")
Back to Journals » Cancer Management and Research » Volume 12
ZNF671 Inhibits the Proliferation and Metastasis of NSCLC via the Wnt/β-Catenin Pathway
Authors Zhan W , Li Y, Liu X, Zheng C , Fu Y
Received 24 October 2019
Accepted for publication 31 December 2019
Published 24 January 2020 Volume 2020:12 Pages 599—610
DOI https://doi.org/10.2147/CMAR.S235933
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Wei Zhan, 1 Yuzhe Li, 2 Xuhui Liu, 1 Changlong Zheng, 1 Yongmei Fu 1
1Department of Emergency, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510000, People’s Republic of China; 2Department of Clinical Laboratory, The Third Affiliated Hospital of Sun Yat-Sen University, Lingnan Hospital, Guangzhou 510080, People’s Republic of China
Correspondence: Yongmei Fu
Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510000, People’s Republic of China
Email [email protected]
Changlong Zheng
The Third Hospital of Sun Yat-Sen University, Guangzhou 510630, China
Email [email protected]
Background: Lung cancer is the most common cancer in the world and is the main cause of cancer-related death. Revealing the potential mechanism of malignant characteristics of lung cancer is urgent for treating this disease effectively. Zinc finger protein 671 (ZNF671) is a member of the largest transcription factor family in the human genome. The role of ZNF671 in non-small-cell lung cancer (NSCLC) remains unknown. The purpose of this study was to investigate the function and mechanism of ZNF671 in NSCLC.
Methods: ZNF671 expression in NSCLC cells and tissues were detected by Real-Time PCR, Western blot and TCGA databases. Then, we evaluated the prognostic value of ZNF671 expression in NSCLC using the Kaplan–Meier plotter (KM plotter) and TCGA databases. Moreover, the function of ZNF671 in the proliferation and metastasis of lung cancer was investigated by MTT assay, colony formation assay, in vivo experiment, EdU assay, wound healing assay, transwell assay, and 3D culture assay. Luciferase reporter and subcellular fractionation assays were performed to determine the underlying mechanism of ZNF671-mediated proliferation and metastasis of NSCLC.
Results: ZNF671 expression was significantly reduced in both NSCLC cell lines and clinical specimens compared to that in normal controls. The survival analysis results indicated that the downregulation of ZNF671 significantly correlates with poor prognosis and predicts a shorter overall survival and post-progression survival among NSCLC patients. Ectopic overexpression of ZNF671 dramatically restrains, whereas silencing ZNF671 enhanced, cell proliferation and metastasis of NSCLC. Mechanically, gene set enrichment analysis (GSEA) showed that the expression of ZNF671 was significantly correlated with Wnt/β-catenin signaling. Simultaneously, our results confirm that the overexpression of ZNF671 inhibits cell cycle progression and metastasis by weakening the Wnt/β-catenin pathway, and then downregulating the expression of downstream target genes CyclinD1 and MMP9.
Conclusion: This study found that the overexpression of ZNF671 restrains the proliferation and metastasis of lung cancer through inhibiting Wnt/β-catenin signaling pathway. Furthermore, our current results provide important insights into ZNF671 as an excellent predictive biomarker for NSCLC, thus providing a novel perspective for the treatment of NSCLC.
Keywords: zinc finger protein 671, proliferation, metastasis, Wnt/β-catenin pathway, biomarker
Introduction
According to the World Health Organization, lung cancer is the most common fatal malignant tumor in the world.1 About 85% of lung cancer cases are non-small cell lung cancer (NSCLC), and most of the patients are usually diagnosed at the advanced stage, which result in the prognosis of patients with NSCLC being very poor.2,3 In general, the expected median survival time of advanced stage NSCLC patients is 6 months, and the 5-year survival rate is not more than 15%.4 90% of NSCLC patients’ deaths are associated with metastasis.5 In the metastasis process, subgroup cancer cells gain invasive properties and escape immunosurveillance to entry into the blood circulation and lead to distant organ metastasis, and they selectively arrest at specific organs in a non-random manner.6,7 In recent years, molecular targeting therapy has made great progress in the treatment of NSCLC by enhancing the efficacy of tumor cells and reducing the toxicity to normal cells.8,9 However, the challenge of targeted therapy is acquired drug resistance, which is a crucial problem that needs to be dealt with. Therefore, it is of great clinical value to further investigate the critical biomarkers in the diagnosis and treatment of NSCLC.
Zinc finger protein 671 (ZNF671) is a member of the KRAB-ZFP family of mammalian transcriptional repressors, which contains C2H2-type zinc fingers (ZFs) and a Krüppel associated box (KRAB) domain.10 ZNF671 recruits the KRAB-associated protein-1 and other co-repressors and then regulates cell differentiation, proliferation, apoptosis, and tumor suppression.10 Many studies have demonstrated that ZNF671 is epigenetically inhibited by DNA methylation and functions as a novel tumor suppressor in multiple cancers.11,12 Sun et al reported that overexpression of ZNF671 suppresses cell proliferation and tumorigenicity through slowing down cell cycle progression by regulating the expression of cell cycle factors in NPC.10 Studies published by Kondo et al have shown that ZNF671 plays an inhibitory role in ovarian cancer and that the DNA methylation status of ZNF671 may be an effective biomarker for serous ovarian cancer recurrence.12 However, little is known about the clinicopathologic features, biological functions and mechanism of ZNF671 in NSCLC.
In this study, we firstly systematically analyzed the expression of ZNF671 in NSCLC cells and tissues and found that the expression of ZNF671 was downregulated. The promoter of ZNF671 was hypermethylated in NSCLC tumor tissues using analysis of the TCGA database. Then we used the KM plotter database to evaluate the prognostic role of ZNF671 in NSCLC and found that the downregulation of ZNF671 predicted poor prognosis in NSCLC. Furthermore, we also demonstrated that overexpression of ZNF671 significantly reduced the proliferation and metastasis of NSCLC, whereas silencing ZNF671 led to the reverse effects. Mechanistically, the overexpression of ZNF671 remarkably inhibited Wnt/β-catenin signaling pathway. Collectively, our results provide important insight into ZNF671 as a promising biomarker of NSCLC and thus provide a novel prospect for the treatment of NSCLC.
