6 mg), and 6 (20 mg). Finally, fractions 53–57 obtained with n-hexane/EtOAc (3:1) were purified using isocratic elution with 10% MeOH in DCM as eluent and afforded compounds 7 (40 mg) and 8 (7 mg).
Antibacterial Activity
All isolated compounds were studied for in-vitro antibacterial activity against three standard human bacterial pathogens, namely Staphylococcus aureus (S. aureus, ATCC 25923), Escherichia coli (E. coli, ATCC 25922) and Pseudomonas aeruginosa (P. aeruginosa, ATCC 27853) collected from Ethiopian Public Health Institute (EPHI). Experiments were done at the microbiology laboratory of the medical laboratory department, Haramaya University, in collaboration with microbiologists. The degree of susceptibility of each bacterial strain to the isolated compounds was evaluated by using the agar medium disc-diffusion technique as per the standard protocols of Clinical and Laboratory Standards Institute (CLSI).22 That is, about 3–5 fresh colonies with similar morphology of each bacterial species were aseptically transferred into a saline solution using a sterile inoculating loop, and bacterial turbidity was adjusted in reference to 0.5 McFarland standard solution (108 CFU/mL). The Mueller Hinton Agar (HiMedia) medium was prepared as per the manufacturer’s instruction. The bacterial suspension was then streaked by swapping with cotton swap onto Petri plates containing the medium. Sterilized Whatman No. 1 filter paper discs (6 mm in diameter) were prepared using a puncher to hold samples. A stock solution of each isolated compound (1 mg/mL) was prepared in 4% DMSO. Thereafter, four different solutions, 500, 300, 100, and 50 μg/mL, of each compound were prepared from their corresponding stock solutions. Standard antibiotic disc of chloramphenicol (30 µg/disc) and DMSO solvent were served as positive and negative controls, respectively. Each solution (100 µL), including the positive and negative controls, was loaded onto separate paper discs (6 mm in diameter), placed discs onto the Petri plates with the bacterial culture inoculated MHA, and incubated at 37 ℃ for 18–24 hr. Inhibition zones were monitored by the paper disc’s clear area and measured by caliper (in mm).22,23 The experiment was done in duplicate aseptically, and the result was expressed as mean ± standard deviation using SPSS (version 20).
Molecular Docking Studies of the Isolated Compounds
The docking calculations results include binding energy (kcal/mol), hydrogen bond distances, and pictorial representation of viable docked poses. The protein E. coli DNA gyrase B (PDB ID: 6F86), PqsA (PDB ID: 3T07) and S. aureus pyruvate kinase (PK) (PDB ID: 5OE3) were used as target proteins. AutoDock Vina 4.2 (MGL tools 1.5.7) was used to dock the isolated compounds (1–6) into active sites of the target proteins, following reported standard protocol.24 ChemOffice tool (Chem Draw 16.0) was used to prepare the chemical structures of the compounds (1–8) with appropriate 2D orientations, and ChemBio3D was used to obtain the minimized energy of each compound. Then, the energy minimized ligand molecules were then used as input for AutoDock Vina to carry out the docking simulation.
Crystal structures of the receptor protein molecules were obtained from the Protein Data Bank (PDB). The proteins were prepared using protein preparation protocol,25 applying default parameters (remove water molecules and cofactors). The target protein file was generated by leaving the associated residue with protein by using Auto Preparation of target protein file AutoDock 4.2 (MGL tools 1.5.7). A grid box size of 46×46×46 Å pointing in x, y, and z directions with a grid point spacing of 0.375 Å was considered. Lamarckian genetic algorithm (LGA) program with an adaptive whole method search in the Auto Dock was chosen to calculate the different ligand conformers. The potential binding energy between ligand and protein was obtained using the docking Lamarckian genetic algorithm (LGA) program with an adaptive whole method search in the AutoDock Vina. Nine different conformations were generated for each ligand scored using AutoDockVina scoring functions. The conformations with the most favorable (least) free binding energy were selected for analyzing the interactions between the target receptor and ligands by Discovery studio visualizer and PyMOL. The ligands are represented in different colors, H-bonds and the interacting residues are represented in stick model representation.
In silico Pharmacokinetic Analysis
Pharmacokinetic parameters were evaluated for the isolated compounds to investigate their drug candidate chances. The ADMET properties were evaluated with the aid of SwissADME, an online ADME prediction tool (http://www.swissadme.ch). The drug-likeness of the compounds was predicted by adopting the Lipinski’s rule of five.26 Structures of the isolated compounds (1–6) were converted to their canonical simplified molecular-input line-entry system (SMILE) and submitted to SwissADME and PreADMET tool to estimate in-silico pharmacokinetic parameters.27 The toxicity profile of the studied compound (1–6) was predicted using ProTox-II Web tool.28 The selection of the compounds as drug candidates was determined by a parameter called drug score. The higher the drug score value, the higher the compound’s chance is considered a drug candidate.29
Radical Scavenging Activity
The antioxidant activity of each of the isolated compounds from the root of C. cyphopetalum was evaluated based on the scavenging activity of the stable 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical.30,31 Each sample was separately dissolved in methanol and serially diluted to give 200, 100, 50, and 25 µg/mL concentrations. Freshly prepared 0.04% DPPH solution in methanol was added to each concentration. The mixture was shaken and incubated in an oven at 37℃ for 30 min. The absorbance of the resultant solution was measured at 517 nm using UV-Vis spectrophotometry. Ascorbic acid with a similar concentration of test sample was used as the positive control.
The DPPH radical scavenging activity of each of the tested compounds was reported by percentage inhibition using the formula %DPPH Inhibition = [(Acontrol –Asample)/Acontrol]×100, where Acontrol is the absorbance of DPPH solution and Asample is the absorbance of the test sample (DPPH solution plus compound).24,25 The DPPH radical scavenging activity of the compounds was also expressed as IC50, the concentration of the test compound to give a 50% decrease of the absorbance from that of the control solution.
Density Functional Theory Calculations
Currently, density functional theory (DFT) has become a general tool for understanding and predicting the behavior of a broad range of biochemical phenomena applicable to compute the electronic structure of molecules and it enables the determination of properties based on the evaluation of electron density.32
DFT computations of the isolated compounds were performed by using the B3LYP functional,33–35 and the 6–31++G (d,p) basis set.35 Solvent effects were treated using the polarizable continuum model, and methanol as a solvent. All the DFT calculations were performed using the Gaussian 16 program package.36 Frequency calculations were performed for each optimized geometry to confirm that all the optimized geometries correspond to a real minimum. The optimized structures of the compounds were used to determine the molecular electrostatic potential map (MEP), and highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). Based on the energies of HOMO and LUMO of the compounds, the chemical reactivity parameters such as energy gap (ΔE = ELUMO − EHOMO), electronegativity (χ = − ½ (EHOMO + ELUMO), electronic chemical potential (μ = ½ (EHOMO + ELUMO) = − χ), global chemical hardness (η = ½ (ELUMO − EHOMO), global softness (σ = 1/2η), global electrophilicity index (ω = μ2/2η), nucleophilicity index (Nu = 1/ω), dipole moment, and natural atomic charges (NAC) calculations were carried out at the same level of theory.37–39
Results and Discussion
Characterization of the Isolated Compounds
The dichloromethane/methanol (1:1) and methanol root extracts of C. cyphopetalum were separately subjected to gravity column chromatography over silica gel to afford one new spongiane diterpenoid lactone, 3-hydroxyisoagatholactone (1), together with seven known compounds isolated from the plant for the first time. The structure of the known compounds including β-sitosterol (2),40 ε-viniferin (3),41,42 trans-resveratrol (4),43,44 gnetin H (5),45,46 tricuspidatol A (6),47,48 ε-viniferin diol (7),49 and parthenostilbenin B (8)6 were characterized by comparison of their spectral data with those values previously reported in the literature (Figure 1, Supplementary Tables 1–7).
