")
Back to Journals » Drug Design, Development and Therapy » Volume 8
A new recombinant factor VIII: from genetics to clinical use
Authors Santagostino E
Received 26 August 2014
Accepted for publication 13 October 2014
Published 12 December 2014 Volume 2014:8 Pages 2507—2515
DOI https://doi.org/10.2147/DDDT.S73241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Elena Santagostino
Angelo Bianchi Bonomi Hemophilia and Thrombosis Center, Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy
Abstract: Advances in recombinant technology and knowledge about coagulation factor VIII (FVIII) are building a platform for new therapeutic options in patients with hemophilia A. The development of turoctocog alfa, a novel, high-purity, third-generation, B-domain truncated recombinant FVIII, has been produced and formulated without the use of animal-derived or human serum-derived components, in the wake of understanding of the new biochemical characteristics of FVIII, namely its protein structure, and glycosylation and sulfating patterns. Culture conditions and a five-step purification process have been developed to optimize the safety of turoctocog alfa. The results of two pilot clinical trials using turoctocog alfa confirmed high safety levels, with no patient developing inhibitors during the period of observation. The purpose of this review is to describe briefly the molecular and biological properties of turoctocog alfa, together with details of its clinical development, with emphasis on the needs of patients with hemophilia A.
Keywords: hemophilia A, recombinant factor VIII, turoctocog alfa, purification, inhibitor, safety
Letter to the editor has been published
Introduction
Coagulation factor VIII (FVIII) is the nonenzymatic cofactor of activated factor IX (FIXa) in the activation of factor X (FX).1 When proteolytically activated, FVIIIa interacts with FIXa to form a tight noncovalent complex on the membrane phospholipids of activated platelets that binds to and converts FX to the activated proteinase form (FXa).2,3
Hemophilia A is an X chromosome-linked bleeding disorder caused by mutations in the gene coding for FVIII.1 Patients with mild, moderate, and severe disease have a deficiency of FVIII activity in plasma with levels of 5%–40%,3 1%–5%,2,3 and <1%,2,3 respectively. Patients with severe hemophilia A are at risk of uncontrolled and often spontaneous hemorrhages into joints, muscles, or internal organs, or excessive bleeding after injury or surgery.1 Recurrent bleeding episodes may lead to progressive arthropathy and muscle contractures, often associated with chronic pain and disability.1
FVIII replacement therapy has been the cornerstone in the treatment of hemophilia and has progressed over time from the use of blood transfusions to the use of cryoprecipitates in the 1960s, plasma-derived FVIII (pdFVIII) concentrates in the 1970s, and recombinant products in the 1990s.4,5 Manufacture of recombinant FVIII (rFVIII) evolved over decades and provided products that were classified depending on whether animal-derived or human-derived proteins were used during manufacturing and in the final formulation.6
This article reviews the molecular aspects relevant for full functionality of rFVIII and translates the advances of a novel rFVIII, turoctocog alfa, in the setting of its specific pharmacological properties and safety profile as assessed in trials involving patients with hemophilia A.7,8
Recombinant FVIII products
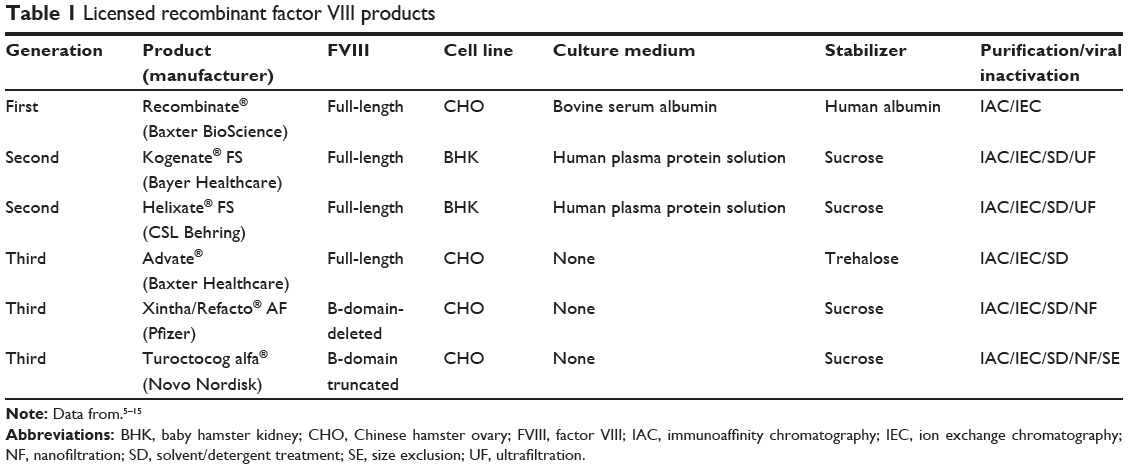
rFVIII products were developed to improve the safety of pdFVIII concentrates. Three different generations of rFVIII products are currently available, including:6 first-generation products using animal-derived proteins in the cell culture medium and human serum albumin in the final formulation to stabilize FVIII; second-generation products using human-derived proteins in the culture medium but with no albumin added in the final formulation; and third-generation products manufactured with no animal or human proteins other than FVIII either during processing or in the final formulation.