Materials and Methods
Bioinformatics Analysis
RNA seq data from the lung Pan-Cancer study of The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) were used to analyze the expression and methylation level of ZNF671 in normal and tumor lung tissues. The survival of 1926 lung cancer patients with different ZNF671 expression levels was analyzed using the online Kaplan–Meier analysis (http://kmplot.com/analysis/). Gene Set Enrichment Analysis (GSEA) was performed by gsea-3.0.jar (http://software.broadinstitute.org/gsea/index.jsp) using the NCBI/GEO/GSE3141 dataset.
Cell Lines
Non-small-cell lung cancer cell lines, including H1299, H292, Calu-3, A549, H460, H1975 and H520 were cultured in RPMI 1640 medium (Gibco, Grand Island, NY, USA). 293FT cells were cultured in DMEM (Gibco). The immortalized lung epithelial cell line BEAS-2B was cultured in BEBM medium (LONZA, Basel, Switzerland). All the cells were purchased commercially from American Type Culture Collection (ATCC, Manassas, VA, USA).
For the establishment of stable cell lines, plasmids (psin-EF2-puro-ZNF671, and the control vectors) were transfected in 293FT to produce lentivirus. A549 and H460 cells were infected with the indicated lentivirus for 48 h and then selected with medium containing puromycin (1μg/mL). All stable cell lines were selected with the indicated antibiotics at a high concentration for over 1 week and maintained in medium containing a low concentration of the indicated antibiotics.
Plasmids and siRNA Transfection
The ORFs of ZNF671 and β-catenin were cloned into the mammalian expression vector pcDNA 3.1 (Invitrogen), and pcDNA3.1 vector was used as the control. siRNAs targeting ZNF671 were obtained from GenePharma Co., Ltd. (Shanghai, China); siRNA#1 targets ZNF671-Homo-626 cDNA (sense strand: CCUUACACCUGGCUAAAUATT; antisense strand: UAUUUAGCCAGGUGUAAGGTT) and siRNA#2 targets ZNF671-Homo-279 cDNA (sense strand: GGAAGAAUGGGAGCUUCUUTT; anti- sense strand: AAGAAGCUCCCAUUCUUCCTT). Transfection of plasmids or siRNA was performed using Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions.
RNA Extraction, Reverse Transcription (RT) PCR and Real Time PCR
Total RNA from cultured cells was extracted using the Trizol reagent (Invitrogen, Carlsbad, CA) as the manufacturer instructed. cDNAs were amplified and quantified in ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA) using dye SYBR Green I (Molecular Probes, Invitrogen, CA Carlsbad, CA). The primers were selected as the following: ZNF671 forward, 5ʹ-GACTTAGACCTGGTTGTTGG-3ʹ and reverse, 5ʹ- GTATTTAGCCAGGTGTAAGGT-3ʹ10 Expression data were normalized to the geometric mean of housekeeping gene GAPDH (forward: 5ʹ-ACCACAGTCCATGCCATCAC-3ʹ and reverse: 5ʹ-TCCACCACCCTG TTGCTGTA-3ʹ) to control the variability in expression levels and calculated as 2−[(Ct of gene) – (Ct of GAPDH)], where Ct represents the threshold cycle for each transcript.
Western Blot
Western blot was performed according to standard methods as described previously,13 using anti-ZNF671 antibody (1μg/mL, Abcam), anti-p-GSK3β (Ser9) (1:1000, Cell Signaling), anti-GSK3β (1:1000, Cell Signaling), anti-β-catenin (1:1000, Cell Signaling), anti-Cyclin D1 (1:2000, Cell Signaling), and anti-MMP9 (1:1000, Cell Signaling). Anti-α-Tubulin (1:5000, Cell Signaling) was used as the loading control.
MTT Cell Viability Assay
Cells were seeded in 96-well plates at a density of 2×103 cells/well. At each time point, cells were stained with 100 μL sterile MTT dye (0.5 mg/mL, Sigma) for 4 h at 37 °C, followed by removal of the culture medium and addition of 100 μL of dimethyl sulphoxide (Sigma). The absorbance was measured at 490 nm wavelength. Each experiment was performed in triplicates.
Colony Formation Assay
Cells were plated in 6-well plates (5× 102 cells per well) and cultured for 10 days. The colonies were stained with 1% crystal violet for 30s after fixation with 4% formaldehyde for 5 min. Colonies were counted and results were shown as the fold change compared to vector cells.
Tumor Xenografts
Male BALB/c nude mice aged 4–6 weeks old were purchased from the Medical Experimental Animal Center of Guangdong Province (Guangzhou, China) and randomly divided into two groups (n = 6 per group). 2×106 indicated cells were suspended in 200 μL PBS, and then subcutaneously injected into the dorsal flank of the nude mice. Tumor volume was measured with external caliper every 3 days and calculated according to the equation (L ×W2)/2. The mice were killed 27 days after inoculation and the tumors were excised and subjected to pathologic examination. All experimental procedures involving animals follow the Guide for the Care and Use of Laboratory Animals (NIH publication nos. 80–23, revised 1996) and follow the institutional ethical guidelines for animal experiments. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Third Affiliated Hospital of Sun Yat-sen University. The retention and use of animals comply with national and local laws and regulations.
EdU Incorporation and Immunofluorescence
Cells were plated on coverslips (Thermo Fisher Scientific) and incubated overnight. The cells were incubated with 10μM 5-ethynyl-2ʹ-deoxyuridine (EdU) for 2 h and stained with Azide 488 (Beyotime) after fixation with 4% formaldehyde. Coverslips were mounted with ProLong Diamond Anti-fade reagent with DAPI (Invitrogen). Gray level images were acquired under a laser scanning microscope (Axio Imager.Z2, Carl Zeiss Co. Ltd.).
Wound Healing, Cell Migration Assays
Indicated cells were plated to confluence in 6-well plates. Streaks were created in the monolayer with a pipette tip. Progression of migration was observed and photographed at 0h and 24h after wounding. Image J software was used to quantify the wound healing area of the indicated cells. And cell migration assays were conducted as previously reported.14
3D Spheroid Invasion Assay
Indicated cells (5 × 103) were seeded in 2% Matrigel-coated 24-well plates, the culture medium was refreshed every other day. Cells forming a 3D spherical structure (spheres) were photographed at 2-day intervals for 10 days.
