")
Back to Journals » Clinical Optometry » Volume 16
Alteration Ocular Motility in Retinitis Pigmentosa: Case–Control Study
Authors Comberiati AM, Lomartire C, Malvasi M , Migliorini R, Pacella F , Malvasi VM, Turchetti P, Pacella E
Received 28 November 2023
Accepted for publication 1 February 2024
Published 22 February 2024 Volume 2024:16 Pages 55—69
DOI https://doi.org/10.2147/OPTO.S446717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Mr Simon Berry
Anna Maria Comberiati,1 Chiara Lomartire,1 Mariaelena Malvasi,1 Raffaele Migliorini,1 Fernanda Pacella,2 Vito Maurizio Malvasi,3 Paolo Turchetti,4 Elena Pacella1
1Department of Sense Organs, Sapienza University of Rome, Rome, Italy; 2Department of Ophthalmology, Carlo Poma Hospital, Mantua, Italy; 3Department of Odontostomatological and Maxillo-Facial Sciences, Sapienza University of Rome, Rome, Italy; 4National Institute for Health, Migration and Poverty (INMP/NIHMP), Rome, Italy
Correspondence: Mariaelena Malvasi, Department of Sense Organs, University of Rome, “Sapienza” via Del Policlinico 155, Rome, 00161, Italy, Tel +393402114859, Email [email protected]
Purpose: To evaluate ocular motility (OM) disorders and strabismus in a sample of patients with retinitis pigmentosa (RP) and a control sample.
Methods: In this cross-sectional retrospective analysis, we studied a sample of RP patients with a mean age of 48.74 years and an average visual acuity of 7/10 based on Snellen optotype and a sample of control patients with similar mean age (49 years [men], 47 years [women]) and sex and an average visual acuity of 9.9/10, with the aim of assessing correlations between alteration of OM and strabismus in RP patients based on age, high refractive defect, or severely impaired binocular vision. The examination followed a protocol of testing for anamnesis and best-corrected visual acuity, as well as a complete eye examination, corneal reflex, cover test, OM, Hess screen, and Lang test.
Results: At the first orthoptic evaluation, 45.16% of patients showed strabismus, 41.93% exotropia (25% of cases intermittent), 3.22% esotropia, and 6.45% vertical deviation. Later evaluation showed strabismus in 25.80% of patients, exotropia in 19.35% (9.67% intermittent), esotropia in 3.22%, and vertical deviation in 3.22%. Assessment of eye motility study showed 51.6% overaction of the inferior oblique and hypofunction of the superior rectus, and 18% overaction of the lateral rectus and hypofunction of the medial rectus. According to our results, alterations in OM and strabismus in RP patients are not correlated with age or high refractive defect. Therefore, motility disorders and strabismus are attributed to a genetic factor to which men are more susceptible.
Conclusion: The incidence of OM disorder was 77.42%, and strabismus was present in 45.16% of patients.
Keywords: retinitis pigmentosa, orthoptic evaluation, strabismus, genetic factor, ocular motility
Introduction
Retinitis pigmentosa (RP) is not a single entity, but a group of disorders that produce gradual loss of vision.1–3 RP affecting more than 1.5 million patients worldwide, RP’s prevalence is approximately 1:4000 (Pagon, 1988),4 although reports vary from 1:9000 (Na et al, 2017)5 to as high as 1:750 (Nangia et al, 2012),6 depending on the geographic location.7
Also called rod–cone dystrophy, RP derives from a primary loss in rod photoreceptors and is followed by a secondary loss in cone photoreceptors.2,3,8,9 In contrast, cone–rod dystrophy is characterized by retinal pigment deposits visible on fundus examination, predominantly localized in the macular region,2,8,9 and causes night blindness, but visual disability and blindness result from cone degeneration.10,11 The mechanism of rod-cell death varies depending upon the gene that is mutated; therefore, RP may be sporadic with a good prognosis, with central vision until later in life, or appear early in life and be inherited in autosomal dominant, autosomal recessive, or X-linked recessive modes.12,13
In the context of RP, some dystrophies share genetically inherited ocular alterations that are associated with eye movement disorders. These eye movement disorders constitute a relevant link in a heterogeneous group of pathological conditions. Clinically, these dystrophies exhibit common features with retinal pigmentary dystrophies. An example of this is observed in RPE65-associated RP, linked to Usher syndrome (US; 2% of cases), and Bardet–Biedl syndrome (BBS; 5% of cases),14,15 where ocular motility alterations constitute a significant element of the clinical presentation, correlated with the mutation.
Loss of the visual field or vision has been estimated in the literature to induce sensory strabismus, and late vision loss induces exotropia and early vision loss.16 In one study, visual field loss and the appearance of strabismus were correlated. The authors concluded that these patients show sensory exotropia in later life, although it is unclear whether patients in the more advanced stages of RP show greater deviation than those in the first stages.17 In addition to this article, only two other articles have highlighted problems concerning ocular motility.
The first is a case report of a patient aged 31 years who presented cyclic exotropia associated with RP,18 while the second is a pilot study that assessed by orthoptic examination the presence of ocular muscle deficits, which was found in 50% of patients.19 The purpose of our study was to evaluate a sample of patients with RP with the aim of analyzing the presence of ocular motility disorders and strabismus and compare it to a control group without RP.
Methods
Two groups of patients were examined by the same operator. The first group consisted of patients with RP, while the second was an age- and sex-matched control group. In this study were included patients who had previously received a confirmed diagnosis of RP through genetic testing performed at other dedicated specialist centers. All selected patients exhibited the same RPE65 genetic mutation, associated with various syndromes within the spectrum of retinal pigmentary dystrophies (RP-related), including US and BBS, which were included in our study. These retinal dystrophies share the presence of the identical RPE65 genetic mutation and manifest phenotypically through a common set of signs and symptoms, characterized by progressive retinal degeneration and primary loss of photoreceptors.
In the literature, it is widely documented that RP can no longer be considered a single hereditary retinal dystrophy caused by a single genetic mutation. Instead, it is identified as a form of genetic retinal dystrophy, which manifests in a broad range of phenotypic heterogeneity, with direct correlations in the genotype–phenotype complex. These correlations have been confirmed through specific genetic investigations.20–23
The groups were visited at the Center of Pediatric Ophthalmology and Strambology of the Eye Clinic, Policlinico Umberto I, La Sapienza University of Rome. This cross-sectional retrospective study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each patient or from parents in underage patients after explanation of the procedure modalities and the benefits and possible risks and/or complications.
