")
Back to Journals » Drug Design, Development and Therapy » Volume 13
YB-1 modulates the drug resistance of glioma cells by activation of MDM2/p53 pathway
Authors Tong H, Zhao K, Zhang J, Zhu J, Xiao J
Received 28 August 2018
Accepted for publication 22 November 2018
Published 14 January 2019 Volume 2019:13 Pages 317—326
DOI https://doi.org/10.2147/DDDT.S185514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Qiongyu Guo
Hui Tong,1,* Kai Zhao,2,* Jingyu Zhang,3 Jinxin Zhu,4 Jianqi Xiao2
1Department of Neurosurgery, Linyi Central Hospital, Linyi, Shandong 276400, People’s Republic of China; 2Department of Neurosurgery, The First Hospital of Qiqihar City, Qiqihar, Heilongjiang 161005, People’s Republic of China; 3Department of Internal Medicine, Jiangpu District Health Center of Huai’an, Huai’an, Jiangsu, 223001, People’s Republic of China; 4Department of Neurosurgery, Lianshui County People’s Hospital, Huai’an, Jiangsu 223400, People’s Republic of China
*These authors contributed equally to this work
Background: Y-box-binding protein-1 (YB-1) is aberrantly expressed in a variety of cancers. However, the biological functional role of YB-1 in glioma is not yet clear.
Methods: The expression of MDM2 and YB-1 was analyzed by real time PCR. Overexpression and knockdown of YB-1 in glioma cells were created by transfection of pcDNA-YB-1 and siRNA against YB-1, respectively. Cell viability was performed by CCK8 assay.
Results: Our findings showed that glioma tissues had higher expressions of YB-1 than that in cancer-free tissues in 54 glioma patients, which were also positively correlated with Murine MDM2 expression. Overexpression of YB-1 or MDM2 renders a drug resistance feature in glioma cell exposed to temozolomide (TMZ), by directly targeting p53. Genetic or chemical inhibition of MDM2 significantly blocked YB-1-modulated response of glioma cells to TMZ. Moreover, inhibition of YB-1 or MDM2 reduced glioma cells metastasis and mortality in mice.
Conclusion: YB-1 facilitates the resistance of glioma cells to TMZ by direct activation of MDM2/p53 signaling and represents a promising molecular target for glioma treatment.
Keywords: glioma, p53, Murine double minute 2, Y-box binding protein-1, drug resistance, temozolomide
Introduction
Glioma is a member of the most prevalent primary brain tumors that severely threatens the central nervous system in adults, with a feature of high aggressiveness and poor prognosis.1,2 Although application of radiotherapy and chemotherapy earns a relatively higher survival rate, recurrence and drug resistance are still a challenge.3,4 Temozolomide (TMZ) is regarded as the first-line chemotherapeutic drug for the treatment of glioma clinically. However, tumor cells generally exhibit innate or acquired resistance against TMZ, and drug resistance seems to be responsible for the therapeutic failure of TMZ in glioma patients.5–7 Therefore, TMZ resistance has been a major obstacle in the treatment of glioma. And, a better understanding of drug resistance is crucial to tumor treatment in glioma.
Y-box binding protein 1 (YB-1) is aberrantly expressed in a variety of cancers, which acting as a human transcription factor participated in the regulation of cancer-associated genes.8,9 Despite the fact that there is evidence indicating that YB-1 plays an important role in cell growth, tumor progression, and multidrug resistance, the clinical significance of YB-1 in glioma has not been entirely understood.10,11 It has been reported that the mechanisms of drug resistance are commonly associated with the overexpression of Murine double minute 2 (MDM2) and p53 gene mutation.4 The protein p53 is a transcription factor that is mainly engaged in the regulation of tumor-suppression process. p53 is often blunted in multiple tumors due to either mutations or functional inhibition by its negative endogenous regulator MDM2.12 Furthermore, MDM2 functionally ubiquitinates p53 through functioning as an E3 ligase promoting proteasomal degradation.13 p53 is considered as essential for nuclear translocation of YB-1. Dunn et al reported that YB-1 induces breast tumor-initiating cells to express CD44 and CD49f, leading to enhanced drug resistance.14 Another study found that YB-1 might be linked to drug resistance of bladder cancer with a TP53 gene abnormality that led to p53 mutation.15 Thus, the aim of the present study was to determine the biological role of YB-1 in drug resistance in glioma and to further investigate the molecular basis underlying the drug resistance.
Methods
Patients and samples
Fifty-four glioma tissue samples were collected from patients who underwent chemotherapy with TMZ at The First Hospital of Qiqihar City from January 2016 to June 2017. The pathological diagnoses and classification were approved by at least two experienced pathologists simultaneously, based on the WHO classification system. Fifty-four gliomas were collected in the current study including 13 grade II and 15 grade III astrocytic tumors, 9 grade II and 7 III oligodendrogliomas, and 10 grade IV glioblastomas. Thirty-seven out of the enrolled 54 glioma patients exhibited TMZ resistance. Patients who showed limited response to TMZ treatment for more than 8 weeks were classified as TMZ resistant. All brain specimens were kept for follow-up testing. This study was approved by the Ethics Committee of The First Hospital of Qiqihar City, and written informed consent was obtained from each subject. The study was carried out in accordance with the principles of the Declaration of Helsinki.
