")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 17
Bioinformatics Study of Flavonoids From Genus Erythrina As Ace2 inhibitor Candidates For Covid-19 Treatment
Authors Herlina T, Akili AWR, Nishinarizki V, Hardianto A , Latip J
Received 22 January 2024
Accepted for publication 3 May 2024
Published 14 May 2024 Volume 2024:17 Pages 61—70
DOI https://doi.org/10.2147/AABC.S454961
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Maria Miteva
Tati Herlina,1 Abd Wahid Rizaldi Akili,1 Vicki Nishinarizki,1 Ari Hardianto,1 Jalifah Latip2
1Department of Chemistry, Universitas Padjadjaran, Jatinangor, West Java, Indonesia; 2Department of Chemical Sciences, Universiti Kebangsaan Malaysia, Bangi, Selangor, Malaysia
Correspondence: Tati Herlina, Email [email protected]
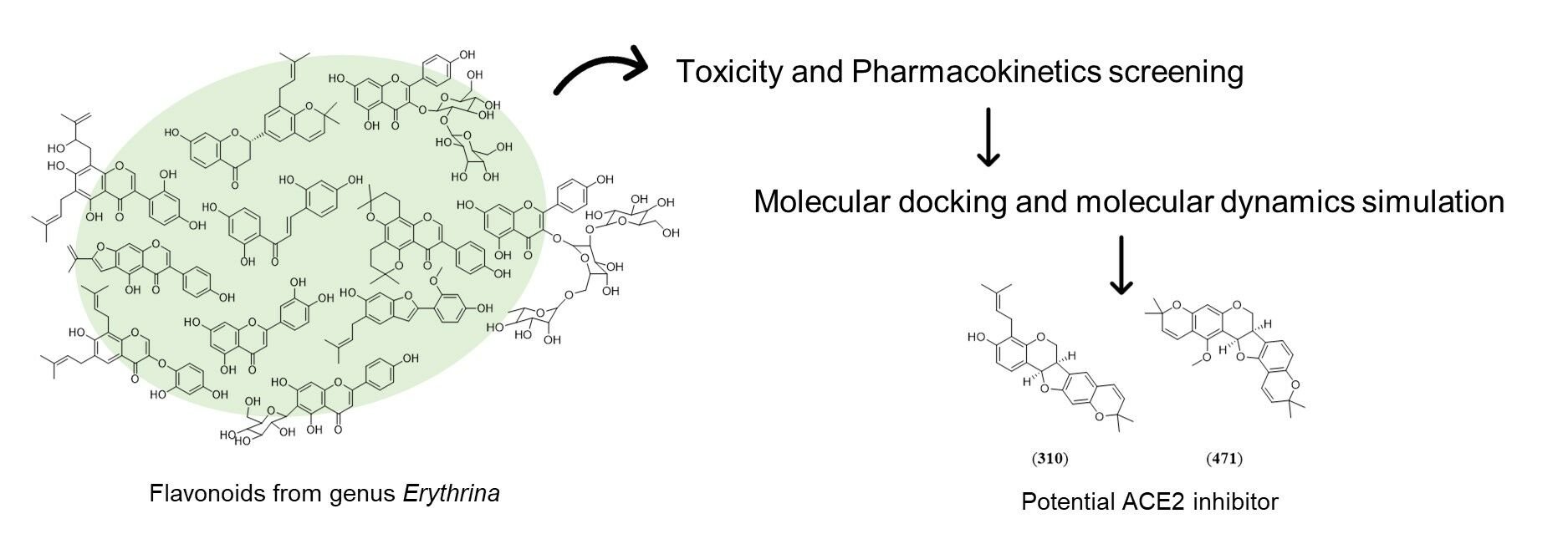
Purpose: This study aimed to screen potential drug candidates from the flavonoids of the genus Erythrina for the Corona Virus Disease 2019 (COVID-19) treatment.
Patients and Methods: A comprehensive screening was conducted on the structures of 473 flavonoids derived from the genus Erythrina, focusing on their potential toxicity and pharmacokinetic profiles. Subsequently, flavonoids that were non-toxic and possessed favorable pharmacokinetic properties underwent further analysis to explore their interactions with the angiotensin-converting enzyme 2 (ACE2) receptor, employing molecular docking and molecular dynamics simulations.