The study protocol was approved by the Ethics Committee of the Azienda Ospedaliero Universitaria Policlinico Umberto I in Rome, and all patients provided informed written consent for both the procedures and data collection. Written informed consent to the publication of personal data in anonymous form was obtained from each participant, in accordance with the principles outlined in the Nuremberg Code and the Belmont Report, and informed consent was given freely, without coercion or bribery of any kind.
A total of 128 RP patients were evaluated A total of 31 (62 eyes) were included, and 93 (186 eyes) healthy patients were examined in terms of the following protocol:
- Anamnesis
- Best-corrected visual acuity
- Complete eye examination: examination of anterior chamber by slit lamp and fundus examination after pupillary dilatation with tropicamide; measurement of intraocular pressure; spectral domain optical coherence tomography (Heidelberg Engineering)
- Corneal reflex (CR)
- Cover test
- Ocular motility
- Hess screen
- Lang test
The inclusion criteria for both groups were:
- Age 6–80 years, with each RP patient matched by healthy patients of same sex and age
- Best-corrected visual acuity between 6/10 and 10/10 (Snellen optotype)
- RP based on a confirmed diagnosis, having undergone specific genetic testing at an external genetics center not affiliated with our hospital
- Diagnosis was also based by anamnesis, fundus examination and optical coherence tomography. These tests included the following patients:
- Patients with the typical non-syndromic form of RP and those with syndromes associated with various types of RP that shared the common RPE65 genetic mutation, exhibiting common phenotypic traits in the RPs US and BBS
- Patients with suspected RP: all were naturally referred after a complete ophthalmological examination, fundus examination, and retinal imaging on optical coherence tomography to a genetics center that conducted specific genetic tests; family history assessment was performed, creating a family tree to discover the presence of RP in other family members (brothers, cousins, and grandfathers) who did not show the disease in other family members; therefore, we did not include the data
The exclusion criteria were the same for both groups:
- Patients who had undergone ocular surgery (including cataract surgery)
- Patients with other eye diseases that compromised visual function (such as cystoid macular edema, cataract, macular hole
- Patients with systemic, vascular, and neurodegenerative disease that could affect orthoptic assessment
- Patients with visual field electronic Humphrey test result <10°
Strabismus was detected using the CR test. To quantify both strabismus and heterophorias, we employed the prismatic alternating cover test. A prismatic bar was used for precise quantification in diopters of ocular deviation.
Binocular vision occurs when both eyes focus on an object, causing the brain to merge the visual input into a singular image. This fusion is essential for stereopsis, which was assessed using the Lang Stereotest II. Complete stereopsis was confirmed if, at a distance of 40 cm, all test figures were discerned: a moon representing a stereopsis of 200 arc seconds (”), a car at 400”, an elephant at 600”, and a monocularly visible star (useful for false negatives or attracting attention). Stereopsis was considered null if only the star were perceived, and coarse if only the star and elephant were detected.
In evaluating ocular motility, we considered the 12 extraocular muscles of both eyes across various gaze positions. A gradient of 0 was assigned for normal ocular motility, +1 for mild overaction, +2 for overaction, and +3 for severe overaction. The same scoring system applied to hypofunction of the contralateral synergist muscle, but with negative values (−3 for severe hypofunction, −2 for moderate hypofunction, −1 for mild hypofunction).
The Hess–Lancaster screen examination was utilized exclusively to determine the presence of concomitant or nonconcomitant strabismus and to validate identified forms of heterophoria. If misalignments were uniform in all nine diagnostic gaze directions, the patient was classified as having concomitant strabismus (and subsequently ruled out). For cases of heterophoria, we ensured it aligned with the findings from the cover test. Conversely, if unequal misalignments were observed in all gaze positions, the strabismus was deemed nonconcomitant.
For statistical analysis, demographic descriptions of both samples, refractive defect analyses, and binocular visual acuity are presented using averages with standard deviations. The prevalence of ocular motility disorders and strabismus within the samples is expressed as percentages. Pearson’s correlation test was applied to examine the correlation between contralateral synergist muscles in individuals with RP: right superior rectus (SR) — left inferior oblique (IO); right inferior rectus (IR) — left superior oblique (SO); left SR—right IO; left IR—ri SO; right lateral rectus (LR)—left medial rectus (MR); right MR—left LR.