Cell lines and drugs
MDM2 inhibitor RG7112 was purchased from AbMole BioScience, Shanghai, People’s Republic of China. Human glioma cell lines U87 (p53wt), DK-MG (p53wt), LN308 (p53null), and U251 (p53mut) were purchased from American Type Culture Collection and cultured in DMEM with 10% FBS (Beyotime, Shanghai, People’s Republic of China) at 37°C in an atmosphere of 5% CO2 and 95% air. TMZ was purchased from Sigma-Aldrich. Aliquots of TMZ were dissolved in dimethyl sulfoxide and stored at −20°C for the following tests. Glioma cell lines U87, DK-MG, and U251 were treated with 25, 50, 100, and 200 μM TMZ for 48 hours.
RNA preparation and quantitative real-time PCR (qRT-PCR)
Total RNA from glioma cell lines and all tissues samples was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Carlsbad, CA, USA) and then reverse transcribed using the Reverse Transcription Kit (Bio-Rad, Minneapolis, MN, USA) according to the manufacturer’s instructions. cDNA amplification was subsequently performed for detecting YB-1 and MDM2 by quantitative real-time PCR (qRT-PCR) using SYBR Green Master mix (Roche Diagnostics) according to the manufacturer’s protocol. The mRNA expression was normalized to the expression of GAPDH mRNA and calculated by using the 2−ΔΔCt method. The PCR primers used are as follows: YB-1 (forward: 5′-AGGCAGGAACGGTTGTAGGT-3′; reverse: 5′-GTCAAGCGTAGGTTGCGGTGATGG-3′); MDM2 (forward: 5′-GGGAGATATGTTGTGAAAGAAGC-3′; reverse: 5′-CCCTGCCTGATACACAGTAACTT-3′); GAPDH (forward: 5′-GTCGGAGTCAACGGATTT-3′; reverse: 5′-GCCATGGGTGGAATCATATTGG-3′).
Cell viability assay
Cell proliferation was detected using the cell counting kit-8 (CCK-8) assay. Cells were seeded in a 96-well culture plate at 1×104 cells/well. Cells were treated with a single dose of TMZ (0, 25, 50, 100, and 200 μM) and exposed to 5 μM RG7112. Ten microliters of CCK-8 were then added to each well and incubated for 4 hours at 37°C. Experiments were replicated at least three times.
Immunohistochemistry
Immunohistochemistry was performed to detect YB-1 and MDM2. Briefly, glioma tissues and cancer-free tissues were fixed using 10% buffered formalin and embedded using paraffin. The deparaffinized tissue sections were transferred to heat-induced epitope retrieval for 10 minutes and then incubated with the primary antibodies (anti-YB-1, 1:5,000, Abcam; anti-MDM2, 1:50, Abcam) at 37°C for 2 hours. Biotinylated secondary antibodies were applied to incubate tissue sections for 30 minutes. The stained cells in ten fields were counted under the microscope (Olympus).
H&E staining
Mice were naturally dead or sacrificed and the lung tissues were excised. The tissues were fixed in 10% formalin for overnight and dehydrated with different concentration of ethanol. Then the tissues were embedded in paraffin and cut into 4 μm thick sections. Sections were mounted on glass slides and stained with H&E. For observation of ectopic tumor metastatic nodes, eight views were randomly selected and counted under the light microscope (Olympus).
Western blotting
Cells were lysed using RIPA lysis buffer (10 mM Tris-HCl [pH 7.4], 0.1% SDS, 1% sodium deoxycholate, 0.15 M NaCl, 1 mM EDTA, and 1% Triton X-100). Lysates were collected and cleared by centrifugation, and the protein concentration was quantified using the bicinchoninic acid method. Protein samples were electrophoresed by 12% SDS-PAGE and then transferred to polyvinylidene fluoride membranes (0.45 μm, Millipore). The membranes were blocked with 5% skim milk, and then incubated overnight at 4°C with the primary antibody (anti-YB-1, 1:1,000, Abcam; anti-MDM2, 1:1,000, Abcam). The membranes were then washed three times with Tris buffered saline with tween and incubated with horseradish peroxidase conjugated goat antirabbit secondary antibody (Beyotime) for 2 hours at room temperature. The bands of all proteins were normalized to the intensity of corresponding bands for β-actin. Protein expression was visualized with a chemiluminescence ECL kit (Beyotime).
Cell transfection
YB-1, MDM2, and control siRNAs were synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, People’s Republic of China) and transfected into cells with 10 nM plasmid containing siRNA using Lipofectamine 2000 transfection reagent (Invitrogen; Thermo Fisher Scientific) according to the manufacturer’s protocol. Assays were performed at 48 hours after transfection. YB-1 was overexpressed in the U87 and DK-MG glioma cell lines, while p53 or p53 mutant was overexpressed in U87 using retrovirus transduction as described previously.16 Ten nanometer DNA oligonucleotides containing the YB-1, p53, and p53 mutant sequences were cloned in plasmid (Invitrogen; Thermo Fisher Scientific) and then transfected into cells. After 24 hours, the transfection efficiency was defined using RT-PCR. The empty plasmid acted as the control.
Statistical analysis
Statistical analysis was performed using SPSS 18.0. The data are presented as mean ± SD or quartiles. Mean comparison between or among groups was conducted by two-tailed t-test or one-way ANOVA. The survival curves were performed using the Kaplan–Meier analysis. P<0.05 was considered statistically significant.