Table 1 reports the characteristics of licensed rFVIII products compared with turoctocog alfa.5–15 rFVIII molecules may be full-length, B-domain-deleted, or B-domain-truncated. The B-domain is generally believed to be unnecessary for coagulant activity; however, the novel properties of this domain in the life cycle of FVIII and in the immune response of hemophilia patients have been gradually revealed.16,17 Moreover, the cellular host system and culture conditions are of the utmost importance for the pattern of post-translational modifications in gene therapy constructs.1 Table 1 also lists a number of methods for inactivation/removal of contaminating pathogens (eg, ultrafiltration, solvent/detergent, nanofiltration) that have been gradually added to successive generations of products to enhance their safety, not only as regards known pathogens but also unknown agents, eg, prions. As part of this progress, development of a new rFVIII molecule by engineering its physicochemical properties is of great interest in the improvement of clinical outcomes.
|
Table 1 Licensed recombinant factor VIII products |
Molecular characterization of rFVIII
Protein structure
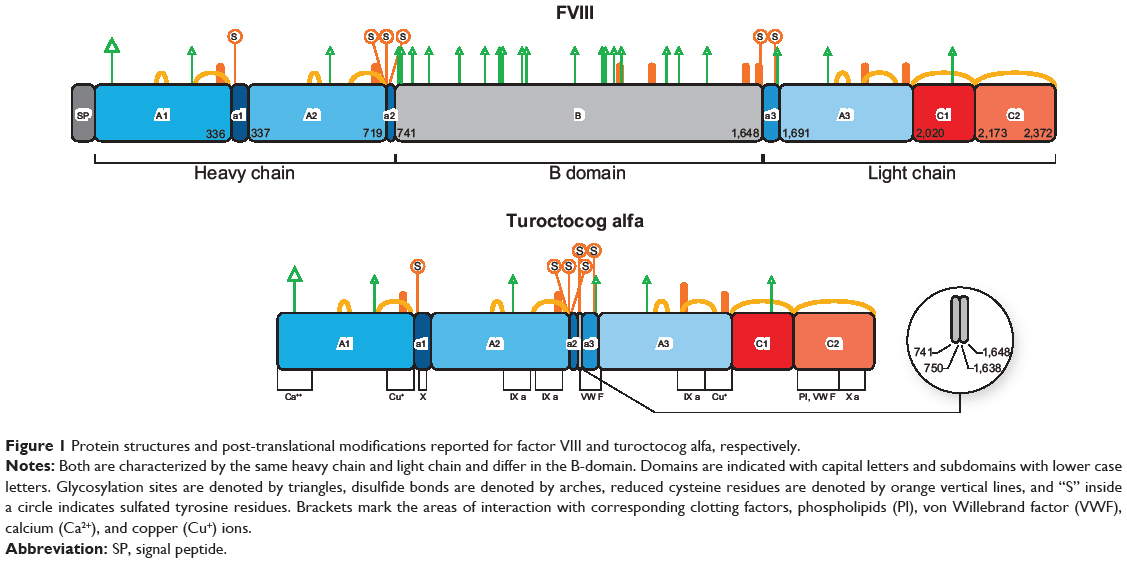
The human FVIII gene is localized on the long arm of the X chromosome and consists of 26 exons and introns, for a total length of 9 kbp in coding sequence.18,19 Sinusoidal endothelial and Kupffer cells in the liver are the major site of FVIII expression.20,21 The gene encodes a large precursor glycoprotein of 2,332 amino acid residues (the maximum length),20 consisting of six structural domains and three acidic subdomains, organized in a heavy chain [A1(a1)A2(a2)B] and light chain [(a3)A3C1C2], as shown in Figure 1.
