")
Back to Journals » OncoTargets and Therapy » Volume 9
YAP induces cisplatin resistance through activation of autophagy in human ovarian carcinoma cells
Authors Xiao L, Shi X, Zhang Y, Zhu Y, Zhu L, Tian W, Zhu B, Wei Z
Received 19 December 2015
Accepted for publication 25 January 2016
Published 16 March 2016 Volume 2016:9 Pages 1105—1114
DOI https://doi.org/10.2147/OTT.S102837
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Lan Xiao,1,* Xiao-Yan Shi,2,* Ying Zhang,1 Ying Zhu,1 Lin Zhu,3 Wang Tian,1 Bing-Kun Zhu,1 Zhao-Lian Wei1
1Department of Obstetrics and Gynecology, First Affiliated Hospital, Anhui Medical University, Hefei, 2Key Laboratory for Molecular Diagnosis of Hubei Province, Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 3Obstetrics and Gynecology Hospital of Fudan University (Shanghai Red House Obstetrics and Gynecology Hospital), Shanghai, People’s Republic of China
*These authors contributed equally to this work
Objective: To identify the role of YAP in cisplatin resistance in human ovarian cancer cells and in the regulation of autophagy in these cancer cells.
Materials and methods: The cisplatin-sensitive OV2008 parental cell line and its cisplatin-resistant variant C13K were cultured. RNA interference was used to knock down the YAP gene. Accumulation of GFP-LC3 puncta was performed by fluorescence microscopy. The formation of autophagosomes was observed by transmission electron microscopy. Drug sensitivity was examined using CCK-8 assay, while apoptosis, the level of intracellular rhodamine 123 and lysosomal acidification were analyzed by fluorescence-activated cell sorting. Acid phosphatase activity was measured using an acid phosphatase-assay kit. Real-time polymerase chain reaction, Western blotting, and immunofluorescence detection were used to detect the protein and messenger RNA expression of YAP, YAP target genes, CCND1, cleaved PARP, and caspase 3, Atg-3 and -5, and the LC3B protein.
Results: YAP signaling may regulate cisplatin resistance in ovarian cancer cells by augmenting cellular autophagic flux. After knockdown of YAP-sensitized C13K cells to cisplatin by inducing a decrease in autophagy, YAP led to an increase in autophagy via enhancement of autolysosome degradation.
Conclusion: YAP-mediated autophagy may play a protective role in cisplatin-resistant human ovarian cancer cells. Therefore, YAP-mediated autophagy should be explored as a new target for enhancing the efficacy of cisplatin against ovarian cancer and other types of malignancies.
Keywords: YAP protein, autophagy, cisplatin resistance, ovarian carcinoma
Introduction
Ovarian cancer, one of the most common gynecologic malignancies, has the highest mortality rate among all gynecologic malignancies.1 Late diagnosis of ovarian cancer and ineffective chemotherapy has resulted in the number of mortalities from ovarian cancer exceeding that of any other cancer of the female reproductive system. Cisplatin resistance is a major obstacle for the successful treatment of ovarian cancer; however, the underlying mechanisms that promote cisplatin resistance in ovarian cancer cells remain to be elucidated.
Previous studies have suggested that autophagy is also a response to certain forms of therapeutic stress, including cytotoxic chemotherapy.2 Autophagy-induced drug resistance in cancer cells has been a challenge to cancer treatment,3 as it functions as an adaptive response that mediates resistance to these treatments.4 However, the role of autophagy is still a topic of intense debate, as it is not clear whether this cellular process functions as a pro-survival or pro-death mechanism to counteract or mediate the cytotoxic effects of anticancer agents.5,6 There is growing evidence for the role of cisplatin treatment in inducing autophagic response in various cancer cells,7–9 including human ovarian cancer cells.10,11 However, the underlying mechanism by which cisplatin induces autophagy is largely unclear. Therefore, exploring the autophagy mechanism of cisplatin resistance is essential for the successful treatment of ovarian cancer.