Statistical Analysis
Statistical analyses were carried out using the SPSS 19.0 statistical software package or GraphPad Prism 6.0 software. Two-tailed, unpaired Student’s t-test or one-way ANOVA test were used for comparisons between groups for statistical significance. A P value < 0.05 was considered statistically significant.
Results
ZNF671 Is Downregulated and Hypermethylated in NSCLC and Correlated with Progression and Poor Prognosis of NSCLC Patients
To determine the potential role of ZNF671 in NSCLC, we first measured ZNF671 expression in NSCLC cell lines, and the results showed that ZNF671 mRNA and protein expression were markedly downregulated in all the tested NSCLC cell lines compared with immortalized lung epithelial cell line BEAS-2B (Figure 1A). Consistently, we detected that ZNF671 expression was lower in six human NSCLC cancer tissues than in their paired adjacent non-tumor tissues (Figure 1B). These results indicated that ZNF671 expression was downregulated in NSCLC cells and tissues. Furthermore, data from the TCGA database also showed that ZNF671 expression levels were dramatically decreased in both lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) tissues compared to the normal control tissues (Figure 1C). In addition, we analyzed the methylation level of ZNF671 in the TCGA database and found that the promoter of ZNF671 was hypermethylated in NSCLC (Figure 1D). These results indicate that ZNF671 protein expression is downregulated in NSCLC and that ZNF671 may play a tumor suppressor role in NSCLC progression and the promoter hypermethylation of ZNF671 mediates its downregulation in NSCLC.
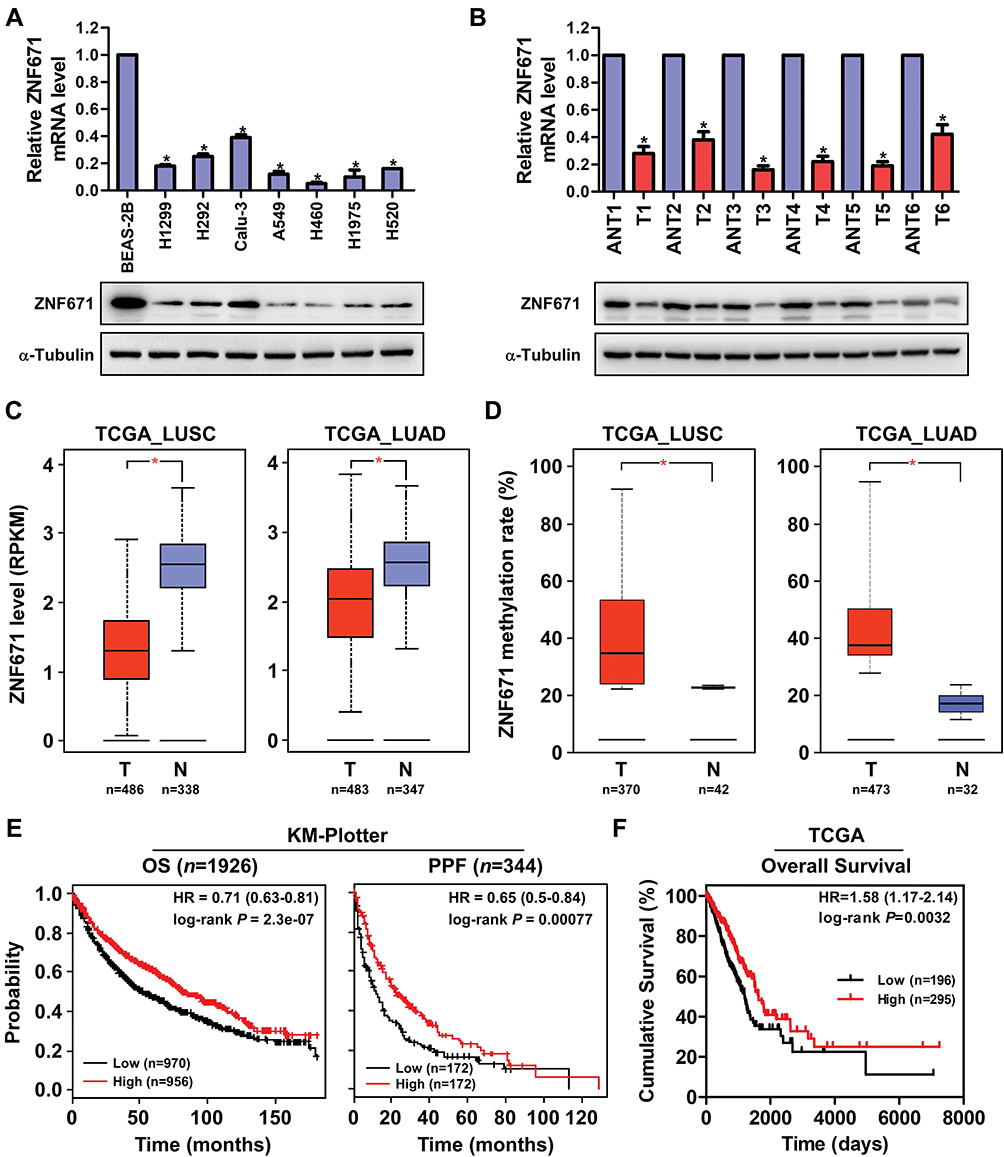 |
Figure 1 ZNF671 is downregulated and hypermethylated in NSCLC and correlated with progression and poor prognosis of NSCLC patients. (A and B) Real-time PCR and Western blot analysis of ZNF671 expression in NSCLC cell lines and paired NSCLC tumor and nontumor tissues. (C) Analysis of the expression of ZNF671 mRNA in NSCLC datasets from the TCGA database. (D) ZNF671 methylation analysis in NSCLC datasets from the TCGA database. (E) Online Kaplan–Meier analyses of OS and PPF of NSCLC patients stratified by ZNF671 expression levels. (F) The OS of NSCLC patients stratified by ZNF671 expression levels in NSCLC datasets from the TCGA database. *P<0.01. |
To further assess the prognostic role of ZNF671 in NSCLC, we explored the relationship between mRNA expression of ZNF671 and patients’ survival using an online KM plotter (http://kmplot.com) and TCGA datasets. KM plotter showed that high ZNF671 mRNA expression was significantly correlated with increased probability of overall survival (HR=0.71; P=2.3e-07) and post-progression survival (HR=0.65; P=0.00077) time for NSCLC patients (Figure 1E). Moreover, the TCGA dataset showed that low ZNF671 expression correlated with worse OS of NSCLC patients (Figure 1F). Overall, the results indicate that epigenetic-mediated downregulation of ZNF671 predicts poor survival in NSCLC.