Results
YB-1 expression is elevated in glioma and positively correlated with MDM2
To explore the functional role of YB-1 and MDM2 in glioma, we first examined the expressions of YB-1 and MDM2 in glioma tissues and cancer-free tissues by immunostaining. We found that the expressions of YB-1 and MDM2 were remarkably positive for glioma tissues compared with that for cancer-free tissues (Figure 1A and B). Next, the mean mRNA levels of YB-1 and MDM2 in 54 glioma tissues and the corresponding cancer-free tissues were analyzed by qRT-PCR. We found that the expressions of YB-1 and MDM2 were significantly increased in glioma tissues compared with that in cancer-free tissues (Figure 1C and D). Subsequently, we tried to determine whether or not YB-1 had a key role in drug resistance of glioma. We found that patients with a history of drug resistance had higher level of YB-1 and MDM2 compared with those drug-sensitive ones (Figure 1C and D). Moreover, the relative expressions of YB-1 were positively correlated with MDM2 expression in glioma tissues (Figure 1E). The above data suggested that YB-1 and MDM2 played crucial roles in drug resistance of glioma.
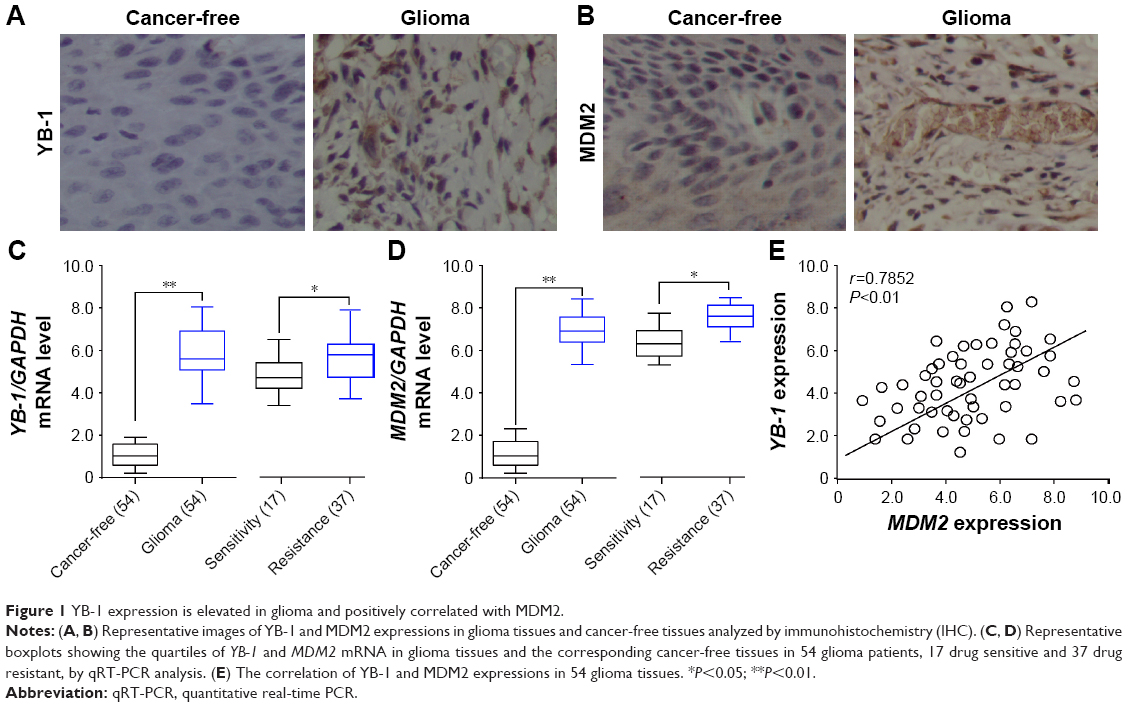 | Figure 1 YB-1 expression is elevated in glioma and positively correlated with MDM2. |
YB-1 mediates glioma cells response to TMZ
Next, we further investigated the effects of YB-1 on drug resistance of glioma. A subset of glioma cell lines U87 and DK-MG were transfected by lentiviral vector containing pcDNA-YB-1 and followed by treatment with various doses of TMZ. We found that YB-1 overexpression significantly promoted TMZ resistance in U87 and DK-MG cells, indicated by a significantly increased percentage of surviving cells (Figure 2A). Subsequently, knockdown of YB-1 expression in U87 and DK-MG cells was genetically achieved using siRNA. It showed that inhibition of YB-1 significantly sensitizes U87 and DK-MG cells to TMZ (Figure 2B). It implicated that YB-1 modulates the response of glioma cells to TMZ. It was reported that MDM2 had a crucial role in drug resistance in multiple cancer types.17 Subsequently, we investigated whether MDM2 participated in YB-1-mediated TMZ resistance in glioma cells. We analyzed expressions of MDM2 in YB-1-sufficient and -deficient U87 and DK-MG cells. We found that overexpression of YB-1 led to an increased expression of MDM2 and a decreased expression of p53 in glioma cells. On the contrary, inhibition of YB-1 resulted in a decreased expression of MDM2 and an increased expression of p53 (Figure 2C and D). This indicated that MDM2 is a direct target of YB-1 in glioma cells.