Results
The sample of patients with RP comprised 31 individuals: 13 women and 18 men with an average age of 48.74 (men 49.61, women 47.53) years (Figure 1A). Most patients (80.64%) had a high percentage of typical RP, while patients with syndromes associated with RP (UHS and BBS) showed an incidence of 19.35%. All demographic data of RP patients are shown in Table 1. The control sample consisted of 93 patients (39 women and 54 men), and each RP patient was matched with three healthy patients of the same sex and mean age (Figure 1B).
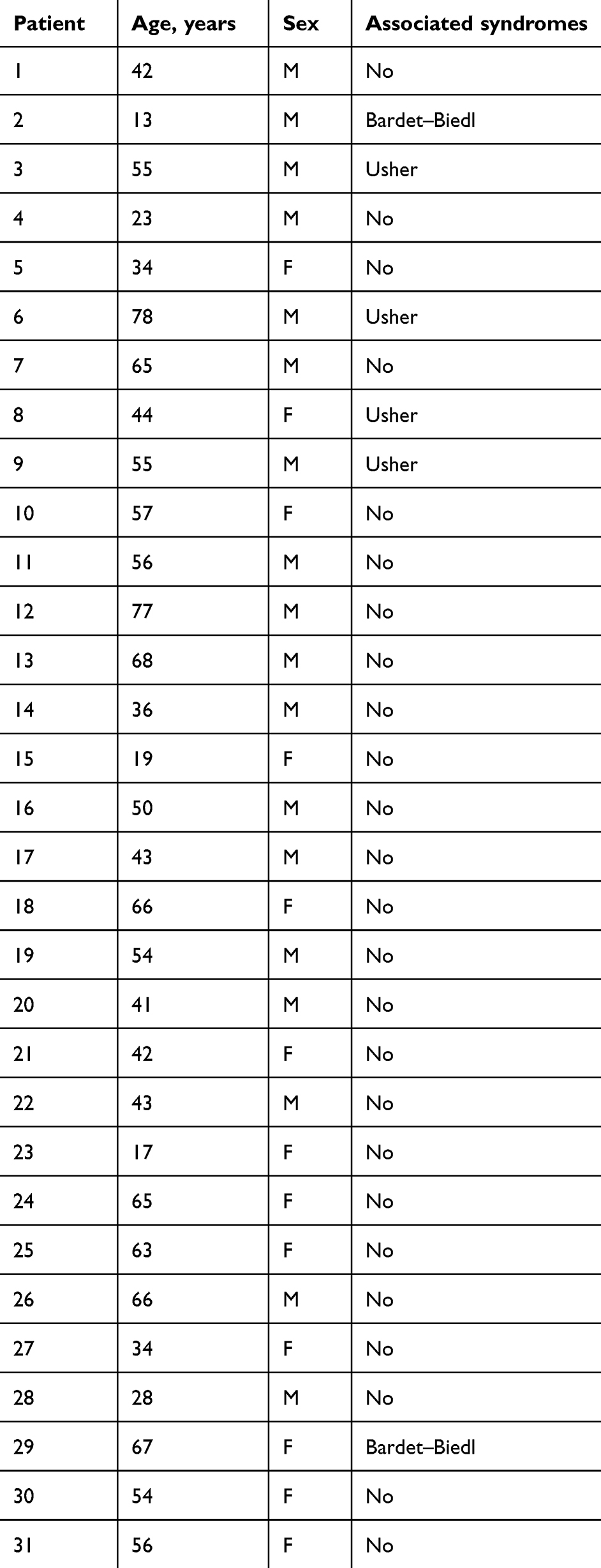 |
Table 1 RP patient characteristics |
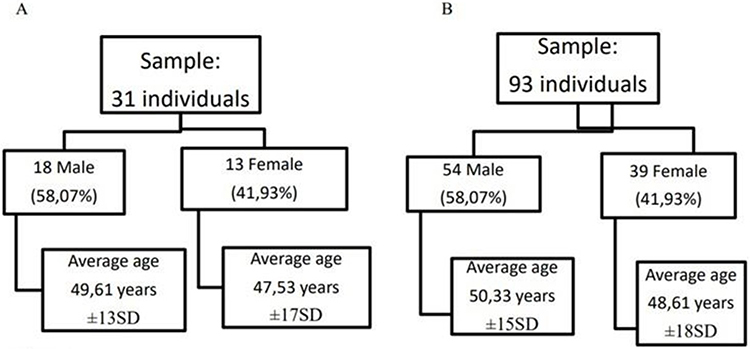 |
Figure 1 Stratification of the sample with RP (A) and control sample (B) by age and sex. |
On evaluation of vision acuity for afar by Snellen optotype, RP patients highlighted isoacuity, with an average of 7/10 with best-corrected visual acuity. Regarding the refractive defects, in our sample emerged an average myopia of of −1.25±1.50 for the right eye and −1±1.25 for the left eye19 and astigmatism of 0.75±0.75 for the right eye and 0.75±1 for the left eye (Figure 2). The control sample had average visual isoacuity of 9.9/10, and in all the sample average myopia was −0.5±0.75 for the right eye and −0.25±0.75 for the left eye, astigmatism 0.75±0.75 for the right eye and 0.75±0.75 for the left eye, and hypermetropia 0.25±0.75 (Figure 3).
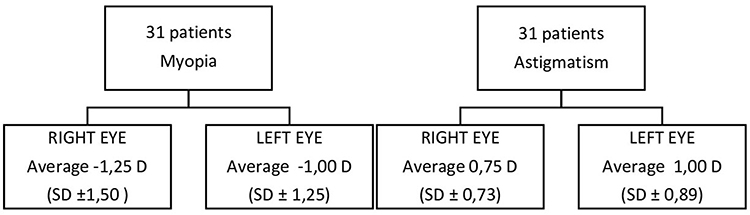 |
Figure 2 Evaluation of refractive errors in RP patients. Notes: The results show that the presence of refractive errors (mild myopia and low astigmatism) does not alter the binocular vision and thus the ocular motility of the patients examined. |
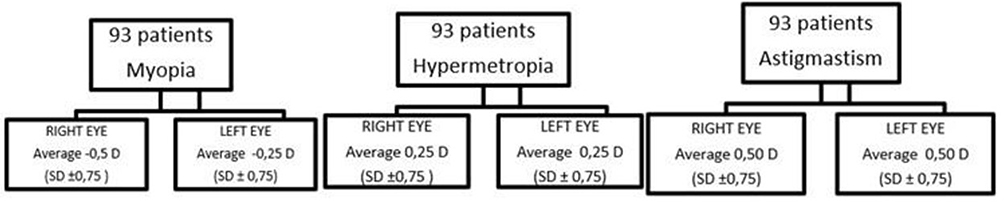 |
Figure 3 Evaluation of refractive errors in control sample. |
Stereoscopic vision was analyzed with use the Lang test II. In RP patients, it was present in 30 of the 31 patients. In only one case of exotropia was the binocular vision compromised and in five patients it was coarse, while in the control sample the stereopsis was complete. Diplopia was not present in either group. These results show that binocular vision was preserved in most of the both sample under analysis.
Alterations in ocular motility in RP patients were present in 77.41% of the sample and involved all the muscles (MR, LR, SR, IR, IO, SO). In the upper-left gaze, overaction of the IO muscle of right eye was severe (+3) in 25.8% and moderate (+2) in 19.3% of cases compared to normal ocular motility (0) in 41.93% of our sample. We also verified the presence of a mild overaction (+1) of 6.45% of cases and a hypofunction of the same percentage, but mild (−1) and moderate (−2) in 3.22% of cases. In the same position of gaze, but in the contralateral eye, we found the SR muscle in the norm (0) in the same percentage and degree of the previous muscle. Alterations were 19.35% of severe hypofunction (−3), 25.8% of moderate hypofunction (−2), and 6.45% of mild hypofunction (−1), and overactions were present in the same proportion, 3.22% of cases, but mild (+1) and moderate (+2).
In the upper-right gaze, we found IO muscle overaction of the left eye to be severe (+3) in 16.12% and moderate (+2) in 25.8% of cases, while normal ocular motility (0) was present in 45.16% of our sample. There was also mild overaction (+1) in 6.45% of cases and mild (−1) and moderate (−2) hypofunction, both in 3.22% of cases. In the contralateral eye, the SR muscle was normal (0, 45.16%), and as regards alterations, we found 16.12% severe hypofunction (−3), 25.8% moderate hypofunction (−2), and 6.45% mild hypofunction (−1). Overaction was present in the same proportion, 3.22% of cases, for mild (+1) and moderate degrees (+2). we found a lower proportion f ocular motility disorders in the left and right gaze positions, but this was statistically significant.
Regarding left lateroversion, the LR of the left eye showed severe overaction (+3) in 3.22% of cases and moderate (+2) in 19.35%. There was also moderate hypofunction (−2) in 3.22%, though it was normal (0) in 74.19%. In the contralateral eye, we found hypofunction of the MR to a severe degree (−3) in 3.22% and moderate (−2) in 19.35% of cases and moderate (+2) overaction in 3.22% of cases, while there was normal function (0) in 74.19% of cases. For right lateroversion, the LR of the right eye showed severe overaction (+3) in 6.45% of cases and moderate (+2) in 16.12%. There was also moderate hypofunction (−2) in 3.22%, while it was normal (0) in 74.19%. In the contralateral eye, we found the MR to have severe hypofunction (−3) in 6.50% and moderate (−2) in 16.12% of cases, while there was also a moderate (+2) degree of overaction in only 3.22% of cases. Normal function (0) was found in 74.19% of cases.