ZNF671 Inhibits Cell Proliferation and Tumorigenesis of NSCLC Cells
To investigate the biological functions of ZNF671 in NSCLC, we selected the lower ZNF671 expression cells to transfect psin-EF2-puro-ZNF671 plasmids, and then established the ectopically overexpressed stable cells in A549 and H460 cells. As shown in Figure 2A, the mRNA and protein expression of ZNF671 was markedly upregulated after psin-EF2-puro-ZNF671 transfection. Interestingly, while we culture these indicated cells, we observed a decreased cell proliferation and diminished growth in the ZNF671-transduced cells compared with the vector control cells; therefore, we subsequently studied these aspects. The result from MTT assay demonstrated that ZNF671 overexpression significantly impaired cell proliferation of indicated cell lines (Figure 2B), and these results were further confirmed by colony formation assay (Figure 2C).
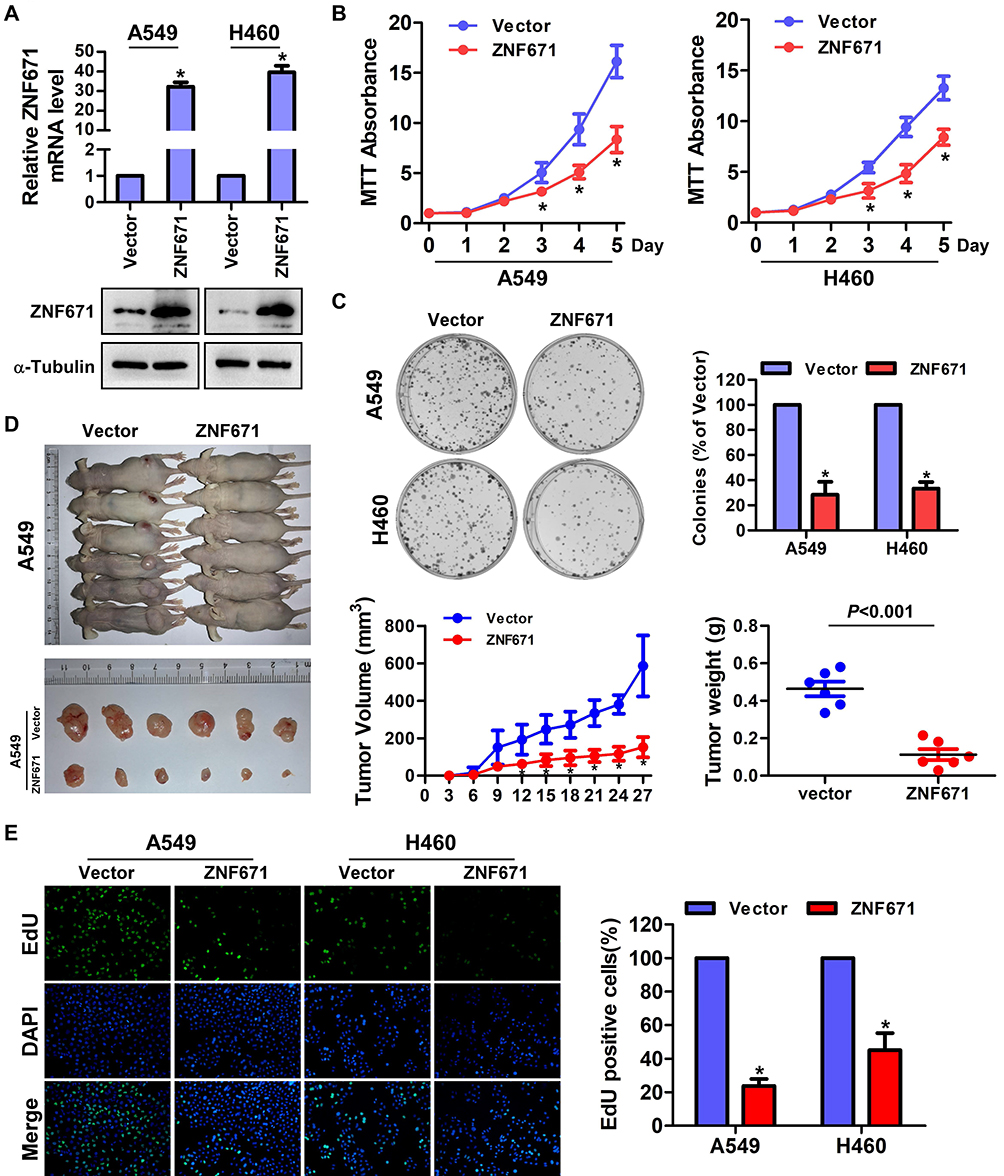 |
Figure 2 ZNF671 inhibits cell proliferation and tumorigenesis of NSCLC cells. (A) Real-time PCR and Western blot analysis of ZNF671 in the indicated cells with stable overexpression of ZNF671. (B) MTT assay analysis of cell growth. Error bars represent the mean ± SD obtained from three independent experiments. *P < 0.05, unpaired t-test. (C) Representative pictures of cell colonies originating from the indicated cells and stained with crystal violet. (D) The indicated cells (2 × 106) were injected subcutaneously into BALB/c nude mice. Tumor volume growth curves (middle) and tumor weights (right) for the tumors formed by the indicated cells. Each bar represents the mean±SD of three independent experiments. Scale bar=1 cm. (E) Representative pictures of EdU staining of the indicated cells. *P<0.01. |
To investigate the effect of ZNF671 on the tumorigenicity of NSCLC, 2×106 of ZNF671-transduced A549 and corresponding vector control cells were subcutaneously inoculated in BALB/c nude mice. As shown in Figure 2D, compared with the vector-control cells, the tumorigenicity of ZNF671-transduced cells was weakened and the tumor growth rate was decreased. Moreover, the tumors formed by ZNF671-transduced A549 cells were smaller and had lower tumor weights than the vector-control tumors. These findings indicate that ZNF671 inhibited the tumorigenicity of NSCLC cells.