|
Figure 1 Protein structures and post-translational modifications reported for factor VIII and turoctocog alfa, respectively. |
Every domain plays a physiological role (see below) throughout the life cycle of FVIII, from biosynthesis to clearance. A particular case is the 908 amino acid residue B-domain, which does not seem to be required for FVIII clotting activity but is important for processing, intracellular transport, and secretion of FVIII protein.16 Today we could not definitely exclude potential post-secretion B-domain role during FVIII activation, platelet binding, and inactivation.16 The B-domain is partially removed from mature FVIII, so that several truncated B-domain variants of FVIII circulate in the bloodstream. Complete or partial deletions of the B-domain are not associated with significant differences in procoagulant properties of these FVIII variants.16
Twenty years of experimental research have shown that engineering B-domain deletions results in increased FVIII secretion in most expression systems.1,16 A variant chosen among progressively finer B-domain truncated forms with a linker peptide of 14 residues has shown useful pharmaceutical properties (ie, improved efficiency of manufacturing, long-term stability); this so-called B-domain-deleted FVIII was developed clinically as a second-generation and third-generation product.22,23
Physiological sulfation of rFVIII
Sulfation is required for full activity of FVIII. When rFVIII is expressed in the presence of chlorate, a potent inhibitor of protein sulfation, or when tyrosine residues are replaced by other amino acids, the functional activity of secreted FVIII is reduced 5-fold.24
Post-translational cellular processing of the FVIII precursor enables sulfation of tyrosine residues in the Golgi apparatus, mediated by a sulfotransferase.25 There are six potential tyrosine sulfation sites on the FVIII molecule, ie, four on the heavy chain (at amino acid residues 346, 718, 719, and 723) and two on the light chain (residues 1664 and 1680).24 All the six sulfation sites are required to modulate FVIII activity (Table 2).26,27 Sulfation of key tyrosine residues is crucial not only for the function of FVIII, but also for its stability and binding to von Willebrand factor (VWF).24,26,27

|
Table 2 Function specificity of sulfated tyrosine residues in factor VIII |
When therapeutic FVIII concentrate (not containing VWF) is infused into patients, complex formation between FVIII and VWF occurs within seconds.28 In the circulating complex, VWF serves as a chaperone, protecting FVIII from premature inactivation (cleavage by FXa and activated protein C), thereby helping to maintain a constant level of circulating FVIII 29,30 Lack of interaction between FVIII and VWF may impair the pharmacokinetics of FVIII. Mutations in the binding sites for VWF (eg, Trp 2229) may induce a conformational change in FVIII, rendering the molecule antigenically distinct from wild-type FVIII.31,32 Therefore, physical interaction between FVIII and VWF may modulate the immunogenicity of FVIII, ie, mask epitopes and prevent entry of FVIII into antigen-presenting cells.32,33 In the absence of binding to WWF, infused rFVIII remains free in the circulation and is therefore more easily targeted by the immune system.33
It has been reported that rFVIII products contain a variable proportion of protein that is unable to bind VWF.34 Recent studies have shown that this may be attributable to the lack of Tyr-1680 sulfation.35,36 In summary, the potential impact of FVIII sulfation and, in particular of tyrosine 1680, on the functionality and immunogenicity of FVIII deserves further attention.
Molecular instability
The FVIII molecule is intrinsically unstable because it has inherently labile structural elements. An example is the amino acid backbone, which has the potential to undergo acid-base and redox reactions and is also prone to temperature-dependent, pH-dependent, and concentration-dependent precipitation.19,37 FVIII concentrates are prone to supramolecular aggregation as a result of their colloidal properties.37 This latest aspect has caused speculation about differential immunogenicity of rFVIII products since baby hamster kidney cells produce more FVIII protein in aggregate form, which could affect tertiary structure and recognition by the immune system.38,39
If left unaddressed, molecular instability could impact the pharmacological properties of FVIII products. To address these issues, molecular level technologies can be employed to ameliorate its physicochemical properties, because design of rFVIII with improved efficacy can be achieved by optimization of drug molecular stability.
Several functional properties can be ascribed to the carbohydrate moieties of a highly glycosylated protein such as FVIII. Glycosylation influences stability and modulates immunogenic properties.40,41 Some glycans carried by rFVIII but not by pdFVIII products may increase the level of antigenicity.42 Stability is also attributed to the conservation of binding sites for metals. It was demonstrated that the stability of a new rFVIII is assured by two metal binding sites, one in domain A3 for Cu+ and one in domain A1 for Zn2+, determined by X-ray fluorescence and X-ray crystallography.43 The intrinsic stability of rFVIII has to be preserved under a number of conditions, from the manufacturing process and lyophilization to storage and continuous infusion. A stable product without alterations in its protein characteristics and a very low propensity to form aggregates is desired.43
Clinical issues with rFVIII treatment
Prophylaxis is considered gold standard for patients with severe hemophilia; even secondary or “tertiary” prophylaxis have been shown to have more beneficial effects than treatment on demand.44 Research on newer rFVIII products is rapidly progressing to meet a number of needs, including but not limited to, the efficacy and safety of prophylactic treatment.45–47
In terms of the main adverse effects, the most serious complication of replacement therapy for hemophilia A is the occurrence of neutralizing alloantibodies (inhibitors) against administered FVIII. Approximately 25%–30% of severely affected patients (FVIII: C<1%) develop inhibitors, typically during the first 20–50 days of exposure.48