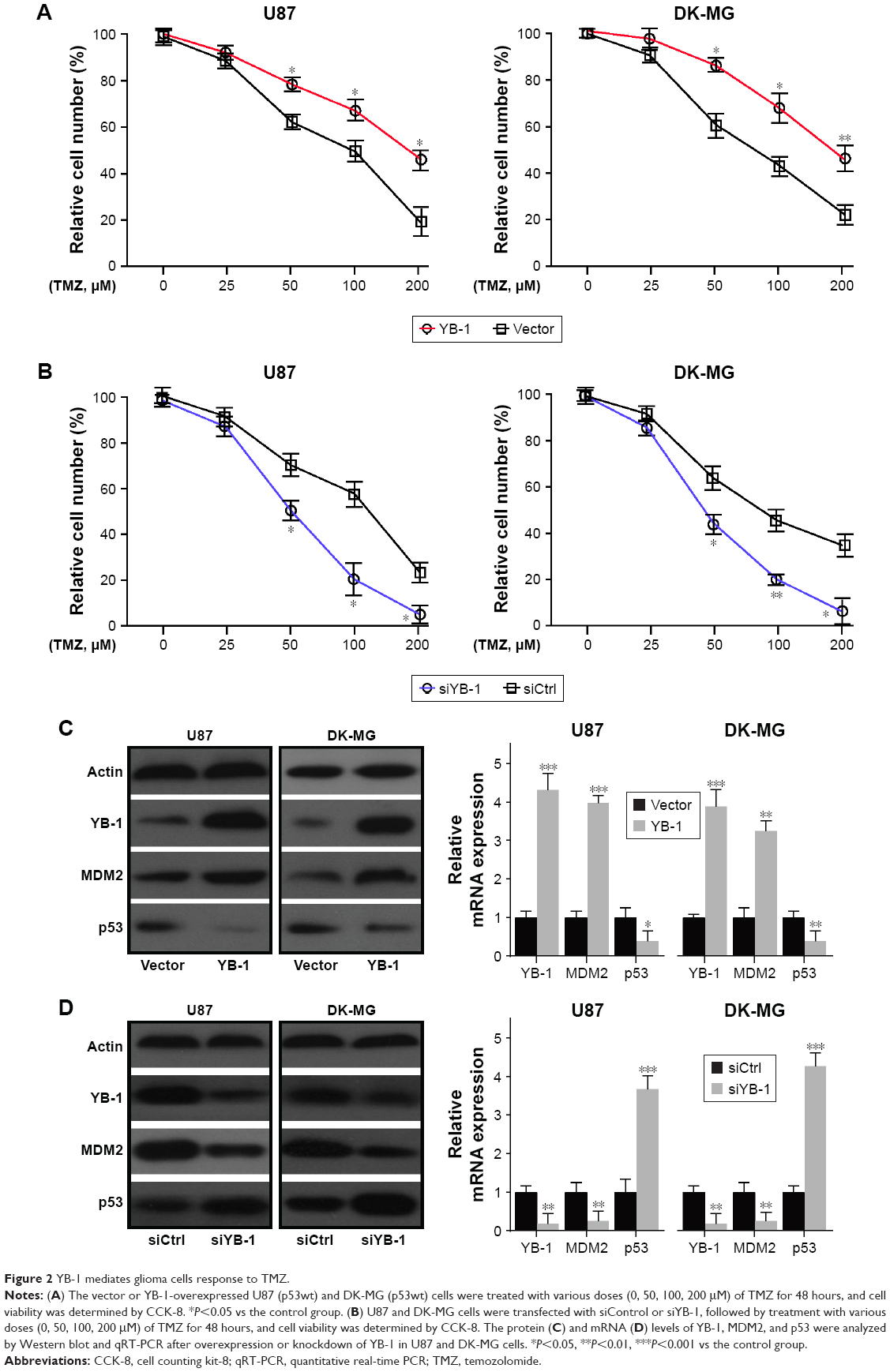 | Figure 2 YB-1 mediates glioma cells response to TMZ. |
Inhibition of MDM2 disables YB-1-mediated TMZ resistance in glioma cells
To further investigate whether MDM2 contributed to drug resistance in glioma, the expressions of MDM2 in U87 and DK-MG cells were genetically inhibited by siRNA methods or chemically inhibited by MDM2 inhibitor RG7112. It showed that inhibition of MDM2 enhanced the sensitivity of glioma cells to TMZ, exhibited by the reduced numbers of viable cells in the MDM2-deficient glioma cells (Figure 3A). Moreover, addition of RG7112 significantly suppressed viability of glioma cells (Figure 3B). It indicated that MDM2 played a crucial role in drug resistance in glioma. Next, to investigate whether MDM2 directly affects YB-1-mediated drug resistance in glioma cells, the expressions of MDM2 in YB-1-overexpressed U87 and DK-MG cells were genetically inhibited by siRNA method. It showed that inhibition of MDM2 dramatically reversed YB-1-mediated drug resistance to TMZ, resulting in a decreased number of viable cells (Figure 3C). In addition, Figure 3D showed that chemical inhibition of MDM2 by the MDM2 inhibitor RG7112 significantly blocked YB-1-mediated TMZ resistance in the U87 and DK-MG cells (Figure 3D). Notably, RG7112 effectively inhibited MDM2 expression, but had limited effects on YB-1 expression (Figure 3D). Based on the above data, we concluded that MDM2, as a downstream effector of YB-1, played a pivotal role in YB-1-mediated TMZ resistance in glioma cells.