Then, to further investigate the role of ZNF671 on the proliferation of NSCLC cells, EdU incorporation assay was performed. As shown in Figure 2E, ZNF671 overexpression dramatically decreased the fraction of EdU-positive A549 and H460 cells in the S-phase fraction compared with their respective control cells. In conclusion, our results indicate that ZNF671 plays a critical role in the proliferation and tumorigenesis of NSCLC.
ZNF671 Reduced Migration and Metastasis of NSCLC
To determine the migratory ability of NSCLC, wound healing assay was performed and the results were shown in Figure 3A. In comparison to the control cells, the migration rate of A549 and H460 cells at 24h post-scratching was significantly restrained in ZNF671-transduced cells (Figure 3A). In addition, transwell assay without matrigel also showed that the migration ability of ZNF671-transduced cells was drastically weakened (Figure 3B). Furthermore, in 3D culture, ZNF671-transduced NSCLC cells grew into less invasive projections compared with the vector cells (Figure 3C). Collectively, our data suggested that ZNF671 greatly contributes to the migration and metastasis ability of NSCLC.
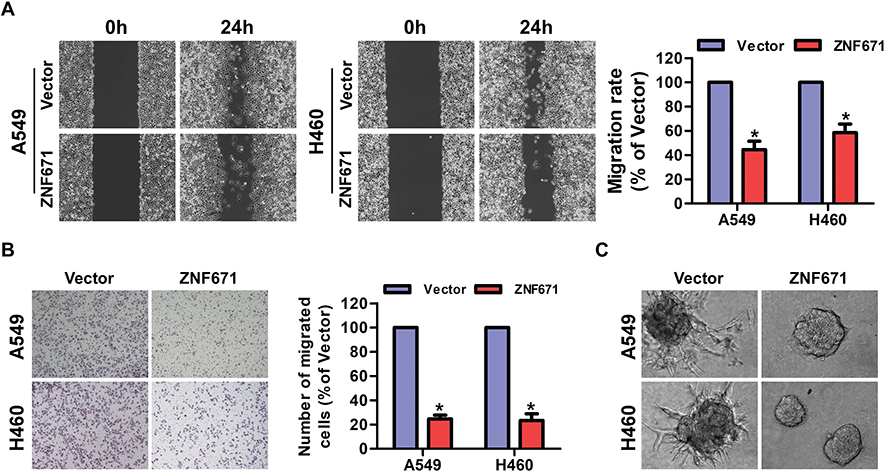 |
Figure 3 ZNF671 reduced migration and metastasis of NSCLC. (A) Representative micrographs of wound healing assay of the indicated cells. Wound closures were photographed at 0 and 24 hrs after wounding. (B) The effect of ZNF671 upregulation on the migration of NSCLC was assessed by a Transwell assay. Results are representative of the average counts from five random microscopic fields. (C) Representative micrographs of indicated cells grown on Matrigel for 10 days in 3D spheroid invasion assay. *P<0.01. |
Knockdown of ZNF671 Restrains the Proliferation and Migration of NSCLC
To further confirm the biological function of ZNF671 in NSCLC, siZNF671#1 and siZNF671#2 siRNA were transfected into Calu-3 cells with high expression of ZNF671. The expression of ZNF671 was detected by Real-time PCR and Western blot, and the result was shown in Figure 4A. MTT assay indicated that the proliferation rate of ZNF671-silenced cells was significantly increased compared with the siNC cells (Figure 4B). Colony formation assay confirmed that silencing ZNF671 promotes the proliferation of NSCLC cells (Figure 4C). In addition, Transwell assay without matrigel showed that the migration ability of ZNF671-silenced cells was drastically promoted (Figure 4D).
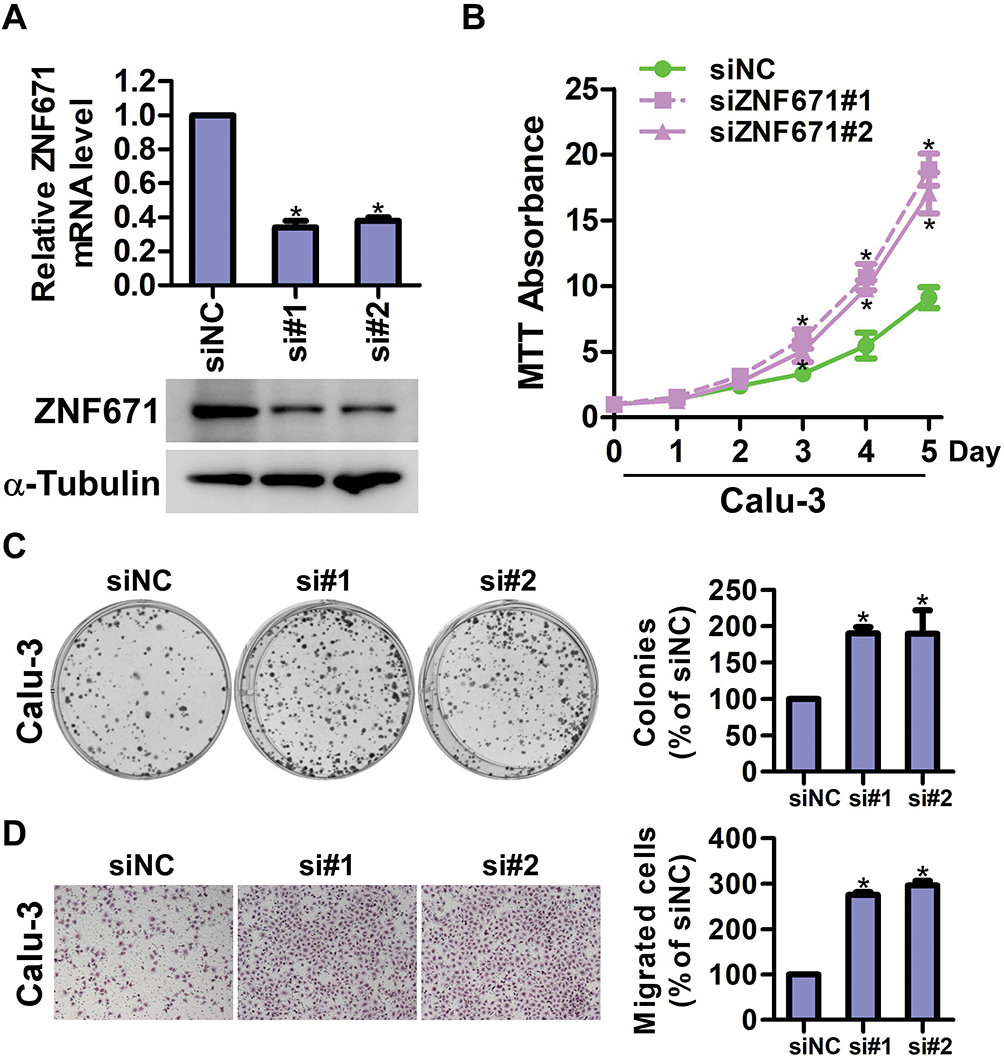 |
Figure 4 Knockdown of ZNF671 restrains the proliferation and migration of NSCLC. (A) Real-time PCR and Western blot analysis of ZNF671 in the indicated cells with silencing of ZNF671. (B) MTT assay analysis of cell growth. Error bars represent the mean±SD obtained from three independent experiments. *P<0.05, unpaired t-test. (C) Representative pictures of cell colonies originating from the indicated cells and stained with crystal violet. (D) The effect of ZNF671 downregulation on the migration of NSCLC was assessed by a Transwell assay. Results are representative of the average counts from five random microscopic fields. *P<0.01. |
Effects of ZNF671 on Wnt/β-Catenin Signaling Pathway in NSCLC