We also evaluated the lower-left and -right positions. For the lower-right gaze, we found overaction of the IR of the right eye to a moderate degree (+2) in 6.45% and mild (+1) in 16.12% of cases. As regards hypofunction, this was present in 3.22% of the cases to a mild (−1) and moderate (−2) degree and a normal function of 70.96%. In the contralateral eye, there was hypofunction of the SO of the left eye to a moderate degree (−2) in 6.45% and mild (−1) in 16.12%. Overaction was moderate (+2) and mild (+1) in 3.22%, and normal function (0) was present in 70.96%. For the lower-left gaze, we found overaction of the IR of the left eye to a moderate degree (+2) in 9.67% and mild (+1) in 12.90%. As regards hypofunction, this was present in 3.22% of the cases to a mild (−1) and moderate degree (−2), with normal function (0) of 70.96%. In the contralateral eye, there was hypofunction of the SO to a moderate degree (−2) in 9.67% and mild (−1) in 12.90%. Overaction was moderate (+2) and mild (+1) in 3.22%, and normal function (0) was present in 70.96% (Table 2).
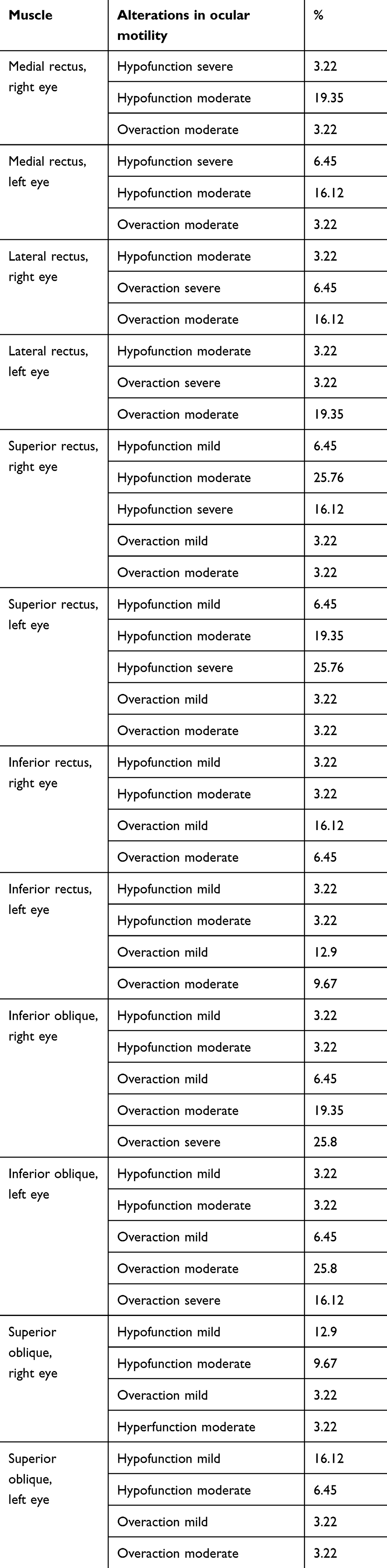 |
Table 2 Evaluation of ocular motility in RP patients |
When evaluating ocular motility, an inversely proportional correlation calculated with Pearson’s correlation (P<-1) is shown when comparing the contralateral synergic muscles, and this allows us to state that there is a direct relationship between RP and ocular motility disorders (Table 3). We conducted an analysis of ocular motility dysfunction, examining distribution based on sex, and obtained noteworthy results. Table 4 illustrates that impairment of the SR and IO muscles is more pronounced in men than women, as depicted in Figure 4. Furthermore, we scrutinized ocular motility dysfunction concerning age, categorizing the sample into two groups: individuals aged ≤50 years and those >50 years. Interestingly, the graphical representation reveals no significant disparity between the two age-groups, as illustrated in Figure 5.
 |
Table 3 Pearson correlations in ocular motility of RP patients |
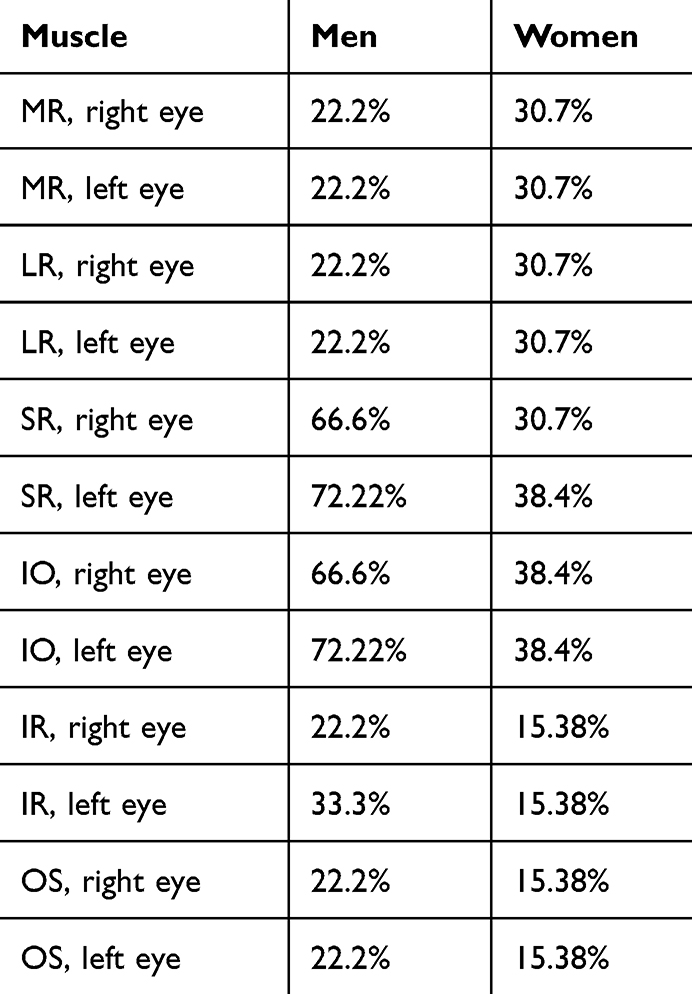 |
Table 4 RP sample: ocular motility dysfunction in men and women |
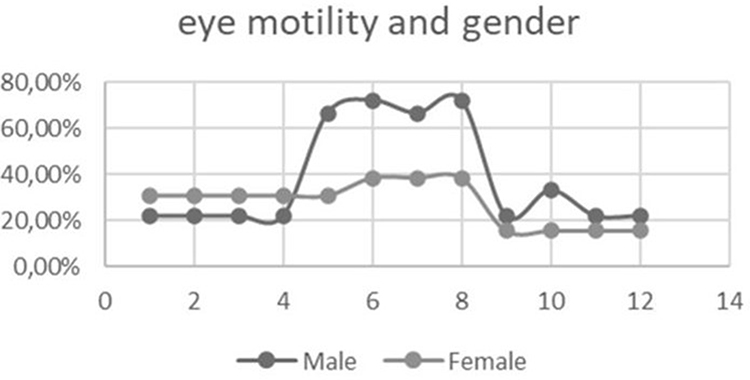 |
Figure 4 Ocular motility dysfunctions by gender. |
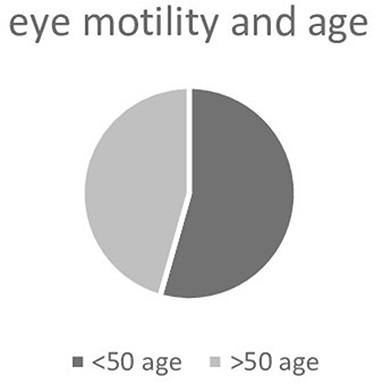 |
Figure 5 Ocular motility dysfunctions by age. |
In the control sample, alterations in ocular motility were present in 19.35% and involved only the MR and LR to a mild degree. For left lateroversion. the LR of the left eye showed mild overaction (+1) in 19.35% of cases, while in 80.64% it was normal (0). In the contralateral eye, we found hypofunction of MR to a mild degree (−1) in 19.35% and normal function (0) in 80.64% of cases. For right lateroversion, the LR of the right eye showed mild overaction (+1) in 19.35% of cases while it was normal (0) in 80.64%. In the contralateral eye, we found the MR had mild hypofunction (−1) in 19.35% and normal function (0) in 80.64% of cases (Table 5).