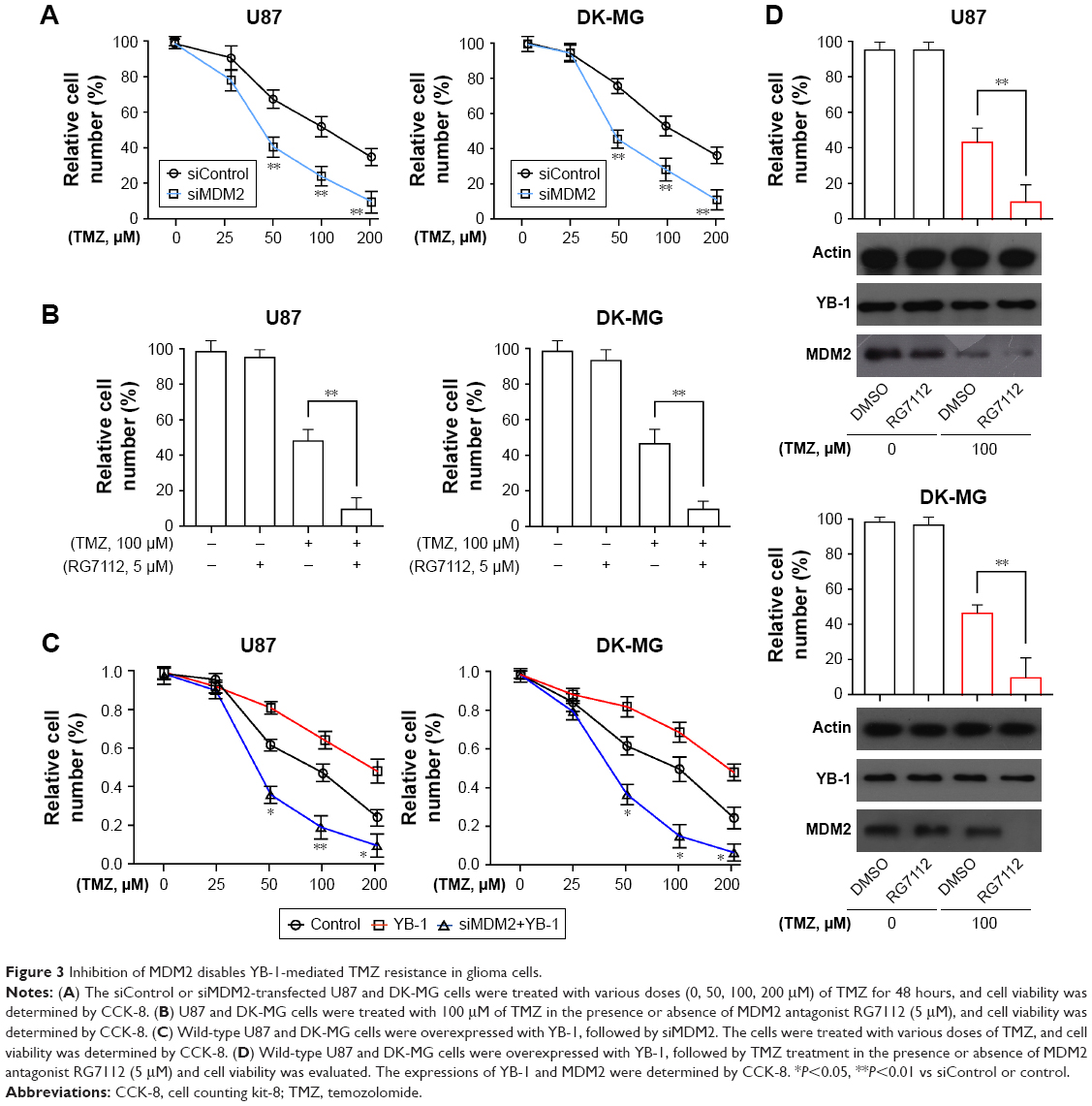 | Figure 3 Inhibition of MDM2 disables YB-1-mediated TMZ resistance in glioma cells. |
YB-1-mediated TMZ resistance in glioma cells in a p53-dependent manner
It is well known that MDM2 is an E3 ubiquitination ligase that directly binds to the transcription domain of p53 for proteasomal degradation.13 Next, to investigate whether TMZ resistance in glioma cells is specifically controlled by p53 status, U87 cells were constructed with a wild-type p53 and a mutant p53 sequence. It showed that overexpression of wild-type p53, not mutant p53, significantly sensitizes glioma cells to TMZ (Figure 4A). Subsequently, the glioma cells U87 (p53wt), LN308 (p53null), and U251 (p53mut) were treated with TMZ to further define the role of p53. Compared with those cells (LN308 and U251 cells) with defective p53 expression, the wild-type p53-bearing U87 cells were more sensitive to TMZ (Figure 4B). Next, we investigated whether YB-1 expression affects chemoresistance to TMZ in p53-mutated glioma cells. We found that YB-1 overexpression had no significant effects on the viability of LN308 cells (Figure 4C). We also detected p53 expressions in glioma tissues by qRT-PCR. We found that glioma tissues had a lower level of p53 than cancer-free tissues. Moreover, the expressions of p53 were significantly reduced in drug-resistant tissues compared with that in drug-sensitive ones (Figure 4D). The above data suggest that YB-1 modulates drug resistance in glioma cells by directly controlling MDM2/p53 pathway (Figure 4E).