Results: Among 473 flavonoids, 104 were predicted to be safe from being mutagenic, hepatotoxic, and inhibitors of the human ether-a-go-go-related gene (hERG). Among these 104 flavonoids, 18 compounds were predicted not to be substrates of P-glycoprotein (P-gp). Among these 18 flavonoids, gangetinin ( 471) and erybraedin D ( 310) exhibit low binding affinities and root mean square deviation (RMSD) values, indicating stable binding to the ACE2 receptor. The physicochemical attributes of compounds 310 and 471 suggest that they possess drug-like properties.
Conclusion: Gangetinin ( 471) and erybraedin D ( 310) may serve as promising candidates for COVID-19 treatment due to their potential to inhibit the ACE2-RBD interaction. This warrants further investigation into their inhibitory effects on ACE2-RBD binding through in vitro experiments.
Keywords: structure-based virtual screening, Erythrina, flavonoids, angiotensin-converting enzyme 2, corona virus disease 2019
Graphical Abstract:
Introduction
The Corona Virus Disease 2019 (COVID-19) pandemic arising from the spread of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has led to an urgent need for effective treatment. In a recent report covering the period from December 11, 2023, to January 7, 2024, the World Health Organization (WHO) noted a significant increase in COVID-19 related hospitalizations and admissions to Intensive Care Units (ICUs). Specifically, hospitalizations saw a surge of 40%, resulting in over 173.000 admissions. Similarly, ICU admissions rose by 13%, with a total of 1.900 admissions. These figures underscore the severity of the ongoing pandemic and the strain it places on healthcare systems worldwide.1 Since the outbreak of COVID-19, there has been a concerted global effort to identify and develop effective treatments for the virus. This has led to the investigation of a wide array of compounds, both new and repurposed, for their therapeutic potential. Among these are remdesivir, favipiravir, cyanorona-20, hydroxychloroquine, chloroquine, CoViTris2020, taroxaz-104, ChloViD2020, teriflunomide, leflunomide, ivermectin, arbidol, and colchicine. However, to date, none have demonstrated conclusive broad-spectrum efficacy against the virus.2
In the field of drug discovery and development, it is important to identify protein targets that can be targeted using potential therapeutic.3 There are mainly there crucial coronaviral proteins that have been targeted for COVID-19 drug development, these are the spike (S) protein, the main protease (Mpro) enzyme and the RNA-dependent RNA polymerase (RdRp). The spike (S) protein plays key role to gain access into the human cell, making this protein as potential nonenzymatic therapeutic target in anti-coronaviral drug design strategy.4 Nevertheless, this protein is unfixed and changeable day by day from one strain to the newer, as observed in the Omicron variant which has at least 36 new mutations in its spike (S) protein, making this protein is no longer attractive target for designing new therapies against SARS-CoV-2 variants.5
ACE2 (angiotensin-converting enzyme 2) has emerged as a potential therapeutic target for COVID-19 treatment. ACE2 plays a crucial role in the entry of SARS-CoV-2 into host cells. The virus binds to the ACE2 receptors on the surface of host cells, thereby facilitating viral entry and infection. Therefore, blocking this binding event or reducing the accessibility of the virus to the ACE2 receptor represents a potential strategy for preventing the spread of COVID-19.6 Additionally, targeting host cell proteins like ACE2 could reduce the chance of the virus developing resistance7 as host-dependent therapies offer advantages by not directly targeting virus proteins, thereby reducing the likelihood of drug-resistant escape mutants.8
In recent years, natural products including flavonoids have gained attention as potent antiviral agents. Flavonoids have been shown to possess antiviral and anti-inflammatory effects, making them promising candidates for the treatment of COVID-19.9 In addition, many scientific studies have shown that flavonoids have an inhibitory effect on ACE2 receptor; among these flavonoids are luteolin, kaempferol, apigenin, and quercetin.10 The genus Erythrina is a group of medicinal plants rich in flavonoids.11,12 These medical plants have long been used to treat frequent parasitic and microbial diseases, inflammation, cancer, and wounds in sub-Saharan Africa,13 suggesting that Erythrina might be a potential source of candidate drugs for COVID-19, especially in terms of their flavonoid compounds.