It has been well established that Wnt/β-catenin signaling pathway participates in cell proliferation, migration and metastasis by modulating the expression of cyclin D1 and MMP9.15,16 To determine whether ZNF671 modulates the cell proliferation, migration, and metastasis via Wnt/β-catenin signaling pathway, gene set enrichment analysis (GSEA) was performed. The results showed that ZNF671 expression negatively correlated with HALLMARK_WNT-BETA_CATENIN_SIGNALING (Genes up-regulated by activation of WNT signaling through accumulation of beta catenin CTNNB1) and PID_BETA_CATAENIN_NUC_PATHWAY (Regulation of nuclear beta catenin signaling and target gene transcription) gene signatures in gene expression profiles of lung cancer patients obtained from the NCBI/GEO/GSE3141 dataset (Figure 5A), indicating that ZNF671 may inhibit the Wnt/β-catenin signaling pathway. Furthermore, TOP/FOP flash assays revealed that overexpression of ZNF671 markedly decreased the transcriptional activation of TCF/LEF in the A549 and H460 cells (Figure 5B). Western blot assay indicated that the protein level of p-GSK3β and β-catenin were strikingly reduced in ZNF671 overexpression cells (Figure 5C). Moreover, the protein level of cyclinD1 and MMP9, downstream genes of Wnt/β-catenin pathway, were also significantly decreased in ZNF671-transduced NSCLC cells (Figure 5C). In addition, the nuclear extraction experiment showed that the overexpression of ZNF671 significantly decreased the nuclear signal of β-catenin. (Figure 5D). To demonstrate the roles of Wnt/β-catenin signaling in ZNF671-transduced NSCLC cells, β-catenin was forced expressed in ZNF671 overexpression NSCLC cells, and the malignancies were determined. As presented in Figure 5E, the declined cell growth ability of ZNF671-transduced cells was reversed by overexpression of β-catenin in indicated NSCLC cells. In addition, the results of transwell assays without matrigel showed that ZNF671-transduced decline of migratory ability in NSCLC was abolished by β-catenin overexpression (Figure 5F). In summary, our results dedicated that the upregulation of ZNF671 restrains the proliferation and metastasis of NSCLC through inhibiting Wnt/β-catenin signaling.
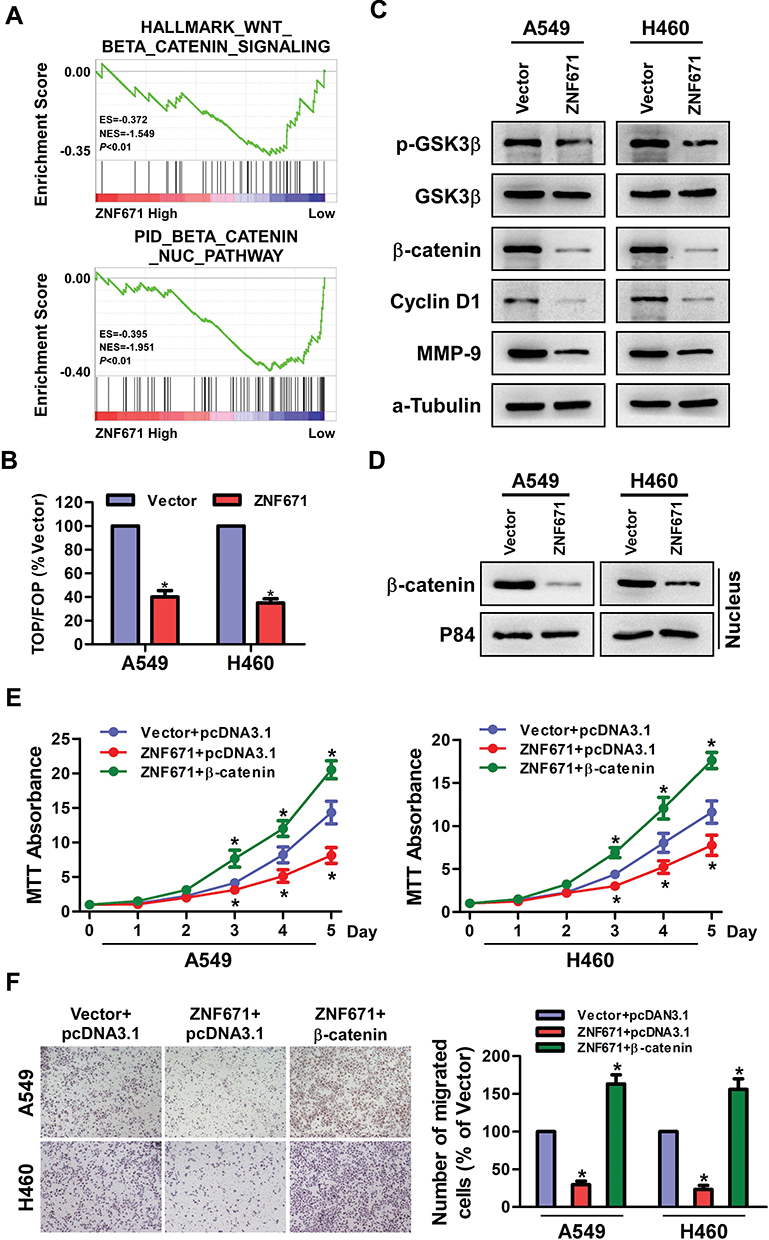 |
Figure 5 Effects of ZNF671 on Wnt/β-catenin signaling pathway in NSCLC. (A) GSEA analysis of the correlation between ZNF671 expression and the HALLMARK_WNT-BETA_CATENIN_SIGNALING and PID_BETA_CATAENIN_NUC_PATHWAY gene signature. (B) TOP/FOP Flash activity of the indicated cells. (C) The cellular protein levels of p-GSK3β, GSK3β, β-catenin, Cyclin D1, and MMP9 in A549 and H460cells were detected by Western blot assay. α-Tubulin was used as the cellular control. (D) Overexpression of ZNF671 changed the nuclear translocation of β‐catenin. Western blot analysis showed the nuclear components of the cells. P84 was used as a loading control. (E) MTT assay was performed to measure the viability of indicated NSCLC cells after upregulated ZNF671 and overexpressed β-catenin. (F) The transwell assay without Matrigel was performed to determine the migratory ability of indicated NSCLC cells after upregulated ZNF671 and overexpressed β-catenin. All experimental data are expressed as mean ± SD (n=3). *P<0.01. |
Discussion
Our current study reports the expression of ZNF671 in NSCLC and its close correlation with clinical patient outcomes. At the molecular level, our mechanistic studies uncover that overexpression of ZNF671 inhibits cancer cell proliferation, migration, and metastasis by serving as a tumor suppressor to weaken Wnt/β-catenin signaling.