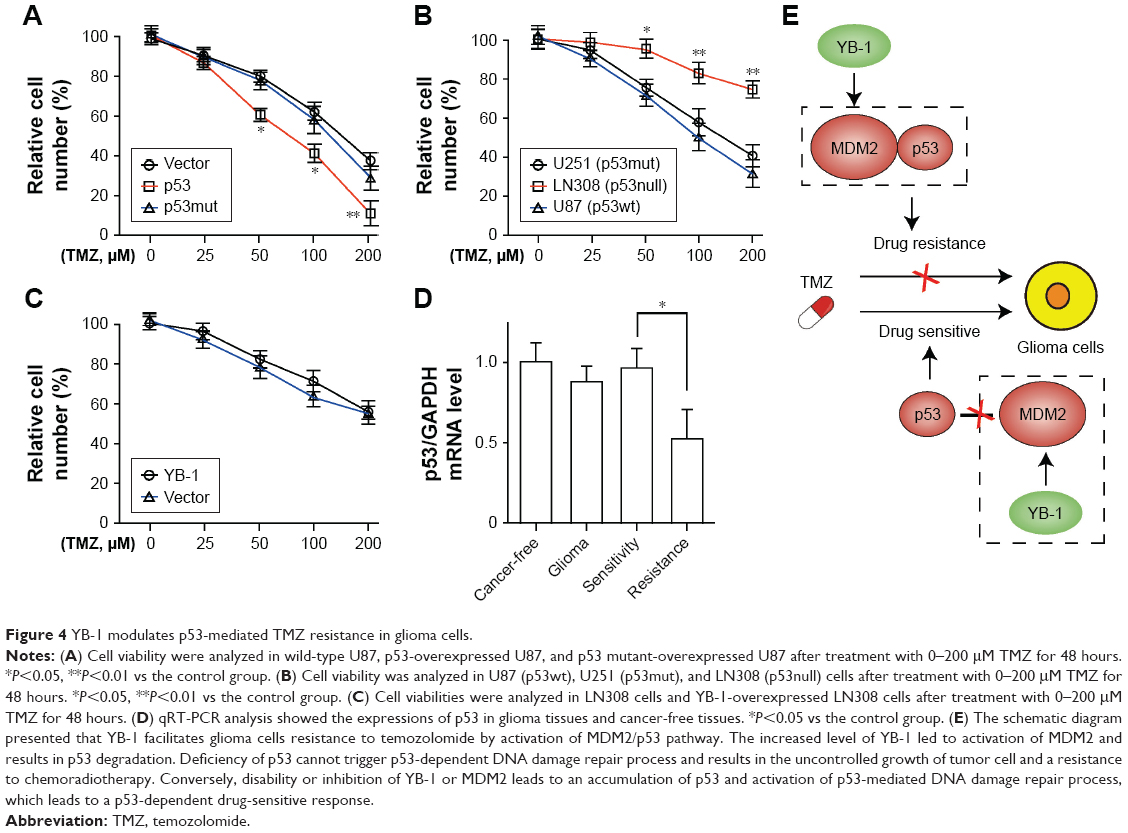 | Figure 4 YB-1 modulates p53-mediated TMZ resistance in glioma cells. |
Inhibition of YB-1 or MDM2 reduced metastatic node and mortality in vivo
Next, we investigated the drug response of YB-1/MDM2 signaling on glioma cells in vivo. The nude mice were injected with wild-type or siYB-1-transfected U87 cells followed by treatment with TMZ. We found that inhibition of YB-1 resulted in a reduction of mice mortality (Figure 5A) compared with the control group. Mice with YB-1 deficiency also had reduced metastatic nodes (Figure 5B) in the lungs as well as the decreased proliferating cells indicated by Ki67 staining (Figure 5C). Next, we performed the same assay by injecting with wide-type or siMDM2-transfected U87 cells. Consistent with our findings, inhibition of MDM2 led to a reduction of overall mortality (Figure 5D), metastatic nodes (Figure 5E), and the proliferating cells (Figure 5F) compared with that in the control group. These above data demonstrated that inhibition of YB-1 or MDM2 in glioma cells reduced tumor metastasis and mortality in mice.
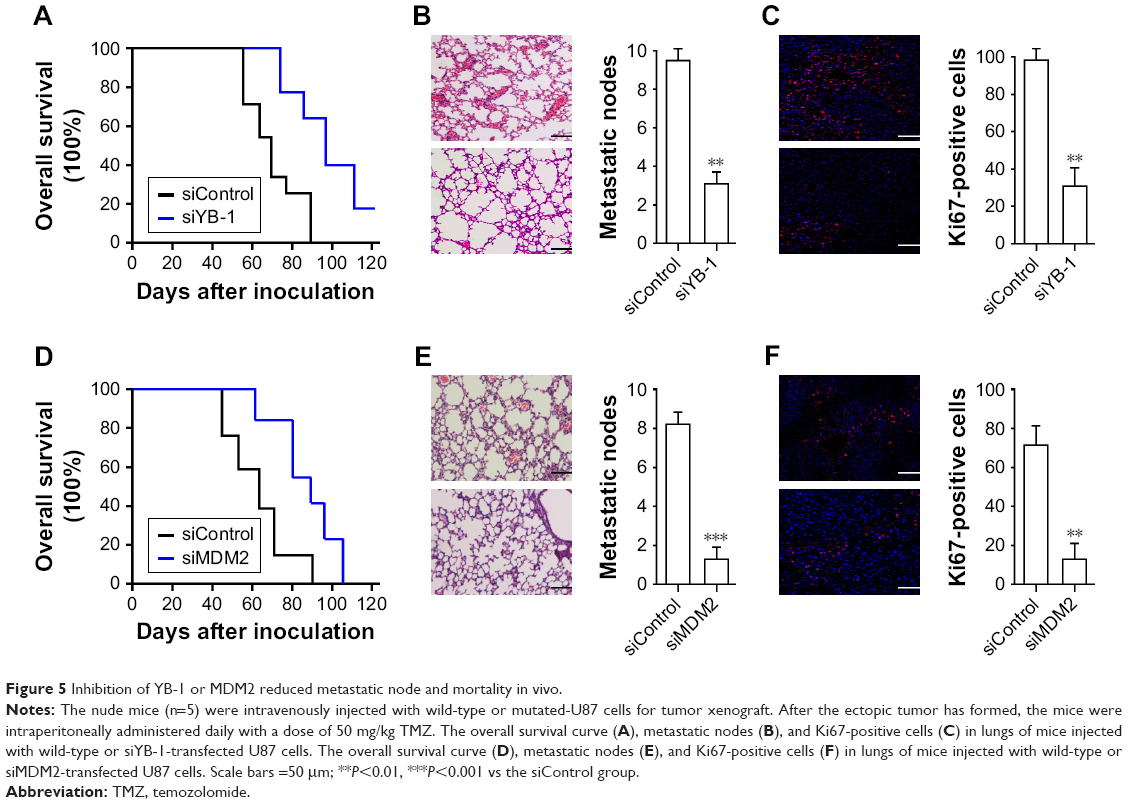 | Figure 5 Inhibition of YB-1 or MDM2 reduced metastatic node and mortality in vivo. |
Discussion
Glioma is a malignant brain tumor commonly found in the central nervous system and the prognosis remains poor due to its biological characteristics.18 It is well known that utilization of chemotherapeutic drug, TMZ, results in an improved survival of patients with glioma and TMZ resistance is still one of the main obstacles for tumor treatment.19 In that situation, a group of drug-resistant genes have been uncovered by bioinformatics tools.20 It has been revealed that TP53, KRAS, and CDKN2A alterations are the most common early events in multiple cancer types including glioma. The current known potential mechanisms of glioma resistance to TMZ include DNA repair, overexpression of EGFR and MDM2, and p53 mutation, etc. Y-box proteins have been linked to RNA translation and DNA repair in a variety of human cancers, but the role of YB-1 regarding this drug resistance remains unclear.21 In this study, we demonstrated that YB-1 modulates p53/MDM2 pathway through direct binding, and knockdown of YB-1 is associated with glioma resistance to TMZ.