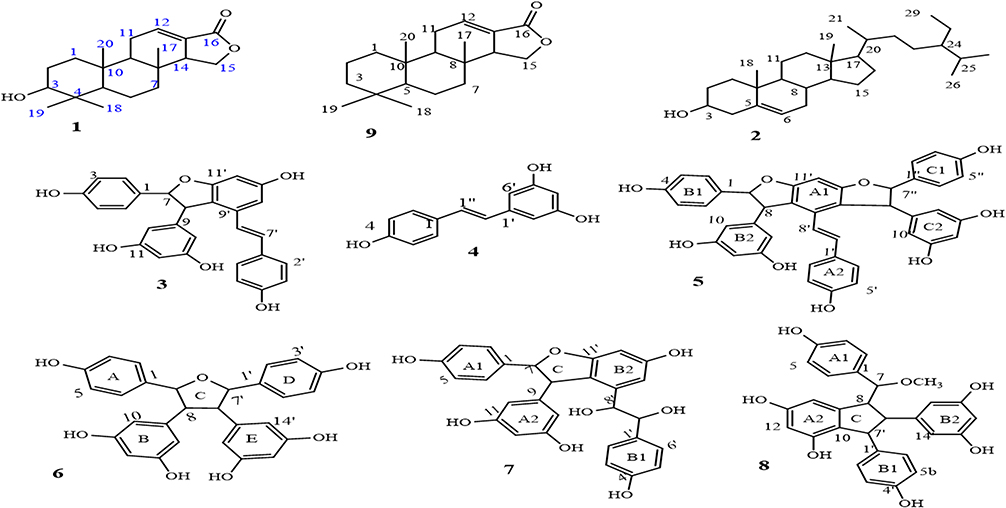 |
Figure 1 Structure of compounds 1–8 and tetracyclic spongiane skeleton (9). |
Compound 1 (3-Hydroxyisoagatholactone)
A yellow solid with the characteristic UV absorption at λmax = 220 nm suggesting π to π* transition of electrons. Its IR spectrum displayed absorption bands at υmax = 1761 and 1687 cm−1attributed to the presence of an α, β-unsaturated γ-lactone ring.50 In addition, the IR spectrum showed bands for hydroxyl (3506 cm−1) and sp3 C–H stretches (2850 cm−1). The 1H/13C NMR data were consistent with published data reported for a spongiane diterpene lactone skeleton (9)50,51 (Table 1 and Figure 1). Its 1H/13C NMR spectrum showed the presence of four methyl signals at δH/δC0.78 (H-17)/14.1, 0.94 (H-18)/28.3, 0.97 (H-19)/22.1, and 0.87 (H-20)/15.1 (Supplementary Figures 1 and 2). The signal at δC 170.3 in the 13C NMR spectra was assignable to the lactone carbonyl at C-16, those at δC 136.4 and 126.9 to the olefinic carbons at C-12 and C-13, respectively (Supplementary Figure 2). In addition, signals at δC 53.9 and 67.2 were assignable to C-14 and C-15, respectively, on γ-lactone ring. The DEPT-135 spectra displayed sex methylene carbon signals (Supplementary Figure 3). The 1H NMR spectra displayed signals for an olefinic proton at δH 6.88 (dd, J = 3.5, 3.5 Hz, H-12), while the diastereotopic oxygenated methylene protons in the lactone ring resonated at δH 4.36 and 4.03 (t, 1H each, J = 9.1, H-15a/b). The signals at δH 3.45 (m, 1H, H-3) suggest an sp3 oxygenated methine proton. The signals at δH 2.27 and 2.78 were assignable to the methylene and methine protons at H-11 and H-14, respectively. The COSY spectra showed correlations between H-2 (δH 1.52) and H-3 (δH 3.42), H-11 (δH 2.31) with H-9 (δH 1.36) and H-12 (δH 6.88), H-14 (δH 2.79) and H-15a (δH 4.36) and H-15b (δH 4.03) (Figure 2, Supplementary Figure 4). The H-C connectivity was determined by the HSQC correlation (Supplementary Figure 5). The HMBC correlations observed between proton at δH 6.88 (H-12) to C-13 (δC 126.9) and C-16 (δC 170.3) (Supplementary Figure 6) were in agreement with α, β-unsaturated ester carbonyl moiety. In addition, the same olefinic proton at δH 6.88 (H-12) showed correlations with C-11 (δC 24.1) and C-14 (δC 51.0). Furthermore, the HMBC spectral data also displayed correlations of δH1.60 (H-1) to C-2 (δC 25.0), C-3 (δC 75.7), and C-10 (δC 37.3), δH1.52 (H-2) to C-1 (δC 32.7) and C-3 (δC 75.7), δH 3.42 (H-3) to C-1 (δC 32.7), C-4 (δC 37.4), C-5 (δC 49.3), and C-19 (δC 22.1), δH 0.78 (H-17) to C-7 (δC 40.6), C-8 (δC 34.4), and C-14 (δC 53.9), δH 0.94 (H-20) to C-1 (δC 32.7), C-9 (δC 51.1), and C-10 (δC 37.0), and δH 0.97 (H-19) to C-3 (δC 75.7), C-4 (δC 37.4), and δOH (5.32) to C-3 (δC 75.7), and C-2 (δC 25.0) (Figure 2, Supplementary Figure 6). Based on the above spectral data analysis and comparison with literature data, compound 1 was elucidated as a hydroxyl derivative of spongiane diterpenoid lactone, 3-hydroxyisoagatholactone (Figure 1, 1), reported herein as a new compound.
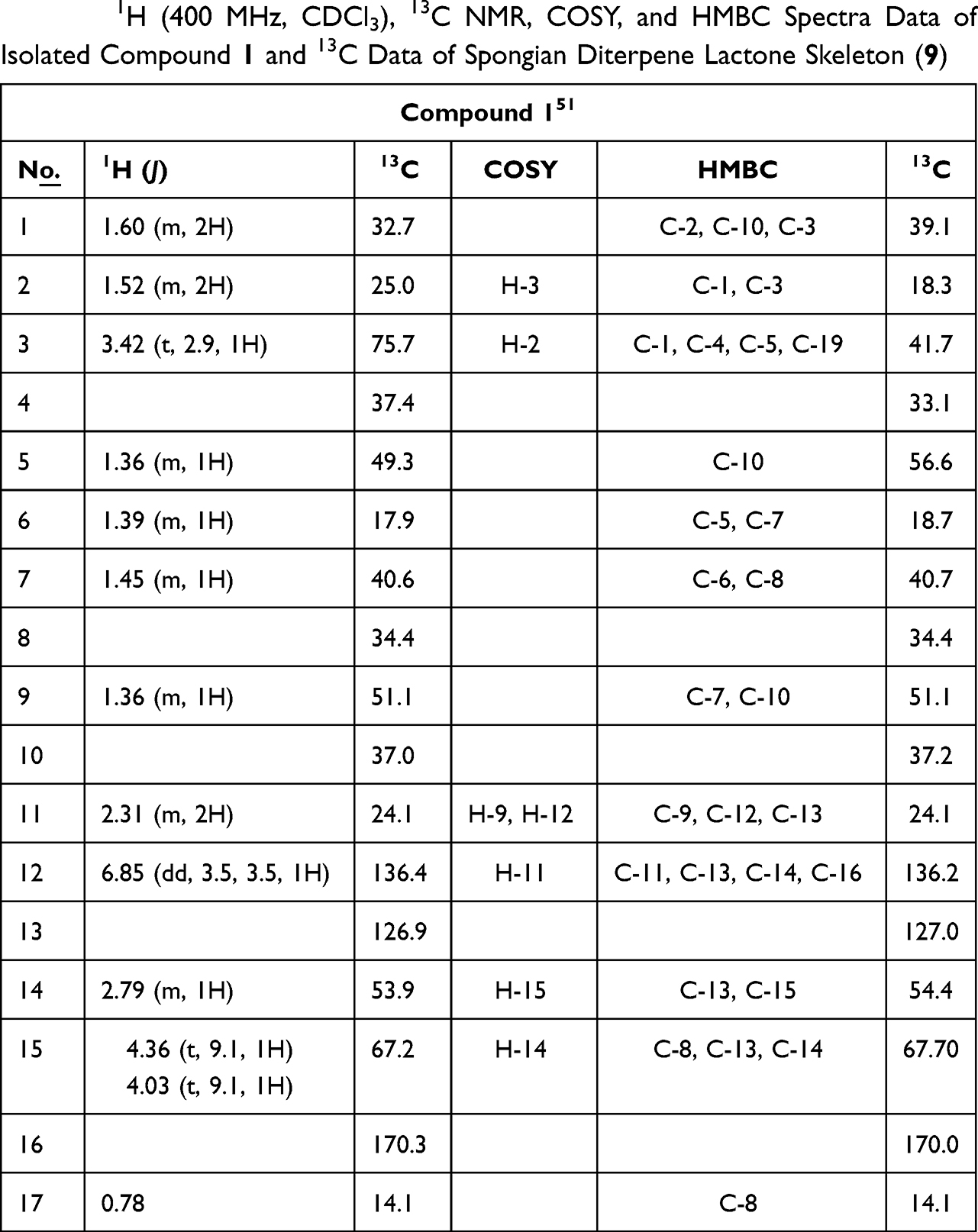 |
Table 1 1H (400 MHz, CDCl3), 13C NMR, COSY, and HMBC Spectra Data of Isolated Compound 1 and 13C Data of Spongian Diterpene Lactone Skeleton (9) |
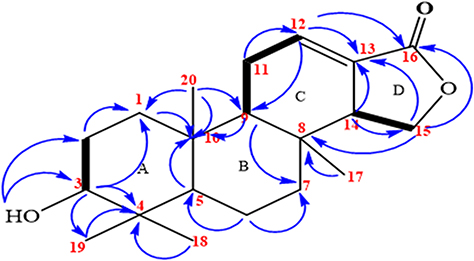 |
Figure 2 Key 1H-1H COSY and HMBC correlations of compound 1. Bolded bonds – 1H-1H COSY, arrows – HMBC. |
Spectra of Known Compounds (2–8)
The spectra data used to establish the structures of isolated compounds 2–8 in this work are tabulated and indicated in Supplementary Tables 1–7, respectively.
Compound 2 (β-Sitsterol)
Pale yellow solid, 1H NMR (400 MHz, CDCl3), δ 5.33 (bs, 1H, H-6), 3.50 (m, 1H, H-3), 2.31 (d, 2H, H-4), 2.04 (m, 2H, H-7), 0.99 (m, 3H, H-21), 0.86 (m, 3H, H-19), 0.83 (m, 3H, H-26), 0.81 (m, 3H, H-29), 0.79 (m, 3H, H-27), and 0.66 (s, 3H, H-18). 13C NMR (101 MHz, CDCl3) δ140.8 (C-5), 121.8 (C-6), 71.9 (C-3), 56.8 (C-14), 56.1 (C-17), 50.2 (C-9), 45.9 (C-24), 42.4 (C-13), 42.2 (C-4), 39.8 (C-12), 37.2 (C-1), 36.2 (C-20), 34.0 (C-22), 31.6 (C-2), 32.0 (C-7), 32.0 (C-8), 36.6 (C-10), 29.2 (C-25), 28.2 (C-16), 26.1 (C-23), 24.8 (C-15), 23.1 (C-28), 21.1 (C-11), 19.9 (C-26), 19.5 (C-27), 19.1 (C-19), 18.8 (C-21), 12.0 (C-29), and 11.9 (C-18).
Compound 3 (ε-Viniferin)
Brown solid, 1H NMR (400 MHz, MeOD) δ 7.16 (d, J = 8.6Hz, 2H, H-2/6), 7.06 (d, J = 8.7 Hz, 2H, H-2’/6’), 6.84 (d, J = 16.4 Hz, 1H, H-7’), 6.79 (d, J= 8.6Hz, 2H, H-3/5), 6.67 (d, J = 8.8 Hz, 2H, H-3’/5’), 6.63 (m, H-14’), 6.59 (d, J = 16.5 Hz, 1H, H-8’), 6.25 (m, 12’), 6.19 (d, J = 3.0 Hz, 2H, H-10/14),6.18 (d, J = 2.0 Hz, 1H, H-12), 5.39 (d, J = 6.6 Hz, 1H, H-7), and 4.37 (d, J = 6.6 Hz, 1H, H-8).13C NMR (101 MHz, MeOD) 162.7 (C-11’), 159.9 (C-11/13), 159.7 (C-13’), 159.4 (C-4), 158.3 (C-4’), 147.3 (C-9), 136.8 (C-9’), 133.8 (C-1), 131.1 (C-1’), 130.3 (C-7’), 128.8 (C-2’/6’), 128.2 (C-2/6), 123.6 (C-8’), 120.0 (C-10’), 116.3 (C-3/5), 116.3 (C-3’5’), 107.6 (C-10/14), 104.3 (C-14’), 102.2 (C-12), 96.8 (C-12’), 94.8 (C-7), and 58.2 (C-8).