YAP, the gene for which is located in the genomic region 11q22, is a transcriptional coactivator that can function as an oncoprotein by interacting with several transcription factors.12 The expression of YAP is amplified in a variety of cancers, including ovarian cancer; high levels of YAP are observed in human liver tumors, and it is a key driver of hepatocellular carcinoma tumorigenesis.13,14 YAP also plays a role in the progression of human colorectal cancer.15 Similarly, a correlation between high YAP expression and poor prognosis has been found in non-small-cell lung cancer and esophageal squamous cell carcinoma.16,17 Recently, YAP expression was shown to be positively correlated with chemotherapeutic drug resistance.18 For example, hepatocellular carcinoma cells with a high level of YAP expression were reported to exhibit doxorubicin resistance.19 Additionally, a high level of YAP localized to the nucleus has been found in colon cancer cells that are resistant to the antimetabolite 5-fluorouracil, as well as castration-resistant prostate tumor samples.20,21 Further, there has been ample evidence to show that upregulation of YAP can induce resistance to the chemotherapeutic drugs taxol and cisplatin in mammalian and ovarian cancer cells.22,23 These studies, however, failed to explore the underlying mechanisms via which YAP participates in cisplatin resistance in ovarian cancer cells.
The recent data indicates that the YAP pathway plays a role in integrating the processes of autophagy and apoptosis during cellular stress.24,25 This finding led us to analyze whether YAP could modulate autophagy in the context of cancer. We hypothesized that the YAP pathway may play a role in cisplatin resistance in ovarian cancer through regulation of autophagy. Illustrating the interaction between YAP and the autophagy pathway may be useful for the development of new potential therapeutic targets in ovarian cancer.
Materials and methods
Cell lines and culture
The cisplatin-sensitive human ovarian cancer cell line OV2008 and its cisplatin-resistant clone C13K were supplied by Dr Wen-Cheng Ding (Cancer Biology Research Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology). Cells were maintained in complete Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere containing 5% CO2. The cisplatin-resistant C13K cells were cultured in RPMI 1640 supplemented with 10% fetal bovine-serum medium containing 1 μmol/L cisplatin to maintain resistance.
The human cell lines (OV2008 and C13K) used in our research is the established human cell lines, from which the identity of donor(s) are not considered human subject and thus institutional approval of use of human cell lines was deemed unnecessary.
Reagents
Cisplatin was purchased from Hansoh Pharmaceutical Co Ltd (Lianyungang, People’s Republic of China [PRC]). Plasmid complementary DNA (pcDNA)3.1–GFP–LC3 was purchased from Yingrun Biotech Co Ltd (Changsha, PRC). Chloroquine was purchased from Sigma-Aldrich Co (St Louis, MO, USA) and used at a concentration of 50 μM to treat cells. Rapamycin was purchased from Cell Signaling Technology Inc (Danvers, MA, USA) and used at a concentration of 20 μM to treat cells.
siRNA transfection
Small interfering RNA (siRNA) against the YAP gene (sc-38637; Santa Cruz Biotechnology Inc, Dallas, TX, USA) was used for loss-of-function experiments. The control siRNA sc-37007 (Santa Cruz Biotechnology) was used as a negative control. Each siRNA (37.5 nM) was transfected into ovarian cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The knockdown of a target gene was verified by Western blotting.
Drug sensitivity assay
Cells were seeded at a density of 1×104 cells per well in 96-well plates. After cellular adhesion, the C13K and OV2008 cells were exposed to various doses of cisplatin (0, 10, 30, and 50 μM) for 48 hours. Each treatment was repeated in four wells. Measurement of viable cell mass was performed using CCK-8 (Beyotime Biotechnology, Shanghai, PRC). In brief, an aliquot of 10 μL of CCK-8 plus 100 μL RPMI 1640 was added to each well and incubated for 2 hours. Absorbance was measured with a microplate reader (model 680; Bio-Rad Laboratories Inc, Hercules, CA, USA) at a wavelength of 450 nm. Each experiment was repeated three times. The concentration of cisplatin that produced a 50% inhibition of growth (IC50) was estimated using the relative survival curve.