Virtual screening is a powerful tool in drug discovery, that uses computational methods to identify potential drug candidates in large compound libraries. This approach involves docking small molecules into the target protein structure to predict their binding affinity and their potential as therapeutic agents.14 In the field of drug discovery and development, virtual screening allows for the identification of potential ligands from a large database of chemical structures using computational processes,15 minimizes the timeline of drug design and discovery by screening a large number of compounds within a short span of time,16 and helps narrow the focus to the most promising candidates, thereby optimizing the use of time, money, and laboratory resources.17 Recent literature has highlighted the application of virtual screening in the discovery of drugs for COVID-19 treatment,16,18 therefore, in this study, we employed structure-based virtual screening to determine the potency of flavonoids from the genus Erythrina as candidate drugs for COVID-19 treatment through the inhibition of the ACE2 receptor.
Materials and Methods
Toxicity and Pharmacokinetics Screening
Pharmacokinetic properties, including absorption, distribution, metabolism, excretion, and toxicity (ADMET), were predicted using the pkCSM web server (http://biosig.unimelb.edu.au/pkcsm). Drug absorption parameters were evaluated in terms of water solubility, Caco-2 permeability, human intestinal absorption, susceptibility to P-glycoprotein substrates, and the ability to inhibit P-glycoproteins I and II. Drug distribution parameters were evaluated in terms of steady-state volume of distribution (VDss), unbound fraction, blood-brain barrier (BBB) permeability, and central nervous system (CNS) permeability. For the metabolism parameter, hits were evaluated in terms of their probability of being substrates or inhibitors of cytochrome P450 enzymes. The excretion properties of the hits were assessed in terms of total clearance and interaction with renal organic cation transporter 2 (OCT2). For toxicity prediction, hits were evaluated in terms of their potential to be mutagenic, hepatotoxic, and inhibitors of human ether-a-go-go-related gene (hERG) ion channels.19
Molecular Docking
The crystal structure of ACE2 with PDBid 6M0J was retrieved from the Protein Data Bank (PDB).20 The 3D structure of the co-crystallized ligand bound to ACE2 was separated using BIOVIA Discovery Studio Visualizer (DS). Based on the prediction of the Marvin Suite 18.21.0 (ChemAxon, Sydney, NSW, Australia) the protonated state of the co-crystallized ligand was adjusted to physiological pH 7.4. Water molecules were removed from the ACE2 structure using BIOVIA Discovery Studio Visualizer v20.1.0.19295 (San Diego, CA, USA; Dassault Systèmes Biovia Corp). The 3D structures of the co-crystallized ligand and ACE2 were then preprocessed in AutoDockTools v1.5.621 and saved in pdbqt format. Redocking of the prepared protein and the co-crystallized ligand was conducted using Autodock4.2. The method is considered valid if the root mean square deviation (RMSD) is less than 2.00 Å.18,22
Molecular Dynamics Simulation
Flavonoids from the genus Erythrina which are potential inhibitors of ACE2 based on the results of virtual screening, Ro5, and ADMET were further analyzed through molecular dynamics (MD) simulation. The atomic charge of flavonoids was calculated using the semi-empirical quantum mechanics Austin model 1 - bond charge correction (AM1-BCC) method in the AmberTools18 antechamber program. Other parameters, such as the bond length, bond angle and dihedral angle, were derived from the General Amber Force Field (GAFF). Modification parameters not contained in the GAFF were generated using the AmberTools18 parmchk program. The parameter ff14SB was used for ACE2, whereas tip3p was used for water and ions such as Na+ and Mg2+. The entire complex system was minimized with a maximum cycle of 2000 times and a non-bonded cutoff of 9.0 Å using the pmemd AMBER program. Furthermore, the production of MD trajectories was analyzed using pmemd and cpptraj programs23 to observe the binding energy values and contributing interactions. RMSD and RMSF were used to study the inhibitory binding potential simulated for 100 ns.
Drug-Likeness
The drug-likeness of potential hits obtained from virtual screening was evaluated using Lipinski’s rule of five (Ro5) on the swissADME webserver (http://www.swissadme.ch/).24
Results
Toxicity Screening
The structures of 473 flavonoids isolated from the genus Erythrina were subjected to in silico toxicity screening using the pKCSM web server.19 The toxicity endpoints evaluated were mutagenicity, hERG inhibition and hepatotoxicity. Mutagenic drugs are pharmaceutical substances that can induce genetic mutations or genotoxic effects in exposed individuals.25 Cardiac arrhythmia is one of the most common unfavorable effects that contribute to medication failure during drug development. This failure is mostly due to the ability of the drug to block hERG cardiac potassium channel.26 Therefore, screening for hERG inhibitor is an essential step in drug development to identify compounds that may cause cardiac arrhythmias by blocking hERG channels. In addition to hERG inhibitors, the hepatotoxicity of drugs is the main cause of drug withdrawal from the pharmaceutical market and interruption in the development of new molecules.27 Among the 473 flavonoids evaluated, 104 were predicted to be safe from being mutagenic, hERG inhibitors, and hepatotoxic (Table S1).