Compound 4 (Trans-Resveratrol)
Brown crystal, 1H NMR (400 MHz, MeOD), 6.04 (d, J = 8.0 Hz, 2H, H-2/6), 5.65 (d, J = 16.1 Hz, 1H, H-1’), 5.49 (d, J = 16.1 Hz, 1H, H-2’), 5.46 (d, J = 8.0 Hz, 2H, H-3/5), 5.16 (s, 2H, 2’/6’), 4.87 (s, 1H, H-4’), 8.02 (4-OH), and 7.85 (3’/5’-OH). 13C NMR (101 MHz, MeOD), 159.5 (C-3’/5’), 158.2 (C-4), 141.2 (C-1’), 130.3 (C-1), 129.3 (C-1’), 128.7 (C-2/6), 126.9 (C-2’), 116.4 (C-3/5), 105.7 (C-2’/6’), and 102.6 (C-4’).
Compound 5 (Gnetin H)
Brown solid, 1H NMR (400 MHz, MeOD) δ 7.15 (d, J = 8.6 Hz, 2H, H-2/6), 7.04 (d, J = 8.8 Hz, 2H, H-2’/6’), 6.92 (d, J = 8.4 Hz, 2H, H-2’/6’), 6.82 (d, J = 16.4 Hz, 1H, H-7’), 6.77 (d, J = 8.6 Hz, 2H, H-3/5), 6.72 (d, J = 8.6 Hz, 2H, H-3’/5’), 6.66 (d, J = 8.6 Hz, 2H, H-3’/5’, 6.58 (d, J = 16.9 Hz, 1H, H-8’), 6.24 (dd, J = 9.3, 3.1 Hz, 1H, H-12), 6.18 (dd, J = 6.8, 2.3 Hz, 1H, H-12’), 6.18 (s, 1H, H-12’), 6.17 (d, J = 2.3 Hz, 2H, H-10/14), 5.93 (d, J = 2.2 Hz, 2H, H-10’/14’), 5.37 (d, J = 6.6 Hz, 1H, H-7), 5.19 (d, J = 6.2 Hz, 1H, H-7’), 4.35 (d, J = 6.6 Hz, 1H, H-8), and 3.79 (d, J = 6.4 Hz, 1H, H-8’).13C NMR (101 MHz, MeOD) δ 162.80 (C-13’), 160.07 (C-11’), 159.77 (C-11), 159.56 (C-13), 158.55 (C-11’/13’), 158.42 (C-4), 157.84 (C-4’), 158.42 (−4’), 147.37 (C-9), 147.35 (C-9’), 137.8 (C-1’), 136.9 (C-1), 133.9 (C-9’), 133.8 (C-1’), 130.4 (C-8’), 130.5 (C-2’/6’), 128.3 (C-2’/6’), 127.6 (C-2/6), 123.7 (C-7’), 120.3 (C-14’), 120.1 (C-10’), 116.4 (C-3, 5), 115.8 (C-3’/5’), 116.2 (C-3’/5’), 106.7 (C-10’/14’), 107.0 (C-10/14), 101.5 (C-12), 101.8 (C-12’), 96.6 (C-12’), 94.9 (C-7), 94.8 (C-7’), 58.3 (C-8), and 57.7 (C-8’).
Compound 6 (Tricuspidatol A)
1H NMR (400 MHz, MeOD) δ 7.27 (d, J = 8.5 Hz, 4H, H-2/6’ 2’/6’), 6.76 (d, J = 8.4 Hz, 4H, H-3/5, 3’/5’), 6.23 (bs, 2H, H-12/12’), 6.02 (s, 4H, H-10/14, 10’/14’), 5.40 (d, J = 6.0 Hz, 2H, H-7/7’), and 3.58 (d, J = 4.4 Hz, 2H, H-8/8’). 13C NMR (101 MHz, MeOD) δ 158.8 (C11/13, 11’/13’), 158.1 (C4, 4’), 142.2 (C9, 9’), 133.9 (C1, 1’), 128.8 (C2/6, 2’/6’), 116.1 (C3/5, 3’/5’), 109.0 (C10/14, 10’/14’), 101.8 (12, 12’), 85.9 (C7, 7’), and 59.0 (8, 8’).
Compound 7 (ε-Viniferin Diol)
1H NMR (400 MHz, MeOD), δ7.14 (d, J = 8.6 Hz, 2H, H-2’/6’), 6.93 (d, J = 8.5 Hz, 2H, H-2/6), 6.71 (d, J = 8.6 Hz, 2H, H-3’/5’), 6.58 (d, J = 8.5 Hz, 2H, H-3/5), 6.23 (d, J = 2.2 Hz, 1H, H-12’), 6.20 (d, J = 2.2 Hz, 1H, H-14’), 6.09 (brs, 1H, H-12), 5.93 (d, J = 2.2 Hz, 2H, H-10/14), 5.18 (d, J = 6.2 Hz, 1H, H-7), 4.07 (d, J = 6.6 Hz, 1H, H-7’), 4.07 (d, J = 6.6 Hz, 1H, H-8’), and 3.78 (d, J = 6.2 Hz, 1H, H-8). 13C NMR (101 MHz, MeOD) δ 162.7 (C-11’), 160.1 (C-11), 159.5 (C-13’), 158.4 (C-4), 157.8 (C-4’), 147.3 (C-9), 146.8 (C-9’), 137.8 (C-1), 133.8 (C-1’), 131.6 (C-2/6), 128.5 (C2’/6’), 126.6 (C-10’), 116.3 (C-3/5), 115.9 (C-3’/5’), 107.5 (C-14’), 107.3 (C-10/14), 101.8 (C-12), 96.6 (C-12’), 94.8 (C-7), 73.2 (H-7’), 72.9 (C-8’), and 57.5 (C-8).
Compound 8 (Parthenostilbenin B)
1H NMR (400 MHz, MeOD) δ7.30 (d, J = 8.6 Hz, 2H, H-2/6), 6.97 (d, J = 8.6 Hz, H-2’/6’), 6.79 (d, J = 8.6 Hz, H-3/5), 6.71 (d, J = 8.6 Hz, H-3’/5’), 6.25 (d, J = 2.4 Hz, H-14), 6.17 (d, J = 2.4 Hz, H-12), 6.14 (d, J = 2.0 Hz, H-12), 6.05 (s, H-10’/14’), 5.43 (d, J = 6.6 Hz, H-7), 4.20 (dd, J = 6.6, 6.3 Hz, H-8’), 4.10 (d, J = 6.5 Hz, H-7’), and 3.59 (dd, J = 6.6, 6.3 Hz, H-8). 13C NMR (101 MHz, MeOD) δ 131.2 (C-1), 128.8 (C-2/6), 116.1 (C-3/5), 157.4 (C-4), 85.9 (C-7), 59.1 (C-8), 142.2 (C-9), 125.6 (C-10), 158.1 (C-11), 103.6 (C-12), 159.4 (C-13), 106.9 (C-14), 133.9 (C-1’), 128.8 (C-2’/6’), 116.1 (C-3’/5’), 156.5 (C-4’), 55.6 (C-7’), 61.0 (C-8’), 150.3 (C-9’), 109.0 (C-10’/14), 158.9 (C-11’/13’), 101.8 (C-12’), and 59.1(OCH3).
Antibacterial Activity
The in vitro antibacterial activities of each of the isolated compounds (1–8) from the roots of C. cyphopetalum were tested against E. coli, S. aureus, and P. aeruginosa pathogens at concentrations of 50, 100, 300, 500, and 1000 μg/mL. The obtained mean inhibition zone diameters (in mm) are presented in Table 2. Compounds 3–8 exhibited growth inhibition (>7 mm inhibition zone diameter) against E. coli at all tested concentrations. The highest mean inhibitory value of 10.45 ± 0.75 mm was exhibited by ε-viniferin (3), followed by gnetin H (5) (10.02 ± 0.13 mm) and parthenostilbenin B (8) (9.52 ± 0.19 mm) against E. coli at concentration of 1000 μg/mL. Compounds 6,7, 4, and 1 exhibited moderate inhibitory effects against against E. coli (8.92 ± 0.04, 8.83 ± 0.02, 8.72 ± 0.0, and 7.32 ± 1.9 mm, respectively) at concentration of 1000 μg/mL. At the lowest concentration of 50 μg/mL, the highest mean inhibition zone against E. coli (8.55 ± 0.45 mm) and S. aureus (9.30 ±1.39 mm) was recorded by compound 3. Compound 5 scored better mean inhibition zone (12.55 ± 0.65 mm) than chloramphenicol (11.76 ± 0.77 mm at 30 μg/mL) against the gram-negative bacterium P. aeruginosa. The bacterium P. aeruginosa was found to be susceptible to all compounds at the concentration of 1000 μg/mL. Gnetin H (5) displayed the largest mean inhibition zone of 12.55 ± 0.65 mm at 50 μg/mL, which is better compared to the inhibition zone displayed by chloramphenicol (11.76 ± 0.77 mm at 30 μg/mL) followed by ε-viniferin diol (7, 9.42 ± 0.42 mm) at 50 μg/mL. Against the gram positive bacterium, S. aureus, the highest activity was recorded by gnetin H (5), followed by ε-viniferin (3), β-sitosterol (2), tricuspidatol A (6), and trans-resveratrol (4) with inhibition zone diameter of 12.45 ± 0.37 mm, 12.22 ± 0.74 mm, 10.93 ± 1.54 mm, 10.79 ± 0.24 mm, 8.62 ± 1.5 mm, respectively, at the concentration of 1000 μg/mL. Compounds 1, 7, and 8 exhibited no activity against S. aureus at all concentrations. At the lowest concentration of 50 μg/mL, except ε-viniferin (3) (9.30 ±1.39 mm) and tricuspidatol A (6) (7.36 ± 0.20 mm), all tested compounds indicated no sign of activity (<7 mm inhibition zone width) against the bacterium S. aureus. Over all, compounds 3–8 exhibited antibacterial activity against E. coli at all tested concentrations, which is in moderate compared to chloramphenicol. Similarly, all the compounds displayed dose-dependent pattern inhibitory activity at all concentration against the bacterium P. aeruginosa, except β-sitosterol (2) at 50 and 100 μg/mL. Gnetin H (5) displayed inhibitory activity higher than that of the standard, chloramphenicol (11.76 ± 0.77 mm at 30 μg/mL) against P. aeruginosa, at all tested concentrations. Against the gram-positive bacterium, S. aureus, gnetin H (5), and ε-viniferin (3) showed growth inhibitory activity in good comparison with that of the standard, chloramphenicol (15.74 ± 1.02 mm). The result of the present finding generally indicates that the isolated compounds displayed an ability to inhibit the growth of the Gram-negative bacterial strains at all tested concentration. Compounds 3 and 6 displayed growth inhibition against Gram-positive bacterium at all concentrations, whereas compounds 2, 4 and 5 displayed promising activity at higher concentrations.