Quantitative real-time PCR analysis
When cells reached a confluence of 90%, they were collected and RNA extracted using Trizol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. The cDNA was synthesized by reverse transcription using the ThermoScript reverse-transcription polymerase chain reaction (PCR) system (Thermo Fisher Scientific) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using SYBR Green PCR master mix (204143; Qiagen NV, Venlo, the Netherlands) in a total volume of 20 μL on the 7900HT fast real-time PCR system (Thermo Fisher Scientific). The primers used were as follows: YAP, 5′-CGCTCTTCAACGCCGTCA-3′ (forward) and 5′-AGTACTGGCCTGTCGGGAGT-3′ (reverse); Cyr61, 5′-ACTTCATGGTCCCAGTGCTC-3′ (forward) and 5′-AATCCGGGTTTCTTTCACA-3′ (reverse); 18s, 5′-GATCCATTGGAGGGCAAGTC-3′ (forward) and 5′-TCCCAAGATCCAACTACGAG-3′ (reverse); CCND1, 5′-TGCCCTCTGTGCCACAGATG-3′ (forward) and 5′-TCTGGAGAGGAAGCGTGTGA-3′ (reverse); CTGF, 5′-GCAGGCTAGAGAAGCAGAGC-3′ (forward) and 5′-ATGTCTTCATGCTGGTGCAG-3′ (reverse); and Beclin1 1, 5′-GGCTGAGGGATGGAAGGGTCTAAG-3′ (forward) and 5′-GTTTCGCCTGGGCTGTGGTAAGTA-3′ (reverse). The levels of messenger RNA (mRNA) were calculated using 2−ΔΔCT and normalized to human 18s mRNA levels.
Western blotting
The cells were lysed in radioimmunoprecipitation lysis buffer (Beyotime), and the protein concentrations were determined. Approximately 60 μg of protein was separated on a 10% sodium dodecyl sulfate polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. The primary antibodies used were as follows: YAP (sc-15407; Santa Cruz Biotechnology), Cyr61 (sc-13100; Santa Cruz Biotechnology), CTGF (sc-101586; Santa Cruz Biotechnology), CCND1 (sc-20044; Santa Cruz Biotechnology), LC3B and Beclin1 (3868S and 3738, respectively; Cell Signaling Technology), cleaved PARP and caspase 3 (9541 and 9661, respectively; Cell Signaling Technology), and Atg-3 and -5 (3415 and 2630, respectively; Cell Signaling Technology).
Transmission electron microscopy
The cells were fixed using 2.5% glutaraldehyde in 0.1 M phosphate buffer for 2 hours at 4°C, and then postfixed in 1% osmium tetroxide for 3 hours. The samples were scraped and pelleted, dehydrated in a graded series of ethanol baths, infiltrated, and embedded in Epon resin. Ultrathin sections (70 nM) were cut using a Leica Ultracut Microtome (Leica Microsystems, Wetzlar, Germany), stained with uranyl acetate for 3 minutes, and examined using a JEM 1400 transmission electron microscope (JEOL, Tokyo, Japan).
Green fluorescent protein–LC3 translocation
GFP-fused LC3 was used to detect autophagy. The C13K and OV2008 cells were transfected with 2 μg of pcDNA3.1-GFP–LC3 using Lipofectamine 2000. After 24 hours, the transfected cells were cultured in the absence or presence of 50 μmol/L cisplatin for 48 hours, and the distribution and fluorescence of GFP–LC3 were visualized by fluorescence microscopy.
Apoptosis analysis
For assessment of apoptosis, an annexin V–fluorescein isothiocyanate (FITC) staining kit was used. The cells were harvested in 0.25% trypsin and washed once with phosphate-buffered saline (PBS). After centrifugation, the cells were stained using the annexin V–FITC/propidium iodide Apoptosis Detection Kit (BD, Franklin Lakes, NJ, USA). Analysis of the apoptotic cells was performed on a flow cytometer (BD). The experiment was performed in triplicate.