Agents that bypass inhibitors, such as recombinant activated factor VII and activated prothrombin complex concentrates, are available to control and prevent bleeding;49 however, their efficacy is suboptimal compared with FVIII replacement therapy. Induction of immune tolerance is the main treatment option for eradicating inhibitors and has high success rates (60%–80%), although it is extremely demanding and costly.50,51 Genetic background and acquired factors interplay and contribute to development of inhibitors.52 Treatment-related determinants include age at first FVIII exposure, treatment intensity (dose and frequency of infusions) and modality (prophylactic or on-demand regimens), and factors linked to concentrate source and type (ie, rFVIII versus pdFVIII, B-domain-deleted versus full-length FVIII).38,51
In recent years, three systematic reviews were carried out to address the issue of immunogenicity of plasma-derived rFVIII and rFVIII in previously untreated patients. However, due to a number of methodological limitations, mainly differences in study design and patient populations, no conclusive and definite answer has been provided.48,53,54
In addition, it has to be considered that the above-mentioned meta-analyses compared products grouped into two classes, although each class included heterogeneous FVIII derivatives; in particular, pdFVIII can be either of high, low, or intermediate purity, or with or without VWF content, while rFVIII can differ in terms of manufacturing process, cell line, and molecular structure (Table 1). Recently, in a large prospective study of previously untreated patients, the second-generation, full-length rFVIII derived from a baby hamster kidney cell line was associated with a higher risk of development of inhibitors than the third-generation, full-length rFVIII derived from a Chinese hamster ovary cell line,55 and this unexpected finding has no plausible biological explanation to date.38
Conflicting results were also provided by meta-analyses of studies carried out in previously treated patients. In this context, some authors have reported an increased incidence of de novo inhibitors with B-domain-deleted rFVIII in comparison with full-length rFVIII.56 In contrast, a subsequent meta-analysis was not able to show any significant difference in the risk of inhibitors associated with use of different rFVIII products in previously treated patients.57
Turoctocog alfa
Current rFVIII products have a high degree of safety and efficacy; however, improvement is still possible. New bioengineering technologies are being actively applied in the search for new recombinant products, and some of them are in preclinical or clinical development.45 Turoctocog alfa is the first of these new products to be approved by the US Food and Drug Administration and European Medicines Agency. Turoctocog alfa is a novel, third-generation, B-domain truncated rFVIII produced and formulated without the use of animal-derived or human serum-derived components.58
Cell line
Chinese hamster ovary cells were transfected with the turoctocog alfa-coding plasmid and selected with the dihydrofolate reductase system, leading to a clonal suspension of producer cells cultivated in medium free of animal components.58 A chemically defined and animal component-free growth medium was used for large-scale fermentation.58
Manufacturing process
The advanced purification techniques utilized for production of turoctocog alfa led to a final product formulated without albumin and animal-derived or human-derived materials. The manufacturing process involves a five-step procedure:5,58
- Solvent/detergent inactivation to eliminate potential viral contaminants endowed with lipid envelopes.
- Immunoaffinity chromatography that allows isolation of pure molecules of rFVIII utilizing a monoclonal F25 antibody directed against the integral A2 domain of the heavy chain, including residue 740 of the amino acid sequence; this step excludes incomplete molecules, particularly those with damage to the heavy chain in the position of greatest instability, 720, being a primary inactivating site by proteolysis.58 A recombinant monoclonal antibody directed against the A2 domain is produced in the same cell line used for production of turoctocog alfa, and it is developed in an animal component- and serum- free process.58
- Ion-exchange chromatography to remove impurities produced by the cell line, and electrical charges to remove impurities in a further purification step.
- Nanofiltration, where a 20 nm pore size filter is used to remove potential viruses, including those with a protein envelope, such as Parvovirus.
- A gel filtration step to provide the pure protein, turoctocog alfa, eliminating aggregated/agglutinated forms of the protein.59
Protein
The primary structure of the turoctocog alfa protein (also called N8) has been identified by a combination of amino acid sequencing, peptide mapping, and mass spectrometry.58 In turoctocog alfa, the 908 amino acid residue wild-type B-domain was reduced to a 21 amino acid residue linker.59 This has the sequence SFSQNSRHPSQNPPVLKRHQR.58 The sequence represents ten amino residues from the N-terminal of the original B-domain linked to 11 amino acid residues from the C-terminal of the B-domain.58 Attention must be paid to the last four amino acid residues (R), which represent the sequence RXXR (where X is a random residue).58 This motif is the site where the protease furin cleaves the single-chain precursor of FVIII, giving rise to a light and heavy chain in the Golgi apparatus.60 Thus, the short but functionally complete B-linker efficiently facilitates the expression of FVIII heavy (A1-A2-linker) and light (A3-C1-C2) chains indistinguishable from those formed from full-length FVIII.58
In addition, the last 20 amino acid residues of the heavy chain represent an acidic subdomain, being the region required for optimal interaction with thrombin and other proteins. In particular, this is the point where FVIII is cleaved by thrombin.1,58 The light chain of N8 was found to consist of two similar species implicated in the formation of tenase.43 Both of these light chain variants are known to be present in pdFVIII.61 These specific features of the primary structure of turoctocog alfa highlight the fact that the molecule was developed with the goal of producing smaller but more efficient bioengineered FVIII.
Glycosylation
Manufacturing of turoctocog alfa was a key advance in bioengineering the glycosylation pathways of the expression system. The aim was to synthesize an rFVIII with the most “humanized” glycosylation possible. The glycoengineering efforts toward improvement of mammalian cell (Chinese hamster ovary) glycosylation has been directed to eliminating immunogenic glycotopes in turoctocog alfa.