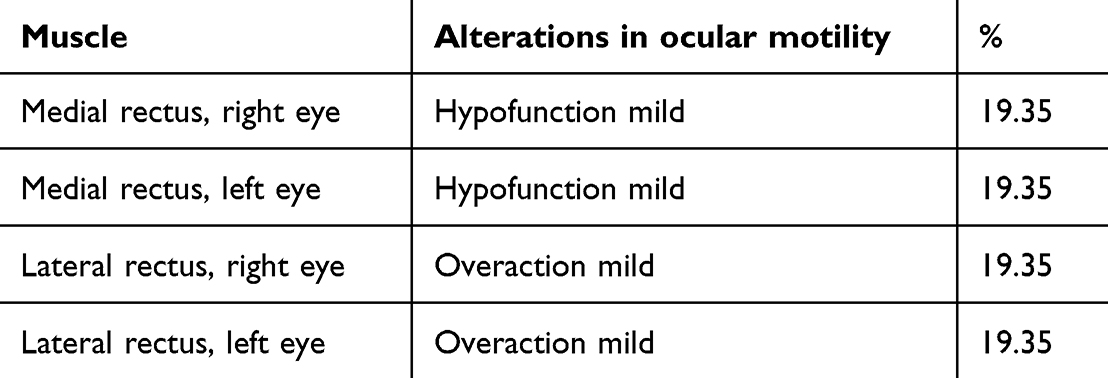 |
Table 5 Evaluation of ocular motility in control sample |
In the RP sample, strabismus was investigated by evaluation of CR from near (33 cm) by use of a target light and afar (3 m) with a letter of Snellen optotype. On evaluation from near, 45.16% (five women, nine men) of patients showed manifest strabismus (or tropia), 25.80% (three women, two men) showed exotropia (manifest divergent strabismus). In 6.45% (two women), this was associated with vertical deviation, in 25.8% (three women, six men) intermittent exotropia, and in 3.22% (one man) esotropia (manifest convergent strabismus). On evaluation from afar, strabismus was present in 16.12% (two women, four men) of patients, 9.67% (one woman, two men) exotropia, 9.67% (one woman, two men) intermittent exotropia, 3.22% (one man) esotropia, and 3.22% (one man) vertical deviation. In the control sample, the CR from near and afar showed 100% orthophoria (Figure 6).
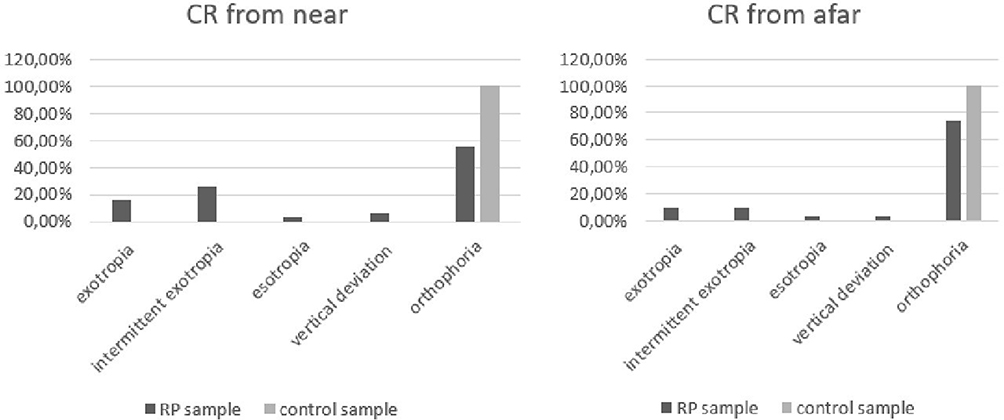 |
Figure 6 CR (corneal reflex) from near and afar in RP patients and control sample. |
Also, we assessed latent deviation by use of the cover test from near (33 cm) with a target light and afar (3 m) with a letter of Snellen optotype, and the deviation was estimated by use of prism bars and measured in prism diopters. In the RP sample, from near (33 cm) without the use of lenses, 93.56% showed divergent deviation (12 women, 17 men), 54.84% vertical deviation (seven women, ten men),24,25 6.44% converging deviation (Figures 7 and 8). On the cover test from near, but this time with the use of lenses, 25.78% of the sample had vertical deviation. For horizontal deviation, 48.4% (six women, ten men) had divergent deviationand 3.22% (one woman) converging deviation. On the cover test from a distance (3 m) without lenses, we found 45.14% with divergent deviation (four women, ten men), 12.90% (two women, two men) vertical deviation, and 3.22% convergent deviation (one man) (Figure 9). From a distance but with lenses, we found 16.11% (one woman, four men) with divergent deviation, 6.45% (two men) vertical deviation,24,25 and no presence of convergent deviation. Also, we found that in patients with UHS and BBS, on the cover test from near without lens, there was exotropiaphoria of 10–14 prism diopters. CRs in control sample from near and afar did not show the presence of strabismus.