Epigenetic regulation can be passed on to offspring without including changes in gene sequences.17,18 Epigenetic methods, such as DNA methylation, nucleosome remodeling, and histone modification are involved in the biological process of maintaining normal development and gene expression.19,20 The accumulation of epigenetic changes plays a vital function in the development of cancer. DNA methylation is an early event in tumorigenesis and plays an important role in the occurrence and development of tumors.21,22 These findings are consistent with previous reports that other ZNF family proteins are frequently found down-regulated by DNA methylation in multiple human cancer types. For example, the Hypermethylation level of ZNF154 promoter leads to the inhibition of gene expression and loss of tumor suppression function, which contributes to the occurrence and poor outcomes in pancreas cancer.23 ZNF545 is frequently downregulated and methylated in multiple carcinomas and suppresses the progression of ESCC by inhibiting NF-κB activation.24 A previous study reported that ZNF671, an epigenetically silenced novel tumor suppressor gene in nasopharyngeal carcinoma, ovarian cancer and urothelial carcinoma. The DNA methylation status of ZNF671 might be a potential predictor and biomarker for the recurrence of multiple human tumors10−12. Up to now, the main research is that the epigenetic regulation of promoter methylation leads to the decrease of ZNF671 expression, while the up-regulation of ZNF671 slows down the cell cycle progression of nasopharyngeal carcinoma cells by regulating the expression of cell cycle factors, thus inhibiting cell proliferation and tumorigenicity.10 However, it has not been reported whether ZNF671 is involved in the regulation of cell function and signaling pathway in NSCLC. Our results revealed that ZNF671 was a tumor suppressor gene with significantly higher methylation in LUSC and LUAD tumor tissues compared to normal tissues. Furthermore, we used online KM plotter analysis to explore the potential prognostic value of ZNF671 in NSCLC. We found that low ZNF671 expression may predict poor survival in NSCLC. To further evaluate the potential biological function of ZNF671 in NSCLC, we conducted a series of experiments on cell proliferation and metastasis and examined that overexpression of ZNF671 could inhibit tumor cell proliferation, cell migration and metastasis in NSCLC, whereas silencing ZNF671 led to the reverse effects. Taken together, these results suggest that ZNF671 restrains the progression and metastasis of NSCLC, similarly to its biological function in other types of cancers.
By far, it has been reported that the over-activation of the Wnt/β-catenin signaling pathway promotes the carcinogenesis and progression of a variety of tumors.25–27 Previous study by Zhao et al demonstrated that the downregulation of XB130 reduces the invasion, metastasis and stem cell self-renewal of breast cancer cells by modulating the EMT process and inhibiting Wnt/β-catenin signaling.28 Furthermore, accumulating evidence has implicated that constitutive activation of Wnt/β-catenin signaling represents one of the most well-recognized cascades modulating tumor invasion and metastasis.29,30 The mutation frequency of the β-catenin gene in HCC somatic cells is 20% to 40%, which usually affects the dominant function of the N-terminal GSK3 and CK1 phosphorylation sites of β-catenin.31,32 β-catenin translocation from cytoplasm to nucleus is detected in 40% to 70% of HCC cases.33,34 In particular, it is worth noting that the activation of Wnt/β-catenin signaling pathway plays a vital role in the process of metastasis of NSCLC.
Conclusion
In our current study, we found that the transcription level and nuclear translocation of β-catenin is inhibited in ZNF671-transduced cells, and meanwhile, the expression of downstream signaling target genes of β-catenin was also decreased, such as cyclinD1 and MMP-9. Moreover, the proliferation and metastasis of NSCLC are significantly restrained in ZNF671-transduced cells. Taken together, our data reveal that tumor suppressor gene ZNF671 modulates the proliferation and metastasis of NSCLC via Wnt/β-catenin signaling.
Acknowledgments
The present study was supported by the Major Program of National Natural Science Foundation of China (grant no. 81500287).
Disclosure
The authors declare that there is no conflict of interest in this work.