Post-translational modifications of turoctocog alfa include N-linked and O-linked glycosylations. Prevalent N-linked glycosylation sites occur at the terminal amide groups of asparagine (Asn) residues present in the Asn-X-Thr/Ser sequence.58,62 O-linked glycosylation refers to linkage of carbohydrate structures at the serine (Ser) or threonine (Thr) residues through the Asn-X-Thr/Ser motif.58,63
Within the A and C domains of FVIII there are N-linked glycosylations at four Asn residues, ie, Asn41, Asn239, Asn1810, and Asn2118. The position of the four N-glycosylation sites known from pdFVIII were all confirmed in turoctocog alfa by mass spectrometry of peptide fragments derived from the tryptic map.58
No significant differences were found between rFVIII and pdFVIII with regard to their oligosaccharide profiles. In both, the glycomap of N-linked glycans characterizes the chains linked to Asn41 and Asn1810 as core fucosylated and biantennary chains complex structures, with one or two sialic acids Asn239 carrying high-mannose, hybrid and complex structures and Asn2118 carrying only high-mannose structures.35,58
The O-linked glycosylation sites of FVIII are prevalent on the B-domain, where at least seven carbohydrate structures have been reported.64 The linker N8 sequence includes one of these sites at Ser in position Ser750.58,60 Curiously, this is the only O-linked glycan for which the precise location and structure on FVIII is known.64 The O-glycan composition of N8 is a doubly sialylated disaccharide, which is a common eukaryotic O-linked glycan.58 This structure was confirmed by consecutive enzymatic digestion of N8. The site is glycosylated in approximately 65% of turoctocog alfa.58
In brief, the oligosaccharide structures of the novel rFVIII and pdFVIII are very similar, with mainly small, quantitative differences, and heterogeneous glycosylation is present in both products.35,58
Sulfation
Analysis of turoctocog alfa for the degree of sulfation demonstrated that all six tyrosine sulfation sites are utilized.58 Detection and quantification of tyrosine sulfation in turoctocog alfa was performed with care because sulfation is a potentially labile modification.35,58 Chromatographic and mass spectrometric techniques after tryptic digestion provided the following results:5 a sulfated tyrosine residue in position 346 of the heavy chain in a single high performance liquid chromatography fraction; three other heavy chain sulfated tyrosines (residues 718, 719, and 723) in three different high-performance liquid chromatography fractions and mass analysis; and the presence of the last two sulfated residues (light chain tyrosine 1664 and 1680) in the individual peptide isolated from tryptic digestion of native N8.
All these six sulfated residues are located in the acidic regions of domains A1, A2, and A3.
Despite technical difficulties with high molecular weight proteins like FVIII, particular attention has been paid to sulfation of tyrosine 1680, because it could impact on binding to VWF.26,36,65 Recently, it has become possible to determine the degree of Tyr1680 sulfation in different rFVIII products (by nanoliquid chromatography coupled with electrospray tandem mass spectrometry).36 The percentage of nonsulfated Tyr1680 ranged from 1% to 6.5% for second-generation rFVIII to up to 15% for third-generation rFVIII, while the percentage of nonsulfated pdFVIII was negligible.26 These results were confirmed in a second study using a proteomics analysis technique (Table 3).35

|
Table 3 Levels of nonsulfated tyrosine in rFVIII |
In summary, turoctocog alfa is sulfated in all tyrosine moieties. This full sulfation was stable for a year in an end-of-shelf life analysis, an indication for physiological stability in circulation.36,66 Moreover, a number of molecular interventions have been shown to stabilize turoctocog alfa against almost all of the major physicochemical instability factors encountered during manufacturing, storage, and clinical use.22,58,65 Improved long-term stability was obtained for turoctocog alfa to accommodate a variety of everyday living conditions in hemophiliac patients and make treatment feasible with ease in any situation.
Clinical development
The clinical development plan for turoctocog alfa, known as the Guardian™ program, has involved several pharmacokinetic, field, efficacy, and safety trials.67–70 Two Phase III trials investigated the safety and efficacy of turoctocog alfa in previously treated patients, ie, Guardian 1 in adult/adolescent patients $12 years and Guardian 3 in pediatric patients aged <12 years.69,70 Overall, the two trials included 210 previously treated patients with severe hemophilia A, involving 150 adults exposed to turoctocog alfa for a mean of 85 (11–172) days and 60 pediatric patients treated for a mean of 60 (range 20–104) exposure days.69,70
The two Phase III Guardian trials specifically evaluated the incidence of inhibitors in previously treated patients, potential immunogenicity and neoantigenicity of rFVIII leading to neutralizing antibodies being the main objective of the studies.69,70 No patient developed inhibitors during either trial.71,72 One adult patient was excluded from the analysis because of a positive inhibitor test at baseline. Interestingly, this patient received 13 infusions of turoctocog alfa with no recurrence of inhibitors.69 Patients in the two trials could continue turoctocog alfa in the ongoing long-term safety and efficacy Guardian 3 extension trial.71
With regard to adverse events other than development of inhibitors, a total of six nonserious events were evaluated by the investigators as possibly or probably related to turoctocog alfa, comprising four in the adult trial (hypertension, sinus tachycardia, and insomnia in a 27-year-old patient; raised liver enzymes in a 37-year-old patient) and two in the pediatric trial (contusion and incorrect dose administration in one patient).69,70
Over and above these pivotal studies in previously treated patients, and as required by regulatory authorities, the Guardian 4 trial is ongoing to assess the safety and efficacy of turoctocog alfa in previous untreated patients.71
Conclusion
The development of turoctocog alfa as a new option for the treatment of hemophilia A was based on advanced bioengineering techniques to address unresolved molecular issues concerning rFVIII products. The novelty of turoctocog alfa required adequate and extensive documentation of clinical safety and efficacy.71 Phase III trials met their primary endpoint regarding the safety of turoctocog alfa, with no induction of FVIII inhibitors.69,70 In the ongoing extension phase of about 3 years, the prophylactic turoctocog alfa regimen resulted in stabilization of bleeding rates at a low level.72 Future studies will add further important information to the database regarding the long-term safety and efficacy of turoctocog alfa in specific patient populations.71
Disclosure
The author acted as a paid consultant and received an unrestricted research grant from Novo Nordisk, and wrote and reviewed the manuscript. Editorial assistance was provided by Airon Communications, Milan, Italy, with financial support from Novo Nordisk, in compliance with international guidelines for good publication practice.