 |
Figure 7 Cover test in RP sample: evaluation of horizontal deviation for near without lens. |
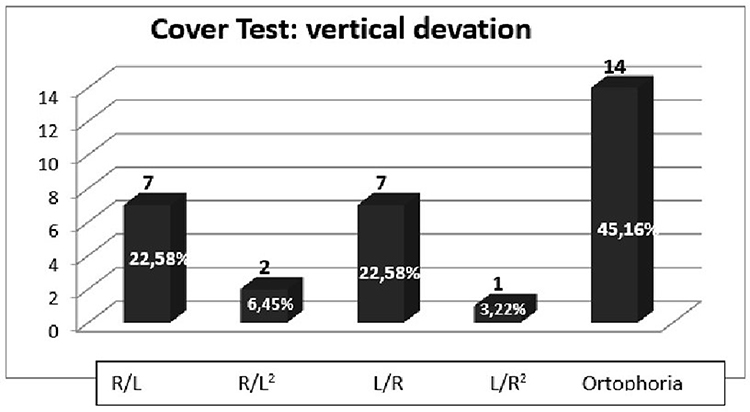 |
Figure 8 Cover test, RP sample: evaluation of vertical deviation for near without lens. |
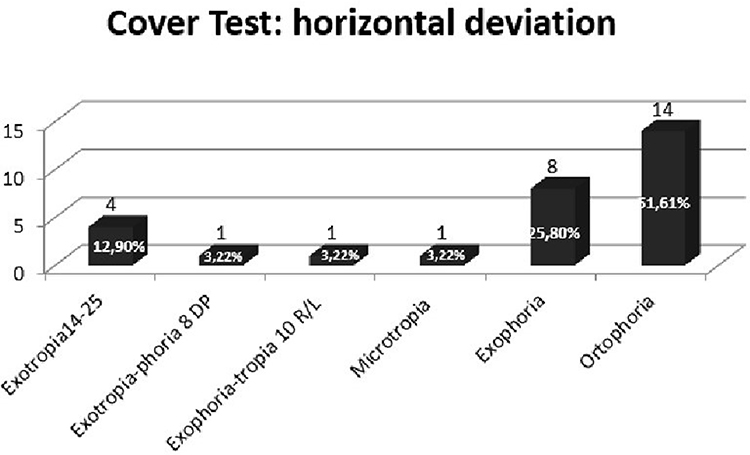 |
Figure 9 Cover test, RP patients: evaluation of horizontal deviation from a distance without lens. |
On the cover test without the use of lenses from near (33 cm), 25.8% (12 women, 12 men) of the sample presented mild divergent deviation and 3.22% (three men) mild converging deviation. From afar, 6.45% (three women, three men) of the sample showed mild divergent deviation and 3.22% (three men) had mild converging deviation. On the cover test with the use of lenses from afar (3 m): 45.16% (30 men, 12 women) of the sample presented mild divergent deviation, 12.90% (six women, six men) mild converging deviation, and from afar, 12.90% (six women, six men) of the sample showed mild divergent deviation and 19.35% (12 men, six women) had mild converging deviation (Figure 10).
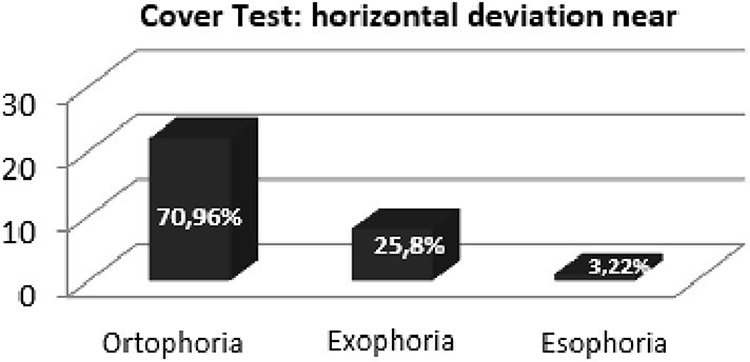 |
Figure 10 Cover test, control sample: evaluation of horizontal deviation for near without lens and the horizontal deviation from afar without lens. |
Discussion
In our RP sample, a significant percentage had typical RP, 80.64%, while patients with syndromes associated with RP (UHS and BBS) represented 19.35%. In this sample, we found on near evaluation that 45.16% had strabismus (or tropia), while on evaluation from afar, strabismus was present in 25.80%. Martinez et al showed that with aging of a population comes an increase in decompensated or paralytic-type strabismus and that paralytic strabismus is the more common, especially after the sixth decade of life.26 RP samples were composed of a similar number of individuals of sex and an average age of 48.74 years, relatively young. This excludes the possibility that the strabismus found originated from age or paralytic decompensation, the latter having been excluded both by carrying out a careful anamnesis and by analyzing ocular motility.
Studies show that advancing age may lead to ocular motility disorders, ie, it occurs in some cases around the age of 40 years due to the decay of the accommodation–convergence component, until it disappears completely around the age of 60 years. On the other hand, as our sample consisted of subjects with an average age of <50 years (49.61 years), we can state that the ocular motility disturbances that are typically age-related in the control group of patients we examined could be minimal or initial. In fact, in the control group, the ocular motility disturbances concerning accommodation–convergence were characterized by mild and unremarkable defects, whereas they were significant in the group of RP patients.
Sensory strabismus is a type of strabismus that is established by organic or functional alteration of the visual system. If this occurs at an early age, we will have a convergent type of strabismus, and otherwise a divergent type.16 Miyata et al showed that low visual acuity and a restricted visual field can facilitate the appearance of sensory-type strabismus in patients with RP.17 From the data obtained, the association between low visual acuity and manifest deviations can be excluded, because there was an isoacuity of 7/10 in both eyes, and in our study a visual field <10° (electronic Humprey) was excluded. Moreover, there was no correspondence between severe refractive defects or anisometropia (medium–low myopia −1±1.5 SD in both eyes, astigmatism 1±1.5 SD in both eyes) and muscular defects. These data exclude the possibility that all strabismus that we found was anisometric because, as demonstrated by Reşat Duman et al, refractive errors that can induce amblyopia and induce strabismus are ≥1 D for hypermetropia, ≥±2 D for astigmatism, and ≥3 D for myopia.27 Strabismus in our study was present in 45.16% from near and 25.80% from afar, and was predominantly of the exotropic type associated with the presence of vertical deviations, unlike a previous study that did not highlight the presence of noteworthy horizontal deviations. Strabismus prevalence was not significant when compared within the RP group, and neither sex, age, nor binocular vision favored its appearance, but upon comparison with sex- and age-matched subjects in the sample group, we can note its absence. The presence of these deviations can be correlated with the fact that in our sample, all the muscles were altered in most subjects (77.41%). In fact, the most significant alterations were:
- 25.80% of the sample presented severe overaction of the IO 20 of the right eye, and in the contralateral eye we found, with the same percentage and severity, hypofunction of the SR.