Pharmacokinetics Prediction
After the toxicity screening of 473 flavonoids, we performed pharmacokinetic evaluation of 104 non-toxic flavonoids through an in silico study. Among the 104 non-toxic flavonoids evaluated for their pharmacokinetic properties, 18 compounds were predicted to not the substrates of P-glycoprotein (P-gp), indicating that the bioavailability of these compounds was not affected by P-gp. These 17 compounds were predicted to have high intestinal absorption ranging from 78% to 98%, whereas the water solubility prediction values of these compounds ranged from −3.201 to −5.967 log mol/L.19 Two of these 17 compounds (compounds 2 and 150) were predicted to have low Caco-2 permeability, with predicted log Papp values of < 9.0×10−6 cm/s. Pharmacokinetic prediction of the selected 18 compounds which were not P-gp substrates are given in Table S2.
Structure-Based Virtual Screening
After evaluating the toxicity potency and pharmacokinetic properties of the 473 flavonoids isolated from the genus Erythrina, 18 flavonoids that were predicted to be safe from the toxicity endpoint being tested and were not the substrate of P-gp, were continued for virtual screening against the ACE2 receptor. The binding affinities and interactions of the 18 compounds are listed in Table 1.
 |
Table 1 Binding Affinity and Interaction of 18 Flavonoids with Amino Acids in ACE2 Receptor |
Molecular Dynamics Simulation
The binding energy evaluation from the 100 ns molecular dynamics (MD) simulation of compound 310 in a 10 ns sliding window is shown in Figure 1. Evidently, the binding energy values obtained from molecular docking are different from those obtained from the MD simulation. From the molecular docking result, compound 310 is predicted to have lower binding energy toward ACE2 than compound 471 (Table 1), whereas the molecular dynamics simulation suggested the opposite (−15.9612 ± 1.9765 and −18.1921 ± 2.6197 kcal/mol respectively for compounds 310 and 471, expressed as median ± SD) The difference of binding energy value obtained from molecular docking and MD simulation may due to the ability of MD simulation to accommodate complete flexibility of ligand and protein, therefore allowing intermolecular interaction adjustment.18,22
 |
Figure 1 Comparative ΔGMMGBSA profiles for compounds 310 and 471 every 10 ns over 100 ns simulation time. Compound 310 shows relatively stable free energy values around −16 Kcal/mol, whereas compound 471 exhibits more fluctuation, indicating differing stabilities in their interactions with the target. |
In addition to the MMGBSA free energy calculation, we also conducted RMSD calculations to evaluate the ACE2 protein stability upon the binding of compounds 310 and 471. The RMSD profile (Figure 2) of the Cα atoms remained stable at average value of 1.59 ± 0.22 Å with interquartile range (IQR) of 0.25 Å for 310-ACE2 complex and 1.65 ± 0.19 Å (IQR 0.23 Å) for 310-ACE2 complex.
 |
Figure 2 Comparative RMSD Profile of ACE2 Complexes with Compounds 310 (left) and 471 (right) over 100 ns simulation time. Both plots demonstrate the stability of their respective ACE2 complexes, with the x-axis indicating the simulation time in nanoseconds (ns) and the y-axis showing the RMSD values in Angstroms (Å). The consistent RMSD values suggest stable binding of the compounds to the ACE2 receptor. |
Druglikeness
Drug-likeness refers to a set of properties and characteristics that make a compound suitable for use as a drug. The concept of drug-likeness is important in the field of drug discovery and development, because it helps researchers identify compounds that have a higher likelihood of success in becoming effective and safe drugs. One commonly used tool for assessing drug-likeness is the rule of five (Ro5), which was introduced by Lipinski et al in 1997.28 The rule of five is a set of criteria based on the physicochemical properties of the orally active drugs. According to the rule of five, a compound is considered drug-like if it meets the following criteria: molecular weight < 500 Da, calculated lipophilicity (Log P) less than 5, number of hydrogen-bond acceptors less than 10, and number of hydrogen-bond donors less than 5. These criteria were developed based on the observation that most of the orally active drugs exhibit these properties.28–30 Based on its physicochemical properties, compound 310 had a molecular weight of 390.48 g/mol (<500 Da), four hydrogen-bond acceptors, one hydrogen-bond donor, and a predicted logP of 4.76; compound 471 had a molecular weight of 418.489 g/mol (<500 Da), five hydrogen-bond acceptors, no hydrogen bond-donor, and a predicted log P of 5.67, therefore compounds 310 and 471 met the drug-like criteria of Lipinski’s rule of five.