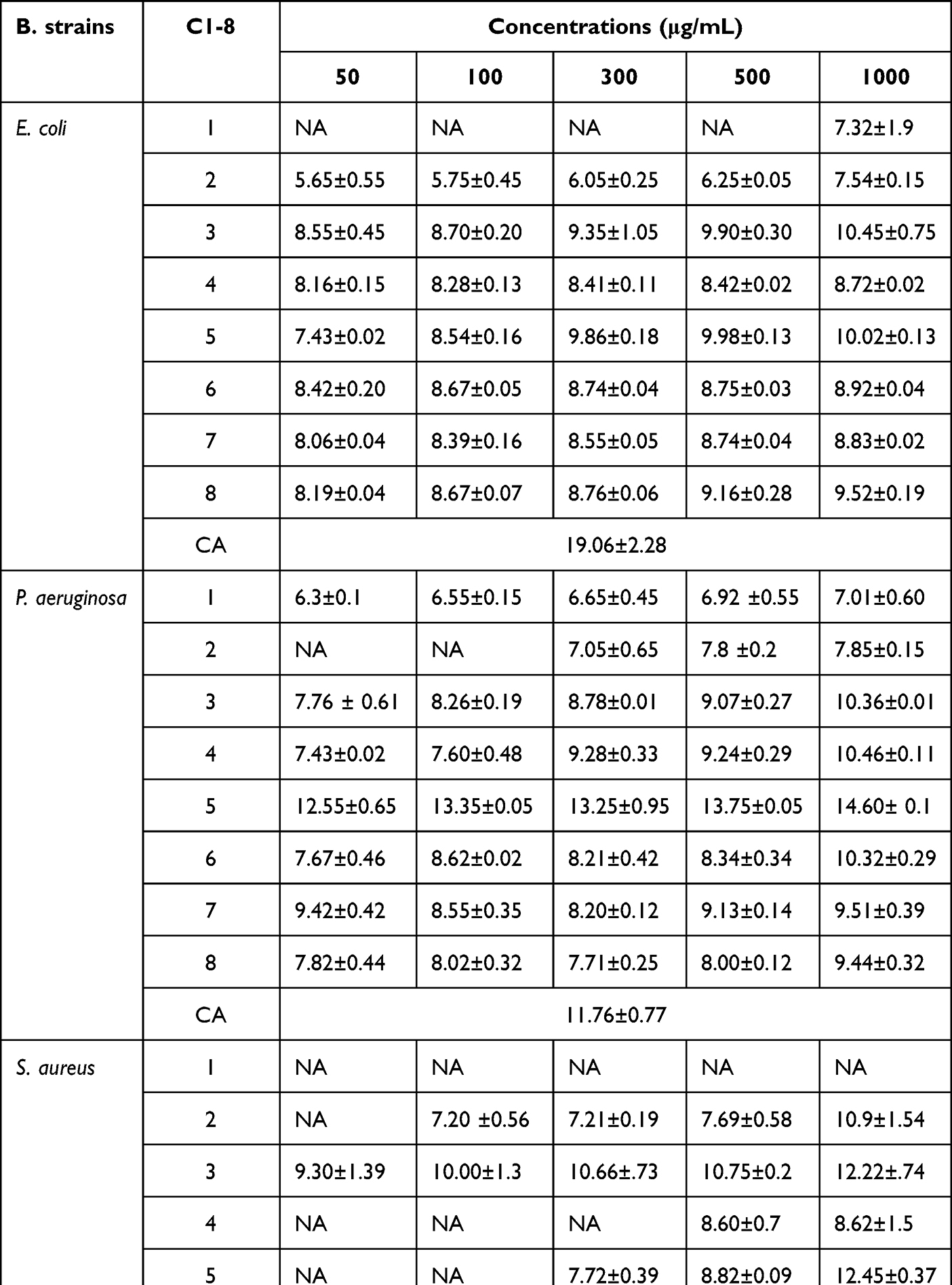 |
Table 2 Antibacterial Activity Inhibition Zone (Mean ± SD, in Mm) of Isolated Compounds Against Standard E. coli, S. aureus, and P. aeruginosa Bacterial Strains |
Molecular Docking Binding Analysis of Isolated Compounds
The molecular docking study was carried out to assess the binding affinity and binding interaction of isolated compounds 1–6 toward target proteins E. coli DNA gyrase B (PDB ID 6F86), PqsA (PDB ID: 5OE3), and S. aureus Pyruvate Kinase (PDB ID: 3T07). The docked compounds (1–6) displayed binding affinity in the range of −6.6 to −7.6 kcal/mol, −6.4 to −8.8 kcal/mol, and −4.4 to −5.8 kcal/mol toward DNA gyrase B, PqsA, and S. aureus PK, respectively (Tables 3–5). All tested compounds (1–6) recorded better binding affinities than the standard drug chloramphenicol (−6.4 kcal/mol) against the protein DNA gyrase B (−6.6 to −7.6 kcal/mol). The highest binding affinity (−7.6 kcal/mol) was recorded by compound 3 followed by compounds 2 and 5 (each −7.4 kcal/mol). The ligand–PqsA interaction of compounds 2–6 displayed binding affinities (−7.0 to −8.8 kcal/mol) equal and/or higher than chloramphenicol (−7.0 kcal/mol). Compound 5–PqsA interaction showed the highest binding affinity (−8.8 kcal/mol), followed by compounds 4 (−7.5 kcal/mol) and 6 (−7.2 kcal/mol). The binding affinities of the ligand–S. aureus PK interactions (−4.6 to −5.8 kcal/mol) were found to be equal to or better than chloramphenicol (−4.6 kcal/mol), except compound 4 (−4.4 kcal/mol). Of these, the strongest binding affinity was displayed by compounds 5 (−5.8 kcal/mol) and 3 (−5.4 kcal/mol). In general, the higher binding affinities results of compound 3 to DNA gyrase (−7.6 kcal/mol), and 5 to PqsA (−8.8 kcal/mol) and S. aureus PK (−5.8 kcal/mol) agrees with the in vitro antibacterial activity result.
 |
Table 3 Molecular Docking Value of Isolated Compounds (1–6) Against E. coli DNA (PDB ID: 6F86) |
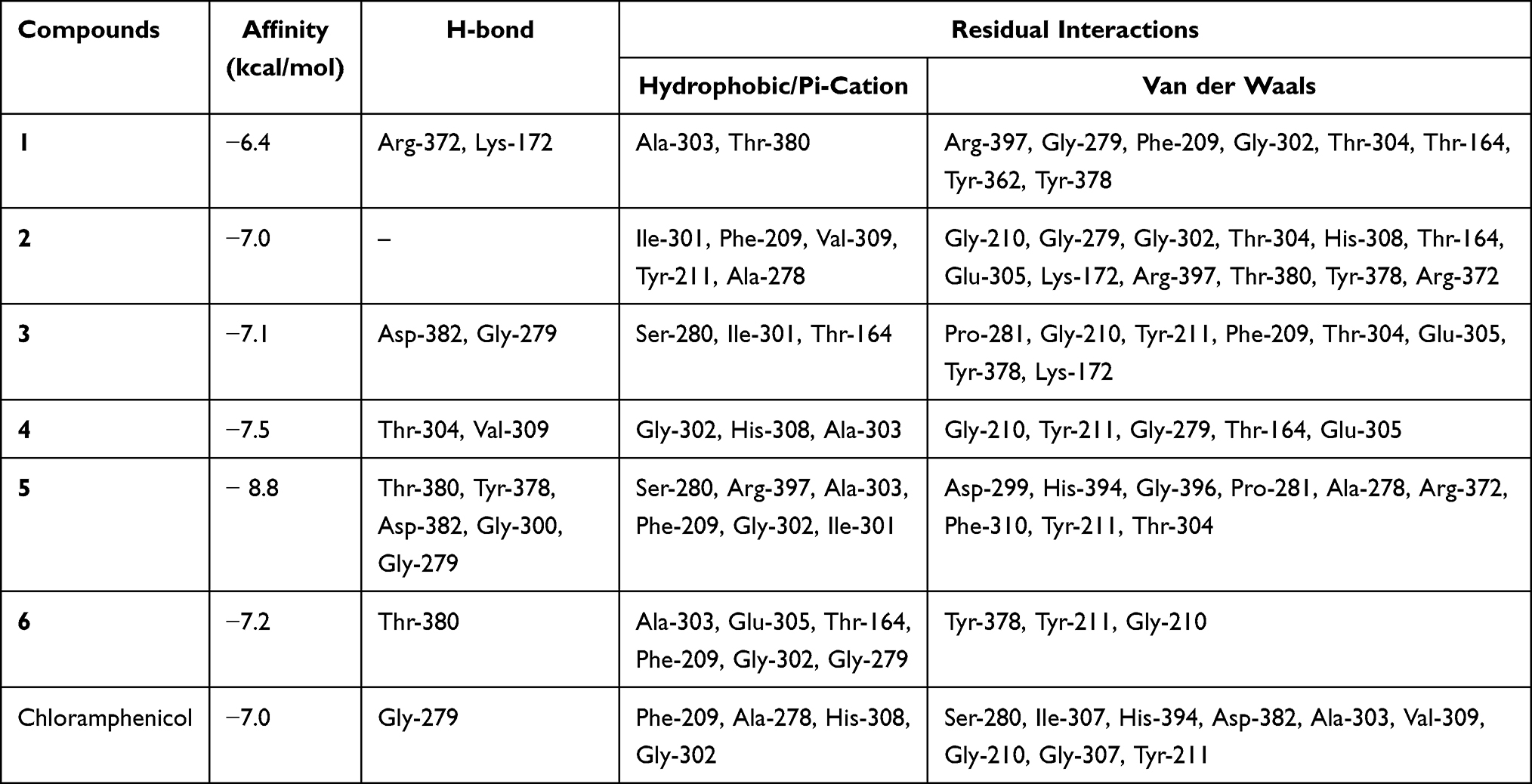 |
Table 4 Molecular Docking Value of Isolated Compounds (1–6) Against N-Terminal Domain of PqsA in Complex with Anthraniloyl-AMP (PDB ID: 5OE3) |
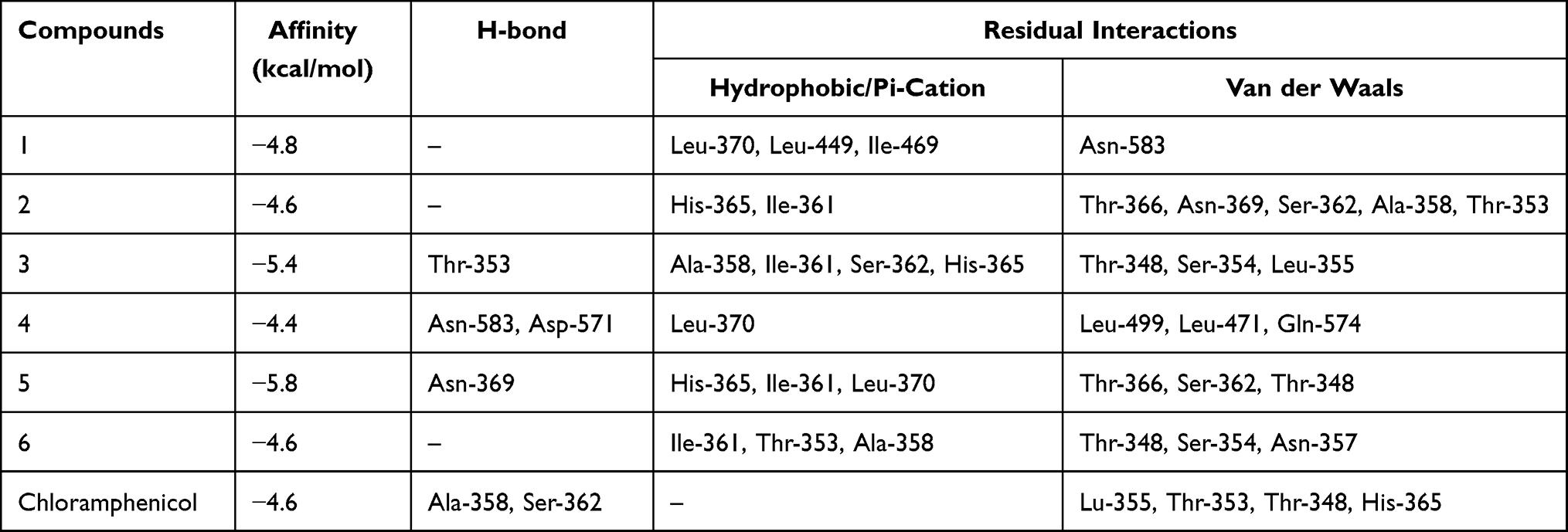 |
Table 5 Molecular Docking Value of Isolated Compounds (1–6) Against S. aureus PK in Complex with a Naturally Occurring Bis-Indole Alkaloid (PDB ID: 3T07) |
In the docking analyses, amino acid residues that are involved in hydrogen bonds, hydrophobic, and van der Waals interactions with the ligands using AutoDock Vina were also mapped (Figures 3–5 and Tables 3–5). According the result, compounds 3–5 displayed hydrogen bond, hydrophobic, and van der Waals interactions through various amino acid resides in each of its ligand-protein complexes. Compounds 1 and 2 displayed hydrogen bond interactions only with protein PqsA (Arg-372 and Lys-172) and