- 25.80% of the sample showed moderate overaction of the IO 20 of the left eye, and in the contralateral, with the same percentage and severity, hypofunction of the SR.
- 19.35% of the cases showed a moderate hypofunction of the MR of the right eye and in the contralateral eye, with the same percentage and severity, overaction of the LR.
- 16.12% of the cases had moderate hypofunction of MR of the left eye and in the contralateral eye, with the same percentage and severity, overaction of the LR.
- 16.12% of the cases presented mild overaction of the IR of the right eye and in the contralateral eye, with the same percentage and severity, and hypofunction of the SO.
- 12.90% of the cases had mild overaction of the IR of the left eye and in the contralateral eye, with the same percentage and severity, and hypofunction of the SO.
By comparing the two sexes, we were able to notice that the eye motility dysfunctions of the SR and OI. affected the men more than the women. Ito et al highlighted how sex could be an indicator of the progression rate of visual function in RP.28 It would be interesting in future studies to expand the sample for a more thorough analysis of this ocular motility dysfunction in men. Comparing these results with the control group, however, we can see in the latter, only two muscles, the MR and LR, had mild dysfunction. This can be explained by there being a physiological loss of convergence, as proved in other studies.29–31 Comparing our results with a previous study,18 we agree that both the OI and RS muscles are the ones most affected by dysfunction.
We thus believe that since RP is a genetically determined pathology in which strabismus can be observed, the ocular motility dysfunctions can be attributed to genetic causes. In a previous study, Migliorini et al found some ocular motility dysfunctions not related to causes such as age or refractive defects18 and as early as 1978, Fishman32 stated that the degree of central loss was milder in cases of autosomal dominant inheritance and more extensive in cases of X-linked recessive inheritance.19
However, this alteration in ocular motility and strabismus does not stem from age, significant refractive defects, or severe impairment of binocular vision. We confirm that these ocular motility disorders can be attributed to a genetic factor. In particular, we observed a higher susceptibility in men to ocular motility dysfunctions in RP. Expanding the sample would be of considerable use to substantiate our hypothesis. Unfortunately, at the moment we do not have specific information on the correlation between the severity of RP and potential alterations in ocular motility. RP is a degenerative genetic eye disease that affects the retina, with symptoms varying from individual to individual, including progressive loss of peripheral vision and difficulty adapting to low-light conditions.
Our study is innovative in exploring the correlation between retinal changes in RP and modifications in ocular motility. However, the current results do not allow us to define specific characteristics of this relationship. It could be hypothesized that ocular motility, controlled by eye muscles, could be influenced by RP, potentially affecting the ability to track objects with the eyes or maintain stable binocular vision, especially in patients with peripheral vision loss and severe compromise in daily life. Nevertheless, it is emphasized that further research and clinical studies are necessary to confirm and deepen these results to obtain more detailed information on the relationship between RP and ocular motility alterations, with the goal of developing preventive measures and early therapies.
Conclusion
This study highlights that early detection of ocular motility alterations in patients with RP can be an early sign of disease progression. RP is characterized by a progressive decline in visual acuity and a narrowing of the visual field.1–4 Therefore, the present study emphasizes the importance of orthoptic evaluation conducted during the initial ophthalmic visit, especially in the presence of a genetic diagnosis, as it can detect muscular alterations influencing strabismus early, which — with the progression of the pathology itself — lead to a dissociation of latent strabismus and a worsening of existing strabismus.29–31 The negative clinical course of inherited retinal dystrophies impacts the patient’s quality of life and autonomy. The association of ocular motility alterations with RP leads to further psychological distress.33 Moreover, our results suggest that early orthoptic assessment in monitoring protocols can be an important indicator of disease progression.
RP is a genetically transmitted disease requiring precise screening, and despite the current lack of specific therapies, potential treatments may emerge through clinical research. Orthoptic assessment, along with ophthalmic examination, allows monitoring disease progression and choosing appropriate therapeutic options. In less advanced cases, prism use can prevent diplopia, while in more severe situations with the development of aesthetic strabismus, surgical intervention may be considered to improve vision without guaranteeing functional recovery.34 Additionally, the importance of psychological support networks is emphasized to address psychosocial distress associated with reduced visual acuity and ocular motility alterations.35
The limited sample size could influence the generalizability of the study results. Therefore, we consider it essential to conduct further research involving a larger number of patients. Current investigative methods for evaluating ocular motility have proven useful in diagnosing and identifying ocular motor alterations in RP patients. The RPE65 gene plays a predominant role in phototransduction and in the biochemical pathways associated with retinal neurotransmission, manifesting with common signs and symptoms in various forms of RP.15
The presence of multigenic hereditary patterns complicates molecular diagnosis, as phenotypic variability is influenced by the complex genetic architectures underlying different forms of RP. Currently, next-generation sequencing is recognized as a powerful tool for mutation screening.20,21 This high-throughput method holds promise as a valuable resource for identifying new genes responsible for diseases and establishing correlations between genotypes and phenotypes, thus contributing to a significant advancement in understanding allele pathogenicity, protein function, and the genetics of the populations involved.20,21 Certainly, advancements in future clinical–instrumental research may contribute to achieving increasingly early diagnoses, enabling timely interventions.
Disclosure
The authors report no conflicts of interest in this work.