References
Orlova NA, Kovnir SV, Vorobiev II, Gabibov AG, Vorobiev AI. Blood clotting factor VIII: from evolution to therapy. Acta Naturae. 2013;5(2):19–39. | ||
Fay PJ. Activation of factor VIII and mechanisms of cofactor action. Blood Rev. 2004;18(1):1–15. | ||
Sichler K, Kopetzki E, Huber R, Bode W, Hopfner KP, Brandstetter H. Physiological fIXa activation involves a cooperative conformational rearrangement of the 99-loop. J Biol Chem. 2003;278(6):4121–4126. | ||
Shapiro AD. Anti-hemophilic factor (recombinant), plasma/albumin-free method (octocog-alpha; ADVATE) in the management of hemophilia A. Vasc Health Risk Manag. 2007;3(5):555–565. | ||
NovoEight (antihemophilic factor [recombinant], prescribing information). Novo Nordisk. Bagsværd, Denmark; 2011. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR__Product_Information/human/002719/WC500157553.pdf. Accessed June 30, 2014. | ||
Franchini M, Mannucci PM. Hemophilia A in the third millennium. Blood Rev. 2013;27(4):179–184. | ||
Haddley K. Turoctocog alfa for the treatment of hemophilia A. Drugs Today (Barc). 2014;50(2):121–131. | ||
Lentz SR, Seremetis S, Staber J, Kulkarni R. Turoctocog alfa and drug development for hemophilia A. Expert Opin Orphan Drugs. 2014;2(4):419–431. | ||
Brooker M. Registry of clotting factor concentrates. 9th ed. Montréal, QC, Canada: World Federation of Hemophilia; 2012. Available from: http://www1.wfh.org/publication/files/pdf-1227.pdf. Accessed October 24, 2014. | ||
Recombinate (antihemophilic factor [recombinant], prescribing information). Deerfield, IL, USA: Baxter Healthcare Corporation; 2010. Available from: http://www.baxter.com/downloads/healthcare_professionals/products/recombinate_pi_5ml.pdf. Accessed June 30, 2014. | ||
Kogenate FS (antihemophilic factor [recombinant], prescribing information). Berlin, Germany: Bayer Schering Pharma AG; 2006. Available from: http://www.abopharmaceuticals.com/ProductSheets/KogenateFS.pdf. Accessed June 30, 2014. | ||
Helixate FS (antihemophilic factor [recombinant], prescribing information). Kankakee, IL, USA: CSL Behring; 2013. Available from: http://labeling.cslbehring.com/PI/US/HelixateFS/EN/HelixateFS-Prescribing-Information.pdf. Accessed June 30, 2014. | ||
ReFacto (antihemophilic factor [recombinant], prescribing information). Madison, NJ, USA: Wyeth Pharmaceuticals Inc; 2005. Available from: http://www.abopharmaceuticals.com/ProductSheets/Refacto.pdf. Accessed June 30, 2014. | ||
Advate [antihemophilic factor (recombinant)] [prescribing information]. Deerfield, IL, USA: Baxter Healthcare Corporation; 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_Product_Information/human/000520/WC500022467.pdf. Accessed June 30, 2014. | ||
Refacto AF (antihemophilic factor [recombinant], prescribing information). New York, NY, USA: Pfizer; 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR__Product_Information/human/000232/WC500049008.pdf. Accessed June 30, 2014. | ||
Pipe SW. Functional roles of the factor VIII B domain. Haemophilia. 2009;15(6):1187–1196. | ||
Grushin K, Miller J, Dalm D, et al. Lack of recombinant factor VIII B-domain induces phospholipid vesicle aggregation: implications for the immunogenicity of factor VIII. Haemophilia. 2014;20(5):723–731. | ||
Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature. 1984;312(5992):326–330. | ||
Solá RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. Bio Drugs. 2010;24(1):9–21. | ||
Hollestelle MJ, Thinnes T, Crain K, et al. Tissue distribution of factor VIII gene expression in vivo – a closer look. Thromb Haemost. 2001;86(3):855–861. | ||
Shahani T, Covens K, Lavend’homme R, et al. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J Thromb Haemost. 2014;12(1):36–42. | ||
Sandberg H, Almstedt A, Brandt J, et al. Structural and functional characteristics of the B-domain-deleted recombinant factor VIII protein, r-VIII SQ. Thromb Haemost. 2001;85(1):93–100. | ||
Recht M, Nemes L, Matysiak M, et al. Clinical evaluation of moroctocog alfa (AF-CC), a new generation of B-domain deleted recombinant factor VIII (BDDrFVIII) for treatment of haemophilia A: demonstration of safety, efficacy, and pharmacokinetic equivalence to full-length recombinant factor VIII. Haemophilia. 2009;15(4):869–880. | ||
Pittman DD, Wang JH, Kaufman RJ. Identification and functional importance of tyrosine sulfate residues within recombinant factor VIII. Biochemistry. 1992;31(13):3315–33325. | ||