E.coli (Glu-50), respectively. Similarly, compound 6 showed H-bond interactions only with DNA gyrase B (Gly-77 and Asn-46 of), and PqsA (Thr-380. This suggests that the scored binding affinities of compounds 1, 2, and 6 accounted for the hydrophobic and van der Waals interactions in each of the ligand-protein complexes, where no hydrogen bond interactions were observed. The ligand binding affinity in interaction of compound 3–gyraseB complex (−7.6 kcal/mol) was stabilized by one hydrogen bond interaction at amino acid residue Asp-73, five hydrophobic interaction at Ile-94, Asn- 46, Ile-78, Arg-76 and Pro-79, and two van der Waals interaction at Ala-47 and Gly-77. The binding affinity in compound 5–PqsA interaction (binding energy of −8.8 kcal/mol) was promoted by hydrogen bond at Thr-380, Tyr-378, Asp-382, Gly-300 and Gly-279, hydrophobic, at Ser-280, Arg-397, Ala-303, Phe-209, Gly-302 and Ile-301, and van der Waals interaction at Asp-299, His-394, Gly-396, Pro-281, Ala-278, Arg-372, Phe-310, Tyr-211 and Thr-304. The interaction of compound 5 with PqsA demonstrated a large number of hydrogen bonds, which boosted its binding affinity. Likewise, the docking result revealed that the recorded binding affinity of compound 5 with the protein S. aureus (−5.8 kcal/mol) was mediated by hydrogen bond, hydrophobic, and van der Waals interactions that are formed across various amino acids.
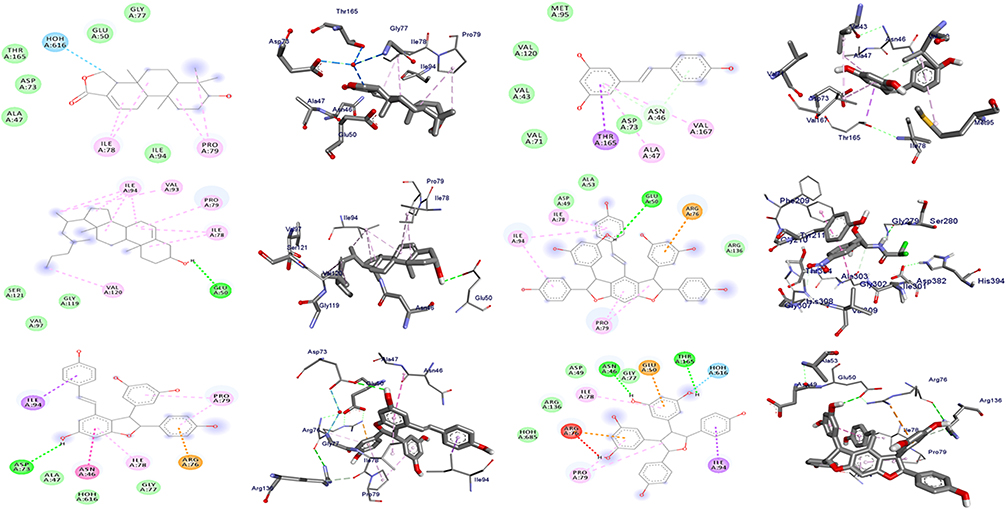 |
Figure 3 The 2D and 3D binding interactions of compounds (1–6) (from top right to bottom left) against DNA gyrase B (PDB ID: 6F86). |
 |
Figure 4 The 2D and 3D binding interactions of compounds (1–6) (from top right to bottom left) against PqsA (PDB ID: 5OE3). |
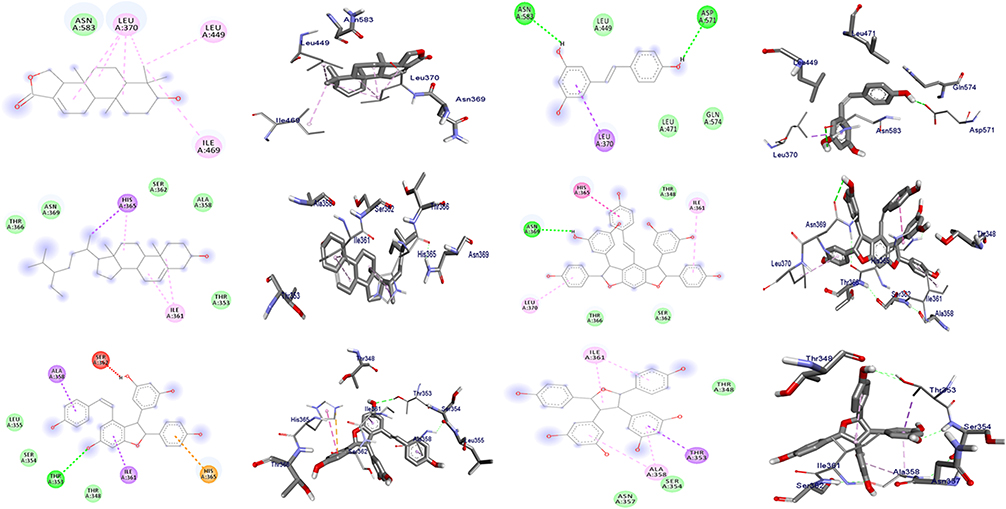 |
Figure 5 The 2D and 3D binding interactions of compounds (1–6) (from top right to bottom left) against S. aurues PK (PDB ID: 3T07). |
Overall, the highest in silico docking binding affinities of compound 3 (−7.6 kcal/mol) toward protein DNA gyrase B and that of compound 5 towards PqsA (−8.8 kcal/mol) and protein S. aureus PK (−5.8 kcal/mol) agree with the in vitro activity displayed by compound 3 against E.coli and compound 5 against P. aeruginosa and S. aureus. In addition, the docked compounds (1–6) scored binding affinities comparable with the standard chloramphenicol. The binding affinity, H-bond and residual amino acid interactions of the six compounds and chloramphenicol are summarized in Tables 3–5. The binding interactions of compounds 1–6 and the standard, chloramphenicol against targeted proteins are displayed in Figures 3–6. Ribbon model shows the binding pocket structure of target proteins with the compounds. Hydrogen bonds between compounds and amino acids are shown as green dash lines, and hydrophobic interactions are shown as pink lines.