YB-1 is one of the members of the cold-shock proteins and acts as a regular transcription factor through binding to double-stranded DNA.22 YB-1 played a major role in promoting cell proliferation and tumorigenesis23,24 and also suppressed the transcription of cell death-promoting genes, such as p53 by binding possible Y-box sequences.25 The expression of YB-1 was correlated with the clinical stage and prognosis of patients in various human cancers. In the present study, we initially demonstrated that YB-1 is strongly expressed in drug-resistant glioma cells. Overexpression of YB-1 could significantly promote drug resistance to TMZ and increase the growth viability of glioma cells, suggesting that YB-1 might have an ability for regulating cellular proliferation, which is in line with findings by Astanehe et al.25 Bergmann26 also indicated that YB-1 protein level is elevated in most human breast cancers, and higher YB-1 levels have been correlated with drug resistance.
p53 is a tumor suppressor encoded by the TP53 gene that could lead to a cessation of cell growth in response to cellular stress. Accumulating evidence from studies have demonstrated that activation of p53 was reported to sensitize the glioma to TMZ.27,28 As a regulator of the p53 pathway, MDM2 protein inhibits the transcriptional activity, nuclear localization, and protein stability of p53. It also reported that also p53 deficiency plays a critical role in glioma resistance to anticancer drugs.29,30 p53–MDM2 interaction is also a potential cancer therapeutic target due to MDM2 overexpression in gliomas.31 RG7112 is a potent MDM2 antagonist, which inhibits the binding between p53 and MDM2 and results in stabilization and accumulation of the wild-type p53.31,32 Our study provides evidence that the overexpression of MDM2 might be associated with drug resistance, which is consistent with the study of Yang-Hartwich33 and Knappskog.34 We also found that RG7112 has antitumor activity exclusively in cell tumors with wild-type p53. The mechanism of drug resistance is quite complex and closely related to genes and transporters. It also reported that YB-1 interacted with p53, leading to suppression of p53 activity and contributing to cell-cycle progression and tumorigenesis.35 Although YB-1 inhibits p53 from causing cell death, YB-1 does not interfere with the ability of p53 to transactivate MDM2.36 Our study demonstrated that YB-1 overexpression in U87 and DK-MG cells could significantly promote MDM2 expression, thus suppressing the level of p53. Therefore, selective change of p53 activity by YB-1 provides an explanation for the relationship of YB-1-mediated drug resistance and poor tumor prognosis. Since inhibition of YB-1 activity downregulates MDM2 expression in human cancer cell lines, the YB-1/MDM2/p53 pathway is a potential molecular target for future cancer therapy. Our study also has several limitations. The precise mechanisms of drug resistance in vivo are yet to be investigated. In addition, the feasibility of YB-1 should be seriously considered in any future study.