Niehrs C, Huttner WB. Purification and characterization of tyrosylprotein sulfotransferase. EMBO J. 1990;9(1):35–42. | ||
Michnick DA, Pittman DD, Wise RJ, Kaufman RJ. Identification of individual tyrosine sulfation sites within factor VIII required for optimal activity and efficient thrombin cleavage. J Biol Chem. 1994;269(31):20095–20102. | ||
Kaufman RJ. Post-translational modifications required for coagulation factor secretion and function. Thromb Haemost. 1998;79(6):1068–1079. | ||
Gilbert GE, Drinkwater D, Barter S, Clouse SB. Specificity of phosphatidylserine-containing membrane binding sites for factor VIII. Studies with model membranes supported by glass microspheres (lipospheres). J Biol Chem. 1992;267(22):15861–15868. | ||
Fay PJ, Coumans JV, Walker FJ. von Willebrand factor mediates protection of factor VIII from activated protein C-catalyzed inactivation. J Biol Chem. 1991;266(4):2172–2177. | ||
Nogami K, Shima M, Nishiya K, et al. A novel mechanism of factor VIII protection by von Willebrand factor from activated protein C-catalyzed inactivation. Blood. 2002;99(11):3993–3998. | ||
Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4(4):558–563. | ||
Dasgupta S, Repessé Y, Bayry J, et al. VWF protects FVIII from endocytosis by dendritic cells and subsequent presentation to immune effectors. Blood. 2007;109(2):610–612. | ||
Gringeri A, Ofosu FA, Grancha S, et al. Understanding FVIII/VWF complex – report from a symposium of XXIX WFH meeting 2010. Haemophilia. 2012;18(3):469–475. | ||
Lin Y, Yang X, Chevrier MC, et al. Relationships between factor VIII: Ag and factor VIII in recombinant and plasma-derived factor VIII concentrates. Haemophilia. 2004;10(5):459–469. | ||
Kannicht C, Ramström M, Kohla G, et al. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res. 2013;131(1):78–88. | ||
Grancha S, Navajas R, Marañón C, et al. Incomplete tyrosine 1680 sulphation in recombinant FVIII concentrates. Haemophilia. 2011;17(4):709–710. | ||
Chi EY, Krishnan S, Kendrick BS, et al. Roles of conformational stability and colloidal stability in the aggregation of recombinant human granulocyte colony stimulating factor. Protein Sci. 2003;12(5):903–913. | ||
Kessler CM, Iorio A. The Rodin (Research Of Determinants of INhibitor Development among PUPs with haemophilia) study: the clinical conundrum from the perspective of haemophilia treaters. Haemophilia. 2013;19(3):351–354. | ||
Pahl S, Pavlova A, Driesen J, et al. In vitro characterization of recombinant factor VIII concentrates reveals significant differences in protein content, activity and thrombin activation profile. Haemophilia. 2013;19(3):392–398. | ||
Kosloski MP, Miclea RD, Balu-Iyer SV. Role of glycosylation in conformational stability, activity, macromolecular interaction and immunogenicity of recombinant human factor VIII. AAPS J. 2009;11(3):424–431. | ||
Lenting PJ, Pegon JN, Christophe OD, Denis CV. Factor VIII and von Willebrand factor – too sweet for their own good. Haemophilia. 2010;16 Suppl 5:194–199. | ||
Schilow WF, Schoerner-Burkhardt E, Seitz R. Charge analysis of N-glycans from human recombinant coagulation factor VIII and human FVIII standards. Thromb Haemost. 2004;92(2):427–428. | ||
Svensson LA, Thim L, Olsen OH, Nicolaisen EM. Evaluation of the metal binding sites in a recombinant coagulation factor VIII identifies two sites with unique metal binding properties. Biol Chem. 2013;394(6):761–765. | ||
Gringeri A, Lambert T, Street A, Aledort L; on behalf of the Adolescent/Adult Prophylaxis Expert Working Group of the International Prophylaxis Study Group. Tertiary prophylaxis in adults: is there a rationale? Haemophilia. 2012;18(5):722–728. | ||
Peyvandi F, Garagiola I, Seregni S. Future of coagulation factor replacement therapy. J Thromb Haemost. 2013;11 Suppl 1:84–98. | ||
Dimichele DM, Blanchette V, Berntorp E. Clinical trial design in haemophilia. Haemophilia. 2012;18 Suppl 4:18–23. | ||
Makris M, Calizzani G, Fischer K, et al. EUHASS: The European Haemophilia Safety Surveillance System. Thromb Res. 2011;127 Suppl 2:S22–S25. | ||
Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9(4):418–435. | ||
Teitel JM, Sholzberg M. Current status and future prospects for the prophylactic management of hemophilia patients with inhibitor antibodies. Blood Rev. 2013;27(2):103–109. | ||