Rhodamine 123 efflux
Cells were incubated with 5 μg/mL of rhodamine 123 (Rh-123) for 30 minutes at 37°C. At the end of incubation, the cells were washed twice with PBS to remove free Rh-123 and maintained in dye-free medium. The fluorescence intensity of Rh-123 in the cells was measured by fluorescence-activated cell sorting using the LSR II flow cytometer (BD) with CellQuest software.
Immunofluorescence assay
The C13K cells were grown on round glass coverslips (Thermo Fisher Scientific) in 35 mm cell-culture dishes. Following a 20-minute fixation with prechilled methanol, the coverslips were washed with PBS, permeabilized with 0.2% Triton X-100–PBS for 15 minutes, and blocked with 2% bovine serum albumin–PBS for 30 minutes. The rabbit monoclonal anti-LC3B (1:50) antibody was incubated with the cells overnight at 4°C. After incubation with the appropriate secondary fluorescent antibody, the cells were stained with propidium iodide and immediately observed under a fluorescence microscope.
Acid phosphatase activity assay
C13K cells that did not receive any treatment, C13K cells treated with control siRNA, and C13K cells treated with YAP siRNA were seeded in six-well plates and cultured for 24 hours. The lysates were assayed using the acid phosphatase-assay kit (CS0740; Sigma-Aldrich) following the manufacturer’s protocol.
Lysosomal acidification assay
Labeling of lysosomes with FITC–dextran was performed according to previously reported procedures.26 Cells that did not receive any treatment, cells treated with control siRNA, and cells treated with YAP siRNA were seeded in six-well plates at a concentration of 1×105 cells/well. The cells were allowed to attach for 6 hours. Subsequently, the medium was discarded and FITC–dextran (5 mg/mL) dissolved in serum-free medium was added to the cells for 30 minutes; this was followed by washing with serum-free medium and subsequent incubation for 90 minutes. After labeling of the lysosomes, the cells were washed with serum-free medium, and complete medium was added to the cells. Fluorescence intensity was measured using a PolarStar Galaxy microtiter plate reader.
Statistical analysis
Results are presented as mean ± standard deviation values. Comparisons between two groups were performed using an unpaired Student’s t-test. P<0.05 was considered to indicate statistical significance.
Results
YAP confers cisplatin resistance to ovarian cancer cells
The cell viability assay showed that the percentage of surviving cells decreased significantly in a dose-dependent manner in the OV2008 cell line. However, the 48-hour IC50 of cisplatin in the OV2008 and C13K cells was 29.8±1.3 μmol/L and 91.3±2.1 μmol/L, respectively (Figure 1A). As shown in Figure 1B, the mRNA level of YAP in the C13K cells was 2.75 times higher than that in the OV2008 cells; the mRNA level of Cyr61 was 3.13 times higher, CTGF was 2.57 times higher, and CCND1 was six times higher. Western blot analysis of the expression of the corresponding proteins confirmed the mRNA findings, as the results showed that YAP, CTGF, Cyr61, and CCND1 were all overexpressed in the C13K cells compared with the OV2008 cells (Figure 1C).