Discussion
The demand for potent antiviral medications that can significantly reduce the transmission, cellular entry, replication, and virulence of SARS-CoV-2 is one of three essential yet unmet needs for the effective management and treatment of COVID-19. This underscores a significant gap in the COVID-19 treatment landscape, emphasizing the urgency to discover and develop drugs to meet this requirement.31 The human ACE2 protein is a focal point in COVID-19 drug development due to its crucial role as the entry point for the SARS-CoV-2 virus into cells. Targeting ACE2, researchers are working to create therapies that can block this interaction, thus preventing the virus from entering and replicating within human cells. Such a strategy could yield effective treatments that combat not only current strains of the virus but also provide protection against future variants.32
Numerous in vitro studies have shown that natural products, such as flavonoids, have potential as ACE2 inhibitors.33–35 The genus Erythrina, a medicinal plant rich in flavonoids,11 could be a potential therapy for COVID-19 through inhibition of the ACE2-RBD interaction. Nevertheless, currently, there is no study focusing on the evaluation of these flavonoids as ACE2 inhibitors. Therefore, we conducted virtual screening to study the potency of flavonoids from the genus Erythrina as ACE2 inhibitors, as described in numerous studies. Through toxicity and pharmacokinetic screening, as well as molecular docking and molecular dynamics simulation studies, we identified two compounds, erybraedin D (310) and gangetinin (471) (Figure 3), among the 437 flavonoids from the genus Erythrina as potential ACE2 inhibitors, exhibiting lower toxicity and favorable pharmacokinetic properties. Erybraedin D (310) and Gangetinin (471) are both types of pterocarpans, a subclass of flavonoid compounds. Erybraedin D (310) has been identified in various species of Erythrina plants, such as the stem bark of E. abyssinica, the root bark of E. eriotriocha, the roots of E. mildbraedii, and E. x bidwillii. Conversely, Gangetinin (471) has been identified in the root bark of the E. sigmoidea species.36
 |
Figure 3 The chemical structure of erybraedin D (310) (left) and ganggetinin (471) (right). |
There are several amino acid residues that are involved in the interaction of ACE2 with the RBD of the SARS-CoV-2 S protein; these are Gln24, Thr27, Lys31, His34, Glu37, Asp38, Tyr41, Gln42, Leu45, Leu79, Met82, Tyr83, Asp90, Gln325, Glu329, Asn330, Lys353, and Gly54.37 The molecular docking study showed that erybraedin D (310) interacts with Glu37 via Pi-anion interaction, whereas gangetinin (471) interacts with Lys31 via hydrophobic interaction (Table 1), which suggests that the binding of both erybraedin D (310) and gangetinin (471) to ACE2 may alter the interaction of ACE2 with the RBD of the SARS-CoV-2 S protein. The molecular dynamics simulation revealed that the binding of both erybraedin D (310) and gangetinin (471) to ACE2 is stable The MMGBSA binding energy calculation from molecular dynamics simulation suggests that the binding of both erybraedin D (310) and gangetinin (471) to ACE2 is favorable, with binding affinities of −16.6551 kcal/mol and −18.9362 kcal/mol, respectively. The binding affinities of both erybraedin D (310) and gangetinin (471) to ACE2 are lower than that of kaempferol reported in the literature,38 which further support that erybraedin D (310) and gangetinin (471) may be potential inhibitors of ACE2.