Coppola A, Di Minno MN, Santagostino E. Optimizing management of immune tolerance induction in patients with severe haemophilia A and inhibitors: towards evidence-based approaches. Br J Haematol. 2010;150(5):515–528. | ||
Ettingshausen CE, Kreuz W. The immune tolerance induction (ITI) dose debate: does the International ITI Study provide a clearer picture? Haemophilia. 2013;19 Suppl 1:12–17. | ||
Kruse-Jarres R. Inhibitors: our greatest challenge. Can we minimize the incidence? Haemophilia. 2013;19 Suppl 1:2–7. | ||
Iorio A, Halimeh S, Holzhauer S, et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma-derived or recombinant factor VIII concentrates: a systematic review. J Thromb Haemost. 2010;8(6):1256–1265. | ||
Franchini M, Coppola A, Rocino A, et al; Italian Association of Hemophilia Centers (AICE) Working Group. Systematic review of the role of FVIII concentrates in inhibitor development in previously untreated patients with severe hemophilia A: a 2013 update. Semin Thromb Hemost. 2013;39(7):752–766. | ||
Gouw SC, van der Bom JG, Ljung R, et al; PedNet and RODIN Study Group. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368(3):231–239. | ||
Aledort LM, Navickis RJ, Wilkes MM. Can B-domain deletion alter the immunogenicity of recombinant factor VIII? A meta-analysis of prospective clinical studies. J Thromb Haemost. 2011;9(11):2180–2192. | ||
Xi M, Makris M, Marcucci M, Santagostino E, Mannucci PM, Iorio A. Inhibitor development in previously treated hemophilia A patients: a systematic review, meta-analysis, and meta-regression. J Thromb Haemost. 2013;11(9):1655–1662. | ||
Thim L, Vandahl B, Karlsson J, et al. Purification and characterization of a new recombinant factor VIII (N8). Haemophilia. 2010;16(2):349–359. | ||
Gagnon P, Cheung CW, Lepin EJ, et al. Minibodies and multimodal chromatography methods: a convergence of challenge and opportunity. Bioprocess Int. 2010;8(2):26–35. | ||
Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92(11):3983–3996. | ||
Lind P, Larsson K, Spira J, et al. Novel forms of B-domain-deleted recombinant factor VIII molecules. Construction and biochemical characterization. Eur J Biochem. 1995;232(1):19–27. | ||
Medzihradszky KF. Characterization of protein N-glycosylation. Methods Enzymol. 2005;405:116–138. | ||
Peter-Katalinic J. Methods in enzymology: O-glycosylation of proteins. Methods Enzymol. 2005;405:139–171. | ||
Mazsaroff I, Yu W, Kelley BD, Vath JE. Quantitative comparison of global carbohydrate structures of glycoproteins using LC-MS and in-source fragmentation. Anal Chem. 1997;69(13):2517–2524. | ||
Leyte A, van Schijndel HB, Niehrs C, et al. Sulfation of Tyr1680 of human blood coagulation factor VIII is essential for the interaction of factor VIII with von Willebrand factor. J Biol Chem. 1991;266(2):740–746. | ||
Nielsen PF, Bak S, Vandahl B. Characterization of tyrosine sulphation in rFVIII (turoctocog alfa) expressed in CHO and HEK-293 cells. Haemophilia. 2012;18(5):e397–e398. | ||
Martinowitz U, Bjerre J, Brand B, et al. Bioequivalence between two serum-free recombinant factor VIII preparations (N8 and ADVATE®) – an open-label, sequential dosing pharmacokinetic study in patients with severe haemophilia A. Haemophilia. 2011;17(6):854–859. | ||
Viuff D, Barrowcliffe T, Saugstrup T, Ezban M, Lillicrap D. International comparative field study of N8 evaluating factor VIII assay performance. Haemophilia. 2011;17(4):695–702. | ||
Lentz SR, Misgav M, Ozelo M, et al. Results from a large multinational clinical trial (Guardian™1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia. 2013;19(5):691–697. | ||
Kulkarni R, Karim FA, Glamocanin S, et al. Results from a large multinational clinical trial (Guardian™3) using prophylactic treatment with turoctocog alfa in paediatric patients with severe haemophilia A: safety, efficacy and pharmacokinetics. Haemophilia. 2013;19(5):698–705. | ||
Laguna P, Vdovin V, Rageliene L, Abad-Franch L, Lindblom A. Overview of a global clinical trial programme with turoctocog alfa, a new recombinant factor VIII: the Guardian™ programme. Presented at the XXIV International Society on Thrombosis and Haemostasis Congress, Amsterdam, The Netherlands, June 29 to July 4, 2013. | ||
Ozelo M, Misgav M, Abdulkarim F, et al. Reductions in annualized bleeding rates over time with turoctocog alfa prophylaxis: 3-year interim results of the Guardian™ 2 extension trial. Haemophilia. 2014;20 Suppl 3:84. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.