 |
Figure 6 The 2D and 3D binding affinity of Chloramphenicol against DNA gyrase B (PDB ID: 6F86), PqsA (PDB ID: 5OE3) and S. aurues PK (PDB ID: 3T07), Left to right. |
In silico Drug-Likeness Prediction
The in silico drug-likeness of the isolated compounds (1–6) was determined based the concept of the Lipinski’s rule of five.26 The chemical structures of compounds 1–6 were converted to their corresponding canonical simplified molecular-input line-entry system (SMILE) and submitted to the SwissADME tool to generate the physicochemical and pharmacokinetic properties of the compounds. The SwissADME prediction outcome showed that compounds 1, 3 and 4 satisfy Lipinski’s rule of five with zero violations (Table 6). All the studied compounds (1–6) recorded lipophilicity (iLogP) value of less than five (range from 1.79 to 4.79), implying their optimal lipophilicity. The number of rotatable bond (NRB) values for the compounds are less than 10, which indicate the conformation stability of the compounds.52 The total polar surface (TPSA) influences permeability and bioavailability of a molecule. Except for compound 5 (160.1), the calculated TPSA value of the tested compounds is found to be less than 140 Å2. The TPSA values of compounds 1, 2 and 3 (46.5, 20.2, and 60.7, respectively) are found to be less enough than the cut-off value (140 Å2), which indicate their excellent absorption in the intestine.
 |
Table 6 In silico Drug-Likeness Predictions of Isolated Compounds Computed by SwissADME |
ADMET Properties
In silico screening of pharmacological properties (ADME) of compounds 1–6 was performed with the SwissADME tool.27 The main ADME parameters concerning pharmacokinetic behavior of the tested compounds are presented in Table 7. The log Kp value of the studied compounds scored within the range of −2.2 to −6.14 cm/s. The more negative the log Kp (with Kp in cm/s), the less skin permeant is the molecule.27 The SwissADME prediction indicated that compounds 1, 3 and 4 have high GI absorption, and compounds 1 and 4 have blood–brain barrier (BBB) permeation. High gastrointestinal (GI) and blood–brain barrier (BBB) permeation can be considered as favorable absorption and distribution properties of drug molecules.52 The ADME prediction outcome also displayed that all compounds under investigation are found to be non-substrate of permeability glycoprotein (P-gp), and hence non-inhibitors of most of the selected cytochromes (CYP). However, compound 4 inhibited CYP1A2, CYP2C9 and CYP3A4, compounds 1 and 3 each inhibited CYP2C9 and CYP3A4, and compound 6 inhibited CYP3A4. The toxicity profile test showed that the LD50 and toxicity class values indicated that none of the compounds has acute toxicity (Table 8). Organ toxicity prediction results indicated that none of the studied compounds (1–6) have hepatotoxicity, mutagenicity, and cytotoxicity. Compounds 1–3 are found to be immunotoxicity. Compound 5 showed carcinogenicity and immunotoxicity. Based on the presented toxicity data, the studied compounds may be the good candidates in this investigation based on ADMET prediction analysis.
 |
Table 7 ADME Predictions of Compounds 1–6, Computed by SwissADME and PreADMET Tool |
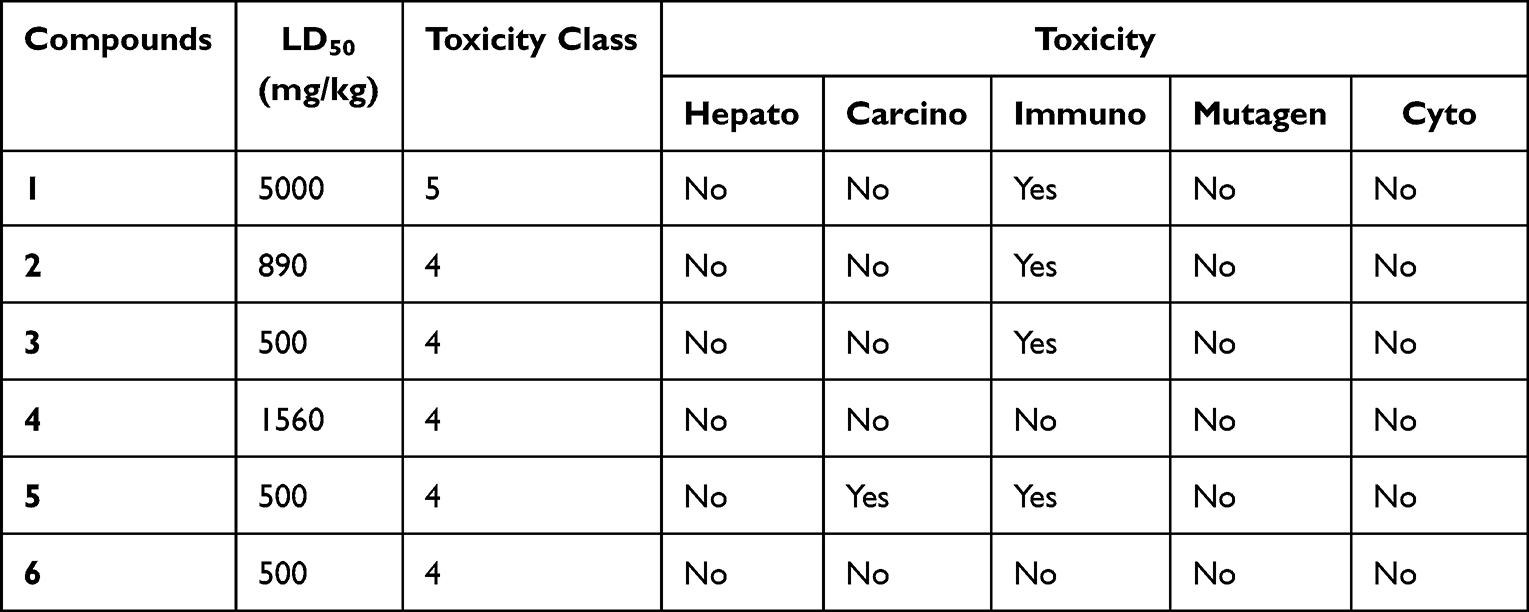 |
Table 8 Prediction of Toxicity of Compounds Computed by ProTox-II |
Radical Scavenging Activity
The radical scavenging activity of each compounds isolated from the root of C. cyphopetalum was evaluated using the DPPH assay, a simple method to evaluate antioxidant activities by measuring absorbance at 517 nm due to the formation of stable DPPH radical.30 At all the tested concentrations of each compound, the purple color of the DPPH solution was changed to yellow color indicating potential as radical scavenger of the compounds. The absorbance of the DPPH radical at 517 nm was also reduced. The DPPH assay result indicated that the compounds showed activity in dose-dependent manner (Table 9). All the tested compounds showed moderate-to-high radical scavenging activity ranging from 55.69% to 76.87%. The highest percentage inhibition was recorded by compound 4 (76.87%) at 12.5 μg/mL, followed by compound 3 (74.30%), and 8 (73.32%), with IC50 value of 0.052 μg/mL, 0.017 μg/mL, and 0.025μg/mL, respectively. Compounds 1 (55.69%) and 2 (59.61%) recorded low radical scavenging activity compared to other compounds in this report, which can be justified due to the higher polarity of the stilbene families (Figure 2).
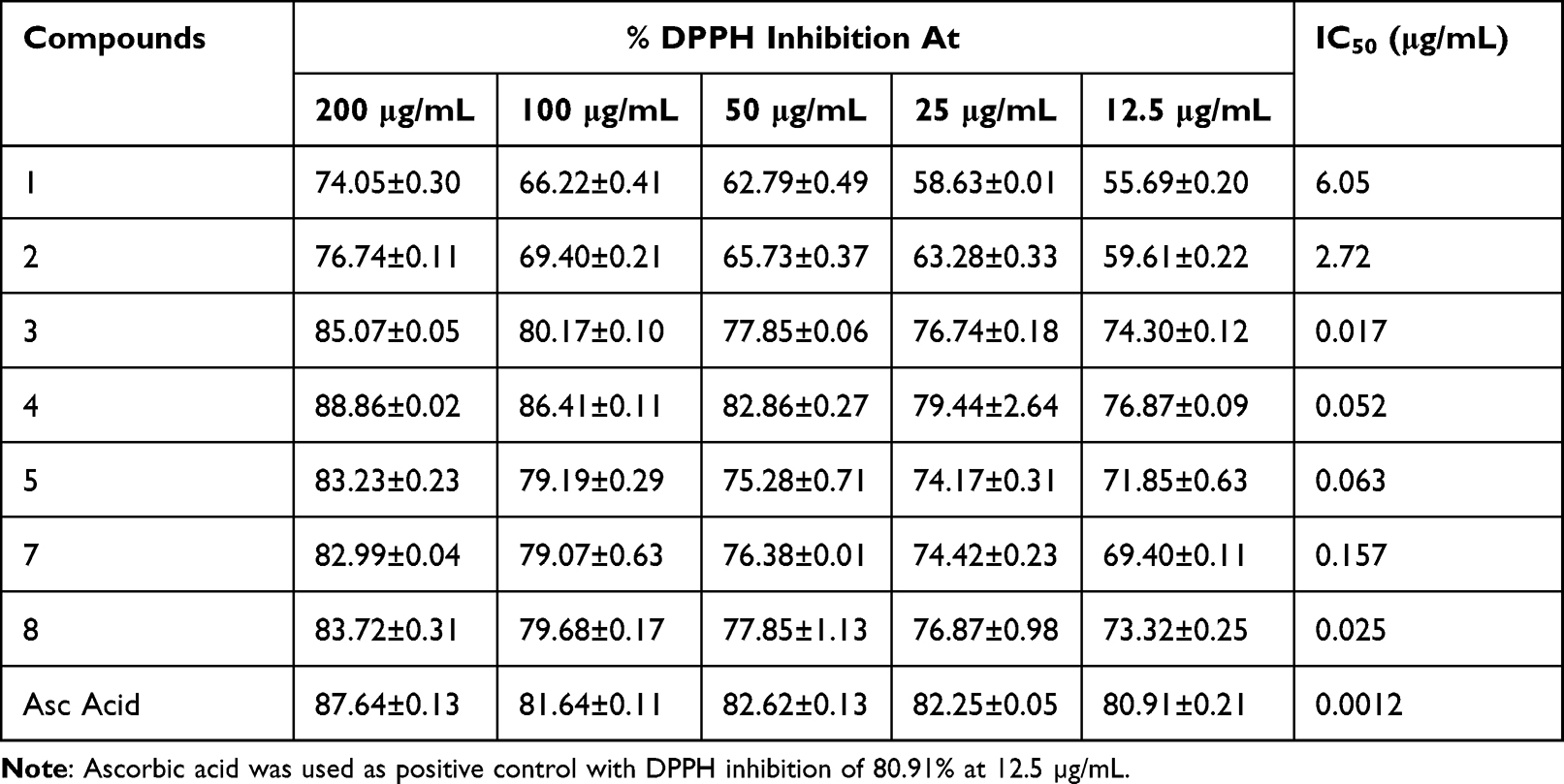 |
Table 9 DPPH Radical Inhibition of Compounds Isolated from the Root of C. cyphopetalum |
DFT Analysis
Fortier molecular orbitals play a crucial role in determining the electron transition, optical properties, and chemical reactivity of compound.32 The energies of frontier molecular orbitals are listed in Table 10. The HOMO–LUMO energy gap is related to the occurrence of intramolecular charge transfer within the system and can be used as an indicator of the biological activity of the system. A large HOMO–LUMO energy gap indicates that greater excitation energies are required for reaction, thus the harder and most stable (less reactive) the molecules are. The calculated energy gaps (eV) of compounds 1–6 were found to be 5.760, 6.181, 4.034, 3.939, 4.125 and 5.502, respectively (Table 10). Compound 4 recorded the lowest energy gap (3.939 eV) indicating its good chemical reactivity, in agreement with its strong scavenging activity. The lowest chemical hardness (1.970 eV) or highest chemical softness (0.254 eV) of compound 4 showed that also indicate that its better reactive that all the studied compounds.