In the early drug discovery process, toxicology screening played a crucial role in evaluating the health risks and potential toxicity of candidate drugs.39 In terms of toxicity screening, the prediction suggested that both erybraedin D (310) and gangetinin (471) are less likely to be hepatotoxic, mutagenic, or hERG inhibitors, meaning that these compounds are less likely to induce genetic mutation,25 cardiac arrhythmia,26 or liver injury which are among the main cause of drug withdrawal from the pharmaceutical market and interruption in the development of new molecules.27
In terms of pharmacokinetics prediction, gangetinin (471) is predicted to have higher water solubility (−4.516 log mol/L) than erybraedin D (310) (−5.636 log mol/L). Both compounds exhibit high Caco-2 permeability (log Papp > 0.9×10−6 cm/s) and high intestinal absorption and neither is the substrate of P-gp which indicates that the absorption of erybraedin D (310) and gangetinin (471) is not affected by the activity of P-gp which can pump xenobiotics back to lumen. These parameters suggest that both erybraedin D (310) and gangetinin (471) may be properly absorbed in the human body. For the distribution parameter, gangetinin (471) is predicted to have lower volume of distribution (0.122 log L/kg) than erybraedin D (310) (0.382 log L/kg) suggesting that gangetinin (471) requires lower dosage to achieve the desired plasma concentration compared to erybraedin D (310).40 Both erybraedin D (310) and gangetinin (471) have low BBB permeability (log BB < −1) and can be easily penetrate the central neural system (log PS > −2).
Drug metabolism is another factor that affects bioavailability. Cytochrome P450 (CYP) enzymes play crucial roles in the bioavailability of various substances. Cytochrome P450 enzymes are responsible for the metabolism of drugs, xenobiotics, and other endogenous compounds. These enzymes are involved in the oxidative metabolism of substances, leading to their biotransformation and elimination from the body.41 Cytochrome P450 enzymatic activities can affect the bioavailability of substances by influencing their metabolism and clearance.42 The pharmacokinetics prediction suggests that both erybraedin D (310) and gangetinin (471) are the substrates of CYP3A4, meaning that their bioavailability might be altered by this enzyme.
Conclusion
In this study, through structure based virtual screening, toxicity and pharmacokinetics prediction of 473 flavonoids from genus Erythrina, we identified erybraedin D (310) and gangetinin (471) as potential ACE2 inhibitors. They showed lower toxicity and favorable pharmacokinetic properties. These pterocarpans, found in different parts of Erythrina plants, may alter the ACE2 interaction with the SARS-CoV-2 spike protein, potentially inhibiting the virus. Molecular docking and molecular dynamics study confirmed their stable binding to ACE2, with favorable binding energies of −16.6551 kcal/mol and −18.9362 kcal/mol, respectively. Additionally, they are predicted to be less likely to cause hepatotoxicity, mutagenicity, or hERG inhibition. Both compounds met the drug-like criteria of Lipinski’s rule of five. Therefore, erybraedin D (310) and gangetinin (471) could be further developed as alternative therapeutic for COVID-19 treatment. Nevertheless, it is imperative that the current findings undergo validation via in vitro experimental procedures.
Acknowledgments
The authors are grateful to Universitas Padjadjaran for providing funds for this research.
Funding
This research was funded by Universitas Padjadjaran through the riset data pustaka dan daring (RDPD) scheme and the Academic Leader Grant (ALG) by Tati Herlina (Grant Number 1549/UN6.3.1/PT.00.2023).
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. World Health Organization. COVID-19 epidemiological update – 19 January 2024. Available from: https://www.who.int/publications/m/item/covid-19-epidemiological-update---19-january-2024.