References
1. O’Neal TB, Luther EE. Retinitis Pigmentosa. In: StatPearls. StatPearls; 2018.
2. Parmeggiani F, Sato G, De Nadai K, Romano MR, Binotto A, Costagliola C. Clinical and rehabilitative management of retinitis pigmentosa: up-to-date. Curr Genomics. 2011;12(4):250–259. doi:10.2174/138920211795860125
3. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi:10.1186/1750-1172-1-40
4. Pagon RA. Retinitis pigmentosa. Surv Ophthalmol. 1988;33(3):137–177. doi:10.1016/0039-6257(88)90085-9
5. Na KH, Kim HJ, Kim KH, et al. Prevalence, age at diagnosis, mortality and cause of death in retinitis pigmentosa in Korea - a nationwide population-based study. Am J Ophthalmol. 2017;176:157–165. doi:10.1016/j.ajo.2017.01.014
6. Nangia V, Jonas JB, Khare A, Sinha A. Prevalence of retinitis pigmentosa in India: the Central India eye and medical study. Acta Ophthalmol. 2012;90(8):e649–50. doi:10.1111/j.1755-3768.2012.02396.x
7. Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–186. doi:10.1016/j.preteyeres.2018.03.005
8. Hamel CP. Cone Rod Dystrophies. Orphanet J Rare Dis. 2007;2:7. doi:10.1186/1750-1172-2-7
9. Sahel J-A, Marazova K, Audo I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb Perspect Med. 2014. doi:10.1101/cshperspect.a017111
10. Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24–37. doi:10.1016/j.preteyeres.2017.08.004
11. Guadagni V, Novelli E, Piano I, Gargini C, Strettoi E. Pharmacological approaches to retinitis pigmentosa: a laboratory perspective. Prog Retinal Eye Res. 2015;2015:1.
12. Priya S, Nampoothiri S, Sen P, Sripriya S. Bardet–biedl syndrome: genetics, molecular pathophysiology, and disease management. Indian J Ophthalmol. 2016;64(9):620–627. doi:10.4103/0301-4738.194328
13. Jay M. On the heredity of retinitis pigmentosa. Br J Ophthalmol. 1982;66(7):405–416. doi:10.1136/bjo.66.7.405
14. Cai CX, Locke KG, Ramachandran R, Birch DG, Hood DC. A comparison of progressive loss of the ellipsoid zone (EZ) band in autosomal dominant and X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55(11):7417–7422. doi:10.1167/iovs.14-15013
15. Malvasi M, Casillo L, Avogaro F, Abbouda A, Vingolo EM. Gene therapy in hereditary retinal dystrophies: the usefulness of diagnostic tools in candidate patient selections. Int J Mol Sci. 2023;24:13756. doi:10.3390/ijms241813756
16. Havertape SA, Cruz OA, Chu FC. Sensory strabismus--eso or exo? J Pediatr Ophthalmol Strabismus. 2001;38(6):327–330. doi:10.3928/0191-3913-20011101-05
17. Miyata M, Oishi A, Ogino K, et al. Relationship between ocular deviation and visual function in retinitis pigmentosa. Sci Rep. 2018;8:14880. doi:10.1038/s41598-018-33211-6
18. Hwang JM, Kim J. Cyclic exotropia associated with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2006;244(11):1549–1551. doi:10.1007/s00417-005-0231-0
19. Migliorini R, Comberiati AM, Galeoto G, Fratipietro M, Arrico L. Eye motility alterations in retinitis pigmentosa. J Ophthalmol. 2015;2015:145468. doi:10.1155/2015/145468
20. Donato L, Alibrandi S, Scimone C, et al. The impact of modifier genes on cone-rod dystrophy heterogeneity: an explorative familial pilot study and a hypothesis on neurotransmission impairment. PLoS One. 2022;17(12):e0278857. PMID: 36490268; PMCID: PMC9733859. doi:10.1371/journal.pone.0278857
21. Scimone C, Donato L, Alafaci C, et al. High-throughput sequencing to detect novel likely gene-disrupting variants in pathogenesis of sporadic brain arteriovenous malformations. Front Genet. 2020;11:146. PMID: 32184807; PMCID: PMC7059193. doi:10.3389/fgene.2020.00146
22. Scimone C, Bramanti P, Ruggeri A, et al. CCM3/SERPINI1 bidirectional promoter variants in patients with cerebral cavernous malformations: a molecular and functional study. BMC Med Genet. 2016;17(1):74. PMID: 27737651; PMCID: PMC5064884. doi:10.1186/s12881-016-0332-0
23. Scimone C, Bramanti P, Ruggeri A, et al. Detection of novel mutation in ccm3 causes familial cerebral cavernous malformations. J Mol Neurosci. 2015;57(3):400–403. PMID: 26115622. doi:10.1007/s12031-015-0606-6
24. Migliorini R, Fratipietro M, Segnalini A, Arrico L. Persistent vertical diplopia after cataract surgery: a case report. Clin Ter. 2013;164(1):e31–3. doi:10.7417/CT.2013.1518
25. Migliorini R, Malagola R, Comberiati AM, Arrico L. Inferior oblique weakening and abnormal head position: controlled myotomy versus recession. J Ophthalmol. 2016. doi:10.1155/2016/1725484
26. Martinez-Thompson JM, Diehl NN, Holmes JM, Mohney BG. Incidence, types, and lifetime risk of adult-onset strabismus. Ophthalmology. 2014;121(4):877–882. doi:10.1016/j.ophtha.2013.10.030
27. Duman R, Atilla H, Çatak E. Characteristics of Anisometropic Patients with and without Strabismus. Turk J Ophthalmol. 2018;48(1):23–26. doi:10.4274/tjo.44342
28. Ito N, Miura G, Shiko Y, Kawasaki Y, Baba T, Yamamoto S. Progression rate of visual function and affecting factors at different stages of retinitis pigmentosa. Biomed Res Int. 2022;2022:7204954. doi:10.1155/2022/7204954
29. Hiroaki O, Kazunori O, Nobuo S, Kazuya Y, Shuhei Y, Shotai K. Decline of vertical gaze and convergence with aging. Gerontology. 2004;50(3):177–178. doi:10.1159/000076777
30. Pickwell LD. The increase in convergence inadequacy with age. Ophthalmic Physiol Opt. 1985;1985:1.
31. Bruce AS, Atchison DA, Bhoola H. Accommodation-convergence relationships and age. Invest Ophthalmol Vis Sci. 1995;36(2):406–413.
32. Fishman GA. Retinitis pigmentosa. Genet Percent Arch Ophthalmol. 1978;96(5):822–826. doi:10.1001/archopht.1978.03910050428005
33. Marsh IB. We need to pay heed to the psychosocial aspects of strabismus. Eye. 2015;29(2):238–240. PMID: 25475233; PMCID: PMC4330292. doi:10.1038/eye.2014.283
34. Al Shehri F, Duan L, Ratnapalan S. Psychosocial impacts of adult strabismus and strabismus surgery: a review of the literature. Can J Ophthalmol. 2020;55(5):445–451. PMID: 33131636. doi:10.1016/j.jcjo.2016.08.013
35. Mojon-Azzi SM, Kunz A, Mojon DS. The perception of strabismus by children and adults. Graefes Arch Clin Exp Ophthalmol. 2011;249(5):753–757. PMID: 21063886. doi:10.1007/s00417-010-1555-y
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.