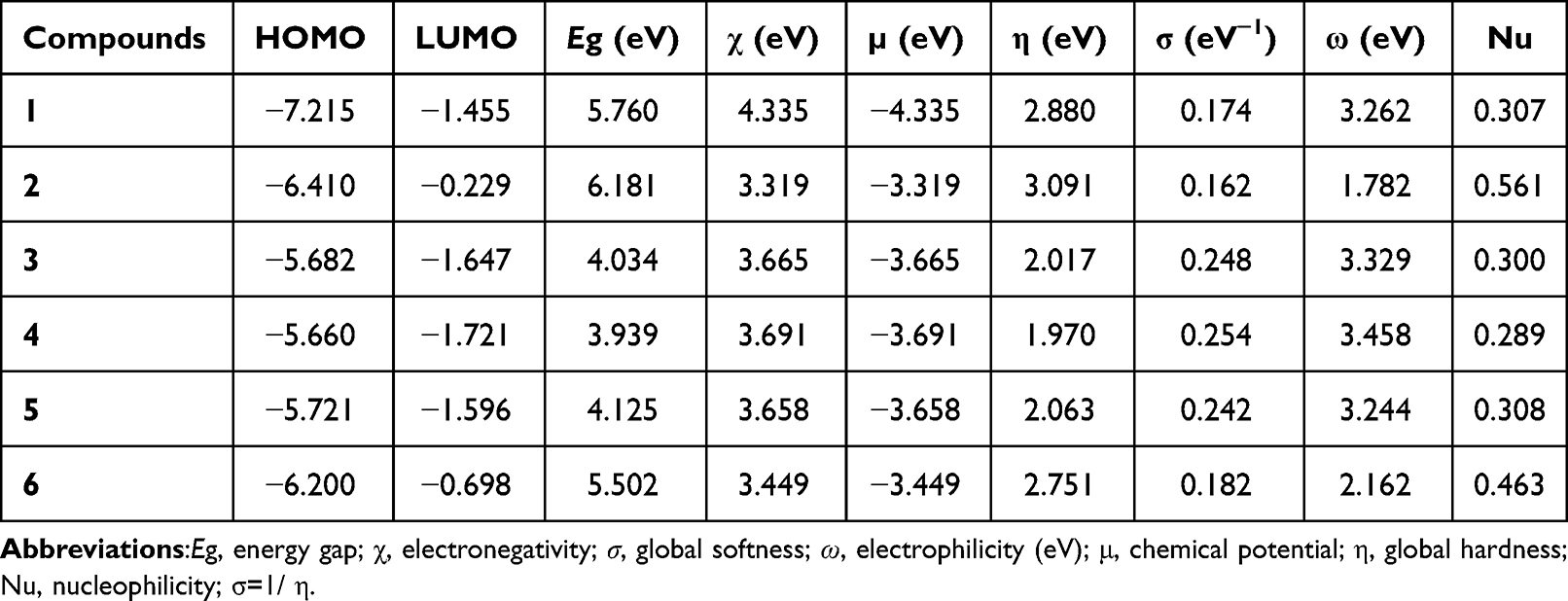 |
Table 10 Quantum Chemical Descriptors of the Ligand and Its Metal Complexes |
Conclusion
In this study, eight compounds were identified from the roots extracts of C. cyphopetalum of which 3-hydroxyisoagatholactone (1) is a new hydroxyl-spongiane diterpenoid lactone. The in vitro antibacterial activity result shows, among the tested compounds, compound 3 exhibited high antibacterial activity against E. coli (8.55 ± 0.45 mm) and S. aureus (9.30 ±1.39 mm), at the lowest concentration of 50 μg/mL, which is low in comparison to the positive control, but undeniably important. Compound 5 was found to be better therapeutic agent (12.55 ± 0.65 mm) than the standard, chloramphenicol (11.76 ± 0.77 mm at 30 μg/mL) against P. aeruginosa. The docking result revealed that compounds 3 (against DNA gyrase) and 5 (against PqsA and PK) displayed better docking scores than chloramphenicol. Compounds 1, 3 and 4 satisfy Lipinski’s rule of five with zero violations. Organ toxicity prediction results indicated that none of the studied compounds (1–6) have hepatotoxicity, mutagenicity, and cytotoxicity. The DPPH assay result indicated that, at concentration of 12.5 μg/mL, compounds 4, 3 and 8 (in increasing order) showed good antioxidant activity compared to that of ascorbic acid. The DFT result showed that compound 4 possesses the lowest energy gap (3.939 eV) indicating its good chemical reactivity, in agreement with its strong scavenging activity. The in silico study analysis and in vitro activity results suggest that compounds 3 and 5 could be considered as potential antibacterial agents against E. coli, P. aeruginosa and S. aureus. Therefore, the in vitro antibacterial activity and molecular docking analysis suggest the potential use of the isolated compounds as antibacterial agents that corroborates the traditional uses of the roots of C. cyphpetalum.
Data Sharing Statement
The NMR spectra used to establish the structures of the novel compound isolated in this work and the tabulated spectra of the known compounds are depicted as Supplementary information. Additionally, further data can be obtained from the corresponding author.
Acknowledgments
We thank Adama Science and Technology University for the research fund with grant number: ASTU/AS-R/003/2020. We are also grateful to the World Academy of Sciences (TWAS) and the United Nations Educational, Scientific and Cultural Organization (UNESCO) for financing this research with funds allocated to the AD research team under the TWAS Research Grant RGA No. 20-274 RG/CHE/AF/AC_G -FR3240314163. Computational resources were supplied by the project ‘e-Infrastruktura CZ’ (e-INFRA CZ ID: 90140) supported by the Ministry of Education, Youth and Sports of the Czech Republic.
Funding
TWAS and ASTU funded part of the work (Grant No. 20-274 RG/CHE/AF/AC_G- FR3240314163 and ASTI/AS-R/003/2020, respectively).
Disclosure
The authors declare that they have no conflicts of interest in relation to this work.
References
1. Sousa F, Figueiredo E, Smith GF. The genus Cyphostemma (Planch.) Alston (Vitaceae) in Angola The genus Cyphostemma (Planch.) Alston (Vitaceae) in Angola. Bradleya. 2011;29(January):79–92. doi:10.25223/brad.n29.2011.a10
2. Teklay A, Abera B, Giday M. An ethnobotanical study of medicinal plants used in Kilte Awulaelo District, Tigray Region of. J Ethnobiol Ethnomed. 2013;9. doi:10.1186/1746-4269-9-65
3. Teklehaymanot T, Giday M, Medhin G, Mekonnen Y. Knowledge and use of medicinal plants by people around Debre Libanos monastery in Ethiopia. J Ethnopharmacol. 2007;111(2):271–283. doi:10.1016/j.jep.2006.11.019
4. Esubalew ST, Belete A, Lulekal E, Gabriel T, Engidawork E, Asres K. Review of ethnobotanical and ethnopharmacological evidences of some Ethiopian medicinal plants traditionally used for the treatment of cancer. Ethiop J Heal Dev. 2017;31(3):161–187.
5. Bitew H, Gebregergs H, Tuem KB, Yeshak MY. Ethiopian medicinal plants traditionally used for wound treatment: a systematic review. Ethiop J Heal Dev. 2019;33(2):1–26.
6. Kim HJ, Saleem M, Seo SH, Jin C, Lee YS. Two new antioxidant stilbene dimers, parthenostilbenins A and B from Parthenocissus tricuspidata. Planta Med. 2005;71(10):973–976. doi:10.1055/s-2005-871229
7. Mahweety JANA. Chemical study on the leaves of Cyphostemma digitatum. PSM Biol Res. 2016;1(2):10–13.
8. Bala A, Kollmann A, Ducrot PH, et al. Cis-viniferin: antifungal resveratrol dehydrodimer from Cyphostemma crotalarioides root. J Phytopathol. 2000;2000:29–32.