In the present study, we demonstrated that YB-1 and MDM2 were overexpressed in glioma, which is associated with drug resistance to TMZ. The YB-1 protein modulates drug resistance of glioma to TMZ by activation of MDM2, and the resultant degradation of p53. The findings suggest that YB-1 facilitates glioma cell resistance to TMZ through the MDM2/p53 pathway, which shed light on the development of YB-1-targeted molecular therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Wang Y, Guan G, Cheng W, et al. ARL2 overexpression inhibits glioma proliferation and tumorigenicity via down-regulating AXL. BMC Cancer. 2018;18(1):599. | ||
Vredenburgh JJ, Desjardins A, Herndon JE, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13(4):1253–1259. | ||
Nishikawa R. Standard therapy for glioblastoma – a review of where we are. Neurolo Med Chir. 2010;50(9):713–719. | ||
Messaoudi K, Clavreul A, Lagarce F. Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov Today. 2015;20(7):899–905. | ||
Sato A, Sunayama J, Matsuda K, et al. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells. 2011;29(12):1942–1951. | ||
Zhang J, Stevens MD, Bradshaw T. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol. 2012;5(1):102–114. | ||
Grek CL, Sheng Z, Naus CC, Sin WC, Gourdie RG, Ghatnekar GG. Novel approach to temozolomide resistance in malignant glioma: connexin43-directed therapeutics. Curr Opin Pharmacol. 2018;41:79–88. | ||
Bargou RC, Jürchott K, Wagener C, et al. Nuclear localization and increased levels of transcription factor YB-1 in primary human breast cancers are associated with intrinsic MDR1 gene expression. Nat Med. 1997;3(4):447–450. | ||
Lin Z, Lu Y, Meng Q, et al. miR372 promotes progression of liver cancer cells by upregulating erbB-2 through enhancement of YB-1. Mol Ther Nucleic Acids. 2018;11:494–507. | ||
Perazzoli G, Prados J, Ortiz R, et al. Temozolomide resistance in glioblastoma cell lines: implication of MGMT, MMR, P-glycoprotein and CD133 expression. PLoS One. 2015;10(10):e0140131. | ||
Mantwill K, Naumann U, Seznec J, et al. YB-1 dependent oncolytic adenovirus efficiently inhibits tumor growth of glioma cancer stem like cells. J Transl Med. 2013;11:216. | ||
Villalonga-Planells R, Coll-Mulet L, Martínez-Soler F, et al. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS One. 2011;6(4):e18588. | ||
Janouskova H, Maglott A, Leger DY. Integrin α5β1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Eur J Cancer. 2012;72(14):3463–3470. | ||
To K, Fotovati A, Reipas KM, et al. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010;70(7):2840–2851. | ||
Yamashita T, Higashi M, Momose S, Morozumi M, Tamaru JI. Nuclear expression of Y box binding-1 is important for resistance to chemotherapy including gemcitabine in TP53-mutated bladder cancer. Int J Oncol. 2017;51(2):579–586. | ||
Shurin MR. Cancer as an immune-mediated disease. Immunotargets Ther. 2012;1:1–6. | ||
Cinatl J, Speidel D, Hardcastle I, Michaelis M. Resistance acquisition to MDM2 inhibitors. Biochem Soc Trans. 2014;42(4):752–757. | ||
Feng Y, Wang L, Liu X, et al. Human corticotrophin releasing factor inhibits cell proliferation and promotes apoptosis through upregulation of tumor protein p53 in human glioma. Oncol Lett. 2018:15(6):8378–8386. | ||
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | ||
Zhang H, Luo S, Zhang X, et al. SEECancer: a resource for somatic events in evolution of cancer genome. Nucleic Acids Res. 2018;46(D1):D1018–D1026. | ||
van Roeyen CR, Scurt FG, Brandt S, et al. Cold shock Y-box protein-1 proteolysis autoregulates its transcriptional activities. Cell Commun Signal. 2013;11(1):63. | ||
Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays. 2003;25(7):691–698. | ||
Berquin IM, Pang B, Dziubinski ML, et al. Y-box-binding protein 1 confers EGF independence to human mammary epithelial cells. Oncogene. 2005;24(19):3177–3186. | ||
Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9(11):785–797. | ||
Lasham A, Moloney S, Hale T, et al. The Y-box-binding protein, YB1, is a potential negative regulator of the p53 tumor suppressor. J Biol Chem. 2003;278(37):35516–35523. | ||
Bergmann S, Royer-Pokora B, Fietze E, et al. YB-1 provokes breast cancer through the induction of chromosomal instability that emerges from mitotic failure and centrosome amplification. Cancer Res. 2005;65(10):4078–4087. | ||
Chen X, Tai L, Gao J, et al. A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation. J Control Release. 2015;218:29–35. | ||
Kim SS, Rait A, Kim E, et al. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano. 2014;8(6):5494–5514. | ||
Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49(1):223–241. | ||
Renner G, Noulet F, Mercier MC, et al. Expression/activation of α5β1 integrin is linked to the β-catenin signaling pathway to drive migration in glioma cells. Oncotarget. 2016;7(38):62194–62207. | ||
Blough MD, Beauchamp DC, Westgate MR, Kelly JJ, Cairncross JG. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J Neurooncol. 2011;102(1):1–7. | ||
Tovar C, Graves B, Packman K, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73(8):2587–2597. | ||
Yang-Hartwich Y, Soteras MG, Lin ZP, et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene. 2015;34(27):3605–3616. | ||
Knappskog S, Lønning PE. P53 and its molecular basis to chemoresistance in breast cancer. Expert Opin Ther Targets. 2012;16(1):S23–S30. | ||
Zauli G, Voltan R, Bosco R, et al. Dasatinib plus Nutlin-3 shows synergistic antileukemic activity in both p53 wild-type and p53 mutated B chronic lymphocytic leukemias by inhibiting the Akt pathway. Clin Cancer Res. 2011;17(4):762–770. | ||
Inoue K, Fry EA, Frazier DP. Transcription factors that interact with p53 and Mdm2. Int J Cancer. 2016;138(7):1577–1585. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.