References
1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–1953. doi:10.1002/ijc.v144.8
2. Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet. 2005;366(9496):1527–1537. doi:10.1016/S0140-6736(05)67625-8
3. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594. doi:10.1016/S0025-6196(11)60735-0
4. Batevik R, Grong K, Segadal L, Stangeland L. The female gender has a positive effect on survival independent of background life expectancy following surgical resection of primary non-small cell lung cancer: a study of absolute and relative survival over 15 years. Lung Cancer. 2005;47(2):173–181. doi:10.1016/j.lungcan.2004.08.014
5. Perlikos F, Harrington KJ, Syrigos KN. Key molecular mechanisms in lung cancer invasion and metastasis: a comprehensive review. Crit Rev Oncol Hematol. 2013;87(1):1–11. doi:10.1016/j.critrevonc.2012.12.007
6. Bauml J, Mick R, Zhang Y, et al. Determinants of survival in advanced non–small-cell lung cancer in the era of targeted therapies. Clin Lung Cancer. 2013;14(5):581–591. doi:10.1016/j.cllc.2013.05.002
7. Sorensen JB, Hansen HH, Hansen M, Dombernowsky P. Brain metastases in adenocarcinoma of the lung: frequency, risk groups, and prognosis. J Clin Oncol. 1988;6(9):1474–1480. doi:10.1200/JCO.1988.6.9.1474
8. Mimeault M, Hauke R, Batra SK. Recent advances on the molecular mechanisms involved in the drug resistance of cancer cells and novel targeting therapies. Clin Pharmacol Ther. 2008;83(5):673–691. doi:10.1038/sj.clpt.6100296
9. Tsuruo T, Naito M, Tomida A, et al. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003;94(1):15–21. doi:10.1111/j.1349-7006.2003.tb01345.x
10. Zhang J, Wen X, Liu N, et al. Epigenetic mediated zinc finger protein 671 downregulation promotes cell proliferation and tumorigenicity in nasopharyngeal carcinoma by inhibiting cell cycle arrest. J Exp Clin Cancer Res. 2017;36(1):147. doi:10.1186/s13046-017-0621-2
11. Yeh CM, Chen PC, Hsieh HY, et al. Methylomics analysis identifies ZNF671 as an epigenetically repressed novel tumor suppressor and a potential non-invasive biomarker for the detection of urothelial carcinoma. Oncotarget. 2015;6(30):29555–29572. doi:10.18632/oncotarget.4986
12. Mase S, Shinjo K, Totani H, et al. ZNF671 DNA methylation as a molecular predictor for the early recurrence of serous ovarian cancer. Cancer Sci. 2019;110(3):1105–1116. doi:10.1111/cas.13936
13. Wu Z, Zhao J, Qiu M, et al. CRISPR/Cas9 mediated GFP knock-in at the MAP1LC3B locus in 293FT cells is better for bona fide monitoring cellular autophagy. Biotechnol J. 2018;13(11):e1700674. doi:10.1002/biot.v13.11
14. Arnold KM, Opdenaker LM, Flynn D, Sims-Mourtada J. Wound healing and cancer stem cells: inflammation as a driver of treatment resistance in breast cancer. Cancer Growth Metastasis. 2015;8:1–13. doi:10.4137/CGM.S11286
15. Yang YT, Wang YF, Lai JY, et al. Long non-coding RNA UCA1 contributes to the progression of oral squamous cell carcinoma by regulating the WNT/beta-catenin signaling pathway. Cancer Sci. 2016;107(11):1581–1589. doi:10.1111/cas.13058
16. Behrens J. Control of beta-catenin signaling in tumor development. Ann N Y Acad Sci.2000;910:21–33. doi:10.1111/j.1749-6632.2000.tb06698.x
17. Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD, Hanson MA. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61(5 Pt 2):5R–10R. doi:10.1203/pdr.0b013e318045bedb
18. Petronis A. Human morbid genetics revisited: relevance of epigenetics. Trends Genet. 2001;17(3):142–146. doi:10.1016/S0168-9525(00)02213-7
19. Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10(7):687–692. doi:10.1093/hmg/10.7.687
20. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–298. doi:10.1038/nrg2005
21. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi:10.1016/j.cell.2012.06.013
22. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16(4):168–174. doi:10.1016/S0168-9525(99)01971-X
23. Hu Y, Qi MF, Xu QL, et al. Candidate tumor suppressor ZNF154 suppresses invasion and metastasis in NPC by inhibiting the EMT via Wnt/beta-catenin signalling. Oncotarget. 2017;8(49):85749–85758. doi:10.18632/oncotarget.20479
24. Ye L, Xiang T, Fan Y, et al. The 19q13 KRAB Zinc-finger protein ZFP82 suppresses the growth and invasion of esophageal carcinoma cells through inhibiting NF-kappaB transcription and inducing apoptosis. Epigenomics. 2019;11(1):65–80. doi:10.2217/epi-2018-0092
25. King TD, Suto MJ, Li Y. The Wnt/beta-catenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. 2012;113(1):13–18. doi:10.1002/jcb.23350
26. Tian Y, Mok MT, Yang P, Cheng AS. Epigenetic activation of Wnt/beta-catenin signaling in NAFLD-associated hepatocarcinogenesis. Cancers (Basel). 2016;8(8). doi:10.3390/cancers8080076
27. Wang J, Zhang K, Wang J, et al. Underexpression of LKB1 tumor suppressor is associated with enhanced Wnt signaling and malignant characteristics of human intrahepatic cholangiocarcinoma. Oncotarget. 2015;6(22):18905–18920. doi:10.18632/oncotarget.4305
28. Xie T, Jiang C, Dai T, et al. Knockdown of XB130 restrains cancer stem cell-like phenotype through inhibition of Wnt/beta-catenin signaling in breast cancer. Mol Carcinog. 2019;58(10):1832–1845. doi:10.1002/mc.23071
29. White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142(2):219–232. doi:10.1053/j.gastro.2011.12.001
30. Chiurillo MA. Role of the Wnt/beta-catenin pathway in gastric cancer: an in-depth literature review. World J Exp Med. 2015;5(2):84–102. doi:10.5493/wjem.v5.i2.84
31. Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5(9):691–701. doi:10.1038/nrg1427
32. Chen J, Rajasekaran M, Hui KM. Atypical regulators of Wnt/beta-catenin signaling as potential therapeutic targets in Hepatocellular carcinoma. Exp Biol Med (Maywood). 2017;242(11):1142–1149. doi:10.1177/1535370217705865
33. Suzuki T, Yano H, Nakashima Y, Nakashima O, Kojiro M. Beta-catenin expression in hepatocellular carcinoma: a possible participation of beta-catenin in the dedifferentiation process. J Gastroenterol Hepatol. 2002;17(9):994–1000. doi:10.1046/j.1440-1746.2002.02774.x
34. Wong CM, Fan ST, Ng IO. beta-Catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92(1):136–145. doi:10.1002/1097-0142(20010701)92:1<136::AID-CNCR1301>3.0.CO;2-R
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.