9. Drumm RBA, Micheal O, Muhammad S. Phytochemistry, pharmacology and perceived health uses of non-cultivated vegetable Cyphostemma adenocaule (Steud. ex A. Rich.) Desc. ex wild and phytochemistry, pharmacology and perceived health uses of non-cultivated vegetable Cyphostemma adenocau. Sci Afr. 2019;2(February):e00053. doi:10.1016/j.sciaf.2019.e00053
10. Chouna F, Rodolphe J, Enta BN, Vonthron-se C, Sewald N, Sewald N. Ceanothane-type triterpenoids from Cyphostemma adenocaule. Arch Pharm Res. 2016. doi:10.1007/s12272-016-0801-1
11. Cao S, Hou Y, Brodie P, et al. Antiproliferative Compounds of Cyphostemma greveana from a Madagascar. Chem Biodivers. 2011;8:643–650. doi:10.1002/cbdv.201000061
12. Udegbunam RI, Udegbunam SO, Anosa GN. Analgesic and anti-inflammatory effects of Cyphostemma vogelii (Hook. f.) Desc. root extract in mice. African J Biotechnol. 2013;12(17):2288–2292. doi:10.5897/AJB12.2230
13. Eleojo O, And DMA, Chiletugo NOF. Hepatoprotective effect of aqueous leave extract of Cyphostemma glaucophilla on carbon tetrachloride induced hepatotoxicity in albino rats. Res J Med Plants. 2012;6:116–122. doi:10.3923/rjmp.2012.116.122
14. Khan R, Saif AQ, Mansour M, Nunes FM, Jordão AM. Antioxidant, antimicrobial and urease inhibiting activities of methanolic extracts from Cyphostemma digitatum stem and roots. Nat Prod Res. 2015;29(April):37–41. doi:10.1080/14786419.2015.1023726
15. Bussa NF, Belayneh A. Traditional medicinal plants used to treat cancer, tumors and inflammatory ailments in Harari Region, Eastern Ethiopia. South African J Bot. 2019;122:360–368. doi:10.1016/j.sajb.2019.03.025
16. Bala AE, Kollmann A, Ducrot P, Majira A, Kerhoas L, Delorme R. Antifungal activity of resveratrol oligomers from Cyphostemma crotalarioides. Pestic Sci. 1999;127(September1998):206–208. doi:10.1002/(SICI)1096-9063(199902)55:2<206::AID-PS871>3.0.CO;2-Z
17. Dagne E. Natural products Database for Africa (NDA). Version 2.0; 2016. Available from: http://www.alnapnetwork.com/SpeciesDetail.aspx.
18. Kefalew A, Asfaw Z, Kelbessa E. Ethnobotany of medicinal plants in Ada’a District, East Shewa Zone of Oromia Regional State, Ethiopia. J Ethnobiol Ethnomed. 2015;11(1):1. doi:10.1186/s13002-015-0014-6
19. Demissew S. A study of the vegetation and floristic composition of southern Wällo. JEthiop Stud. 2019;31(1):159–192.
20. Chekole G. Ethnobotanical study of medicinal plants used against human ailments in Gubalafto. J Ethnobiol Ethnomed. 2017;13–55. doi:10.1186/s13002-017-0182-7
21. Kipkore W, Wanjohi B, Rono H, Kigen G. A study of the medicinal plants used by the marakwet community in Kenya. J Ethnobiol Ethnomed. 2014;10:10–24. doi:10.1186/1746-4269-10-10
22. Balouiri M, Sadiki M, Ibnsouda SK. Methods for in vitro evaluating antimicrobial activity: a review. J Pharm Anal. 2016;6(2):71–79. doi:10.1016/j.jpha.2015.11.005
23. Zaidan MR, Noor Rain A, Badrul AR, Adlin A, Norazah A, Zakiah I. In vitro screening of five local medicinal plants for antibacterial activity using disc diffusion method. Trop Biomed. 2005;22(2):165–170.
24. Galma W, Endale M, Getaneh E, Eswaramoorthy R, Assefa T, Melaku Y. Antibacterial and antioxidant activities of extracts and isolated compounds from the roots extract of Cucumis prophetarum and in silico study on DNA gyrase and human peroxiredoxin 5. BMC Chem. 2021;15(1):1–17. doi:10.1186/s13065-021-00758-x
25. Narramore S, Stevenson CEM, Maxwell A, Lawson DM, Fishwick CWG. New insights into the binding mode of pyridine-3-carboxamide inhibitors of E. coli DNA gyrase. Bioorganic Med Chem. 2019;27(16):3546–3550. doi:10.1016/j.bmc.2019.06.015
26. Lipinski CA, Franc IO, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Acta Petrol Sin. 2012;28(6):1765–1784. doi:10.1016/S0169-409X(96)00423-1
27. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug- likeness and medicinal chemistry friendliness of small molecules. Nat Publ Gr. 2017;(January):1–13. doi:10.1038/srep42717
28. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263. doi:10.1093/nar/gky318
29. Behrouz S, Soltani Rad MN, Taghavi Shahraki B, Fathalipour M, Behrouz M, Mirkhani H. Design, synthesis, and in silico studies of novel eugenyloxy propanol azole derivatives having potent antinociceptive activity and evaluation of their β-adrenoceptor blocking property. Mol Divers. 2019;23(1):147–164. doi:10.1007/s11030-018-9867-7
30. Gülçin I. Antioxidant properties of resveratrol: a structure-activity insight. Innov Food Sci Emerg Technol. 2010;11(1):210–218. doi:10.1016/j.ifset.2009.07.002
31. Braca A, Tommasi N, Bari L, Pizza C, Politi M, Morelli I. Antioxidant principles from Bauhinia t arapotensis. J Nat Prod. 2001;64:892–895. doi:10.1021/np0100845
32. Pandey SK, Khan MF, Awasthi S, Sangwan R, Jain S. A quantum theory of atoms-in-molecules perspective and DFT study of two natural products: trans-communic acid and imbricatolic acid. Aust J Chem. 2017;70(3):328–337. doi:10.1071/CH16406
33. Lee C, Yang W, Parr RG. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. EE Eval Eng. 2011;50(11):36–39.
34. Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem. 1994;98(45):11623–11627. doi:10.1021/j100096a001
35. Becke AD. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J Chem Phys. 1998;2155(August). doi:10.1063/1.462066
36. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 16 Rev. C.01. In: Wallingford CT. Gaussian Inc; 2016.
37. Geerlings P, Proft F. Conceptual DFT: the chemical relevance of higher response functions. Phys Chem Chem Phys. 2008;10:3028–3042. doi:10.1039/b717671f
38. Geerlings P, Proft F, Langenaeker W. Conceptual density functional theory. Am Chem Soc. 2003;1793–1873. doi:10.1021/cr990029p
39. Erdogan T. DFT, molecular docking and molecular dynamics simulation studies on some newly introduced natural products for their potential use against SARS-CoV-2. J Mol Struct. 2021;1242:130733. doi:10.1016/j.molstruc.2021.130733
40. Pierre LL, Moses MN. Isolation and Characterisation of Stigmasterol and β-Sitosterol from Odontonema Strictum (Acanthaceae). J Innov Pharm Biol Sci. 2015;2(1):88–95. doi:10.13140/RG.2.1.3689.7365
41. He S, Wu B, Pan Y, Jiang L. Stilbene oligomers from Parthenocissus laetevirens: isolation, biomimetic synthesis, absolute configuration, and implication of antioxidative defense system in the plant. J Org Chem. 2008;73(2):5233–5241. doi:10.1021/jo8001112
42. Kurihara H, Kawabata J, Ichikawa S, Mizutani J. (−)-ε-Viniferin and related oligostilbenes from Carex pumita Thunb. (Gyperaceae). Agric Biol Chem. 1990;54(4):1097–1099. doi:10.1080/00021369.1990.10870044
43. Commodari F, Khiat A, Ibrahimi S, Brizius AR, Kalkstein N. Comparison of the phytoestrogen trans-resveratrol (3,4′,5- trihydroxystilbene) structures from x-ray diffraction and solution NMR. Magn Reson Chem. 2005;43(7):567–572. doi:10.1002/mrc.1583
44. Mattivi F, Reniero F, Korhammer S. Isolation, characterization, and evolution in red wine vinification of resveratrol monomers. J Agric Food Chem. 1995;43(7):1820–1823. doi:10.1021/jf00055a013
45. Gao Y, He C, Ran R, Zhang D, Li D, Xiao P. The resveratrol oligomers, cis - and trans -gnetin H, from Paeonia suffruticosa seeds inhibit the growth of several human cancer cell lines. J Ethnopharmacol. 2015;169:24–33. doi:10.1016/j.jep.2015.03.074
46. Ito T, Endo H, Shinohara H, Oyama M, Akao Y, Iinuma M. Fitoterapia occurrence of stilbene oligomers in Cyperus rhizomes. Fitoterapia. 2012;83(8):1420–1429. doi:10.1016/j.fitote.2012.08.005
47. Ta TU. A resveratrol Dimer from Parthenocissus tricuspidata. Phytochemistry. 1998;9422(97):1241–1243.
48. Cichewicz RH, Kouzi SA, Hamann MT. Dimerization of resveratrol by the grapevine pathogen Botrytis cinerea. J Nat Prod. 2000;63(1):29–33. doi:10.1021/np990266n
49. Oshima Y, Kamijou A, Ohizumi Y, et al. Novel oligostilbenes from Vitis coignetiae1. Tetrahedron. 1995;51(44):11979–11986. doi:10.1016/0040-4020(95)00757-Y
50. Cimino G, De Rosa D, De Stefano S, Minale L. Isoagatholactone, a diterpene of a new structural type from the sponge Spongia officinalis. Tetrahedron. 1974;30(5):645–649. doi:10.1016/S0040-4020(01)97059-0
51. González AG, Estrada DM, Martín JD, Martín VS, Pérez CC, Pérez R. New antimicrobial diterpenes from the sponge Spongia officinalis. Elsevier. 1984;40:4109to 4113. doi:10.1016/0040-4020(84)85092-9
52. Anza M, Endale M, Science A, Eswaramoorthy R, Science A. Cytotoxicity, antimicrobial activity, molecular docking, drug likeness and DFT analysis of benzo [c] phenanthridine alkaloids from roots of Zanthoxylum chalybeum. Biointerface Res Appl Chem. 2021;(June). doi:10.33263/BRIAC122.15691586
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.