2. Rabie AM. Potent Inhibitory Activities of the Adenosine Analogue Cordycepin on SARS-CoV-2 Replication. ACS Omega. 2022;7(3):2960–2969. doi:10.1021/acsomega.1c05998
3. Li Q, Lai L. Prediction of potential drug targets based on simple sequence properties. BMC Bioinf. 2007;8(1). doi:10.1186/1471-2105-8-353
4. Rabie AM. Future of the current anticoronaviral agents: a viewpoint on the validation for the next COVIDs and pandemics. Biocell. 2023;47(10):2133–2139. doi:10.32604/biocell.2023.030057
5. Rabie AM, Abdalla M. Forodesine and Riboprine Exhibit Strong Anti-SARS-CoV-2 Repurposing Potential: in Silico and In Vitro Studies. ACS Bio and Med Chem Au. 2022;2(6):565–585. doi:10.1021/acsbiomedchemau.2c00039
6. Jia H, Neptune E, Cui H. Targeting ACE2 for COVID-19 Therapy: opportunities and challenges. Am J Respir Cell Mol Biol. 2021;64(4):416–425. doi:10.1165/rcmb.2020-0322PS
7. Hochuli JE, Jain S, Melo-Filho C, et al. Allosteric Binders of ACE2 Are Promising Anti-SARS-CoV-2 Agents. ACS Pharmacol Transl Sci. 2022;5(7):468–478. doi:10.1021/acsptsci.2c00049
8. Goulding LV, Yang J, Jiang Z, et al. Thapsigargin at non-cytotoxic levels induces a potent host antiviral response that blocks influenza a virus replication. Viruses. 2020;12(10):1093. doi:10.3390/v12101093
9. Tutunchi H, Naeini F, Ostadrahimi A, Hosseinzadeh-Attar MJ. Naringenin, a flavanone with antiviral and anti-inflammatory effects: a promising treatment strategy against COVID −19. Phytother Res. 2020;34(12):3137–3147. doi:10.1002/ptr.6781
10. Loizzo MR, Said A, Tundis R, et al. Inhibition of angiotensin converting enzyme (ACE) by flavonoids isolated from Ailanthus excelsa (Roxb) (Simaroubaceae). Phytother Res. 2007;21(1):32–36. doi:10.1002/ptr.2008
11. Fahmy NM, Al-Sayed E, El-Shazly M, Singab AN. Comprehensive review on flavonoids biological activities of Erythrina plant species. Ind Crops Prod. 2018;123:500–538. doi:10.1016/j.indcrop.2018.06.028
12. Son NT, Elshamy AI. Flavonoids and other Non-alkaloidal Constituents of Genus Erythrina: phytochemical Review. Comb Chem High Throughput Screen. 2021;24(1):20–58. doi:10.2174/1386207323666200609141517
13. Kone WM, Solange KNE, Dosso M. Assessing sub-saharian Erythrina for efficacy: traditional uses, biological activities and phytochemistry. Pak J Biological Sci. 2011;14(10):560–571. doi:10.3923/pjbs.2011.560.571
14. Shen C, Ding J, Wang Z, Cao D, Ding X, Hou T. From machine learning to deep learning: advances in scoring functions for protein–ligand docking. Wiley Interdiscip Rev Comput Mol Sci. 2020;10(1). doi:10.1002/wcms.1429
15. Miles JA, Ross BP. Recent Advances in Virtual Screening for Cholinesterase Inhibitors. ACS Chem Neurosci. 2021;12(1):30–41. doi:10.1021/acschemneuro.0c00627
16. Rajendran M, Roy S, Ravichandran K, et al. In silico screening and molecular dynamics of phytochemicals from Indian cuisine against SARS-CoV-2 M Pro. J Biomol Struct Dyn. 2022;40(7):3155–3169. doi:10.1080/07391102.2020.1845980
17. Gimeno A, Ojeda-Montes MJ, Tomás-Hernández S, et al. The light and dark sides of virtual screening: what is there to know? Int J Mol Sci. 2019;20(6):1375. doi:10.3390/ijms20061375
18. Nishinarizki V, Hardianto A, Gaffar S, Muchtaridi M, Herlina T. Virtual screening campaigns and ADMET evaluation to unlock the potency of flavonoids from Erythrina as 3CLpro SARS-COV-2 inhibitors. J Appl Pharm Sci. 2023;13(2). doi:10.7324/JAPS.2023.130209
19. Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–4072. doi:10.1021/acs.jmedchem.5b00104
20. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi:10.1038/s41586-020-2180-5
21. Goodsell DS, Sanner MF, Olson AJ, Forli S. The AutoDock suite at 30. Protein Sci. 2021;30(1):31–43. doi:10.1002/pro.3934
22. Hardianto A, Yusuf M, Hidayat IW, Ishmayana S, Soedjanaatmadja UMS. Exploring the Potency of Nigella sativa Seed in Inhibiting SARS-CoV-2 Main Protease Using Molecular Docking and Molecular Dynamics Simulations. Indonesian J Chem. 2021;21(5):1252. doi:10.22146/IJC.65951
23. Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095. doi:10.1021/ct400341p
24. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1). doi:10.1038/srep42717
25. Okocha RC, Olatoye IO, Adedeji OB. Food safety impacts of antimicrobial use and their residues in aquaculture. Public Health Rev. 2018;39(1). doi:10.1186/s40985-018-0099-2
26. Garrido A, Lepailleur A, Mignani SM, Dallemagne P, Rochais C. hERG toxicity assessment: useful guidelines for drug design. Eur J Med Chem. 2020;195. doi:10.1016/j.ejmech.2020.112290
27. Meunier L, Larrey D. Drug-induced liver injury: biomarkers, requirements, candidates, and validation. Front Pharmacol. 2019;10. doi:10.3389/fphar.2019.01482
28. Shultz MD. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J Med Chem. 2019;62(4):1701–1714. doi:10.1021/acs.jmedchem.8b00686
29. James JP, Ail PD, Crasta L, Kamath RS, Shura MH, S TJ. In Silico ADMET and Molecular Interaction Profiles of Phytochemicals from Medicinal Plants in Dakshina Kannada. J Health Allied Sci NU. 2023. doi:10.1055/s-0043-1770057
30. Matondo A, Dendera W, Isamura BK, et al. In silico Drug Repurposing of Anticancer Drug 5-FU and Analogues Against SARS-CoV-2 Main Protease: molecular Docking, Molecular Dynamics Simulation, Pharmacokinetics and Chemical Reactivity Studies. <![CDATA[Advances and Applications in Bioinformatics and Chemistry]]>. 2022;15:59–77. doi:10.2147/AABC.S366111
31. Rabie AM, Eltayb WA. Potent Dual Polymerase/Exonuclease Inhibitory Activities of Antioxidant Aminothiadiazoles Against the COVID-19 Omicron Virus: a Promising In Silico/In Vitro Repositioning Research Study. Mol Biotechnol. 2023;66(4):592–611. doi:10.1007/s12033-022-00551-8
32. Chamata Y, Jackson KG, Watson KA, Jauregi P. Whey-derived peptides at the heart of the covid-19 pandemic. Int J Mol Sci. 2021;22(21):11662. doi:10.3390/ijms222111662
33. Sui H, Yu Q, Zhi Y, Geng G, Liu H, Xu H. Effects of apigenin on the expression of angiotensin-converting enzyme 2 in kidney in spontaneously hypertensive rats. Wei Sheng Yan Jiu. 2010;39(6):693–6, 700.
34. Hettihewa SK, Hemar Y, Vasantha Rupasinghe HP. Flavonoid-rich extract of Actinidia macrosperma (a wild kiwifruit) inhibits angiotensin-converting enzyme in vitro. Foods. 2018;7(9):146. doi:10.3390/foods7090146
35. Guerrero L, Castillo J, Quiñones M, et al. Inhibition of Angiotensin-Converting Enzyme Activity by Flavonoids: structure-Activity Relationship Studies. PLoS One. 2012;7(11):e49493. doi:10.1371/journal.pone.0049493
36. Son NT, Elshamy AI. Flavonoids and other Non-alkaloidal Constituents of Genus Erythrina: phytochemical Review. Comb Chem High Throughput Screen. 2020;24(1):20–58. doi:10.2174/1386207323666200609141517
37. Han DP, Penn-Nicholson A, Cho MW. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology. 2006;350(1):15–25. doi:10.1016/j.virol.2006.01.029
38. Kashyap P, Thakur M, Singh N, et al. In Silico Evaluation of Natural Flavonoids as a Potential Inhibitor of Coronavirus Disease. Molecules. 2022;27(19):6374. doi:10.3390/molecules27196374
39. Lee MY, Kumar RA, Sukumaran SM, Hogg MG, Clark DS, Dordick JS. Three-dimensional cellular microarray for high-throughput toxicology assays. Proc Natl Acad Sci U S A. 2008;105(1):59–63. doi:10.1073/pnas.0708756105
40. Mansoor A, Mahabadi N. Volume of Distribution. StatPearls Publishing; 2022.
41. Heo JK, Kim HJ, Lee GH, et al. Simultaneous determination of five cytochrome P450 probe substrates and their metabolites and organic anion transporting polypeptide probe substrate in human plasma using liquid chromatography-tandem mass spectrometry. Pharmaceutics. 2018;10(3):79. doi:10.3390/pharmaceutics10030079
42. Tapaninen T, Olkkola AM, Tornio A, et al. Itraconazole Increases Ibrutinib Exposure 10-Fold and Reduces Interindividual Variation—A Potentially Beneficial Drug-Drug Interaction. Clin Transl Sci. 2020;13(2):345–351. doi:10.1111/cts.12716
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.