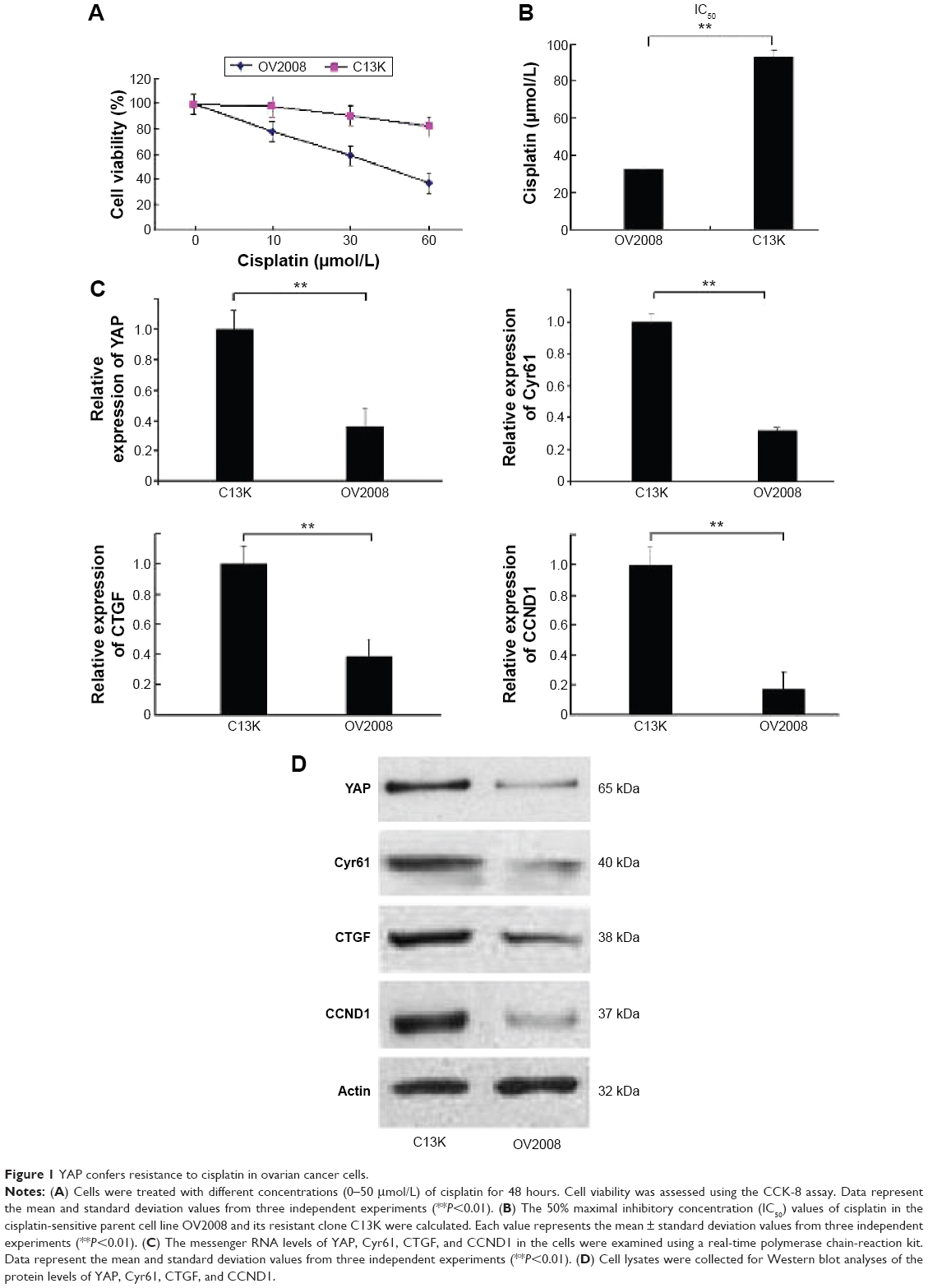 | Figure 1 YAP confers resistance to cisplatin in ovarian cancer cells. |
Cisplatin induces cytoprotective autophagy in cisplatin-resistant ovarian cancer cells
We found that cisplatin treatment preferentially induced autophagy in C13K cells but not in OV2008 cells, as evidenced by upregulation of the mRNA and protein levels of Beclin1 and increased LC3 aggregation and formation of LC3 autophagic vacuoles (Figure 2A–D). Next, C13K and OV2008 cells were pretreated with chloroquine (50 μmol/L) for 24 hours, and then incubated with 50 μmol/L cisplatin for 48 hours. The cell viability of the chloroquine-plus-cisplatin group and the cisplatin group of C13K cells was 37.52%±11.98% and 73.15%±9.85%, respectively. Treatment of OV2008 cells with chloroquine plus cisplatin did not result in a decrease in cell viability compared to treatment with cisplatin alone (Figure 2E). These results suggest that cisplatin induced canonical autophagy, and that induction of autophagy played a protective role in ovarian cancer cells exposed to the cytotoxicity of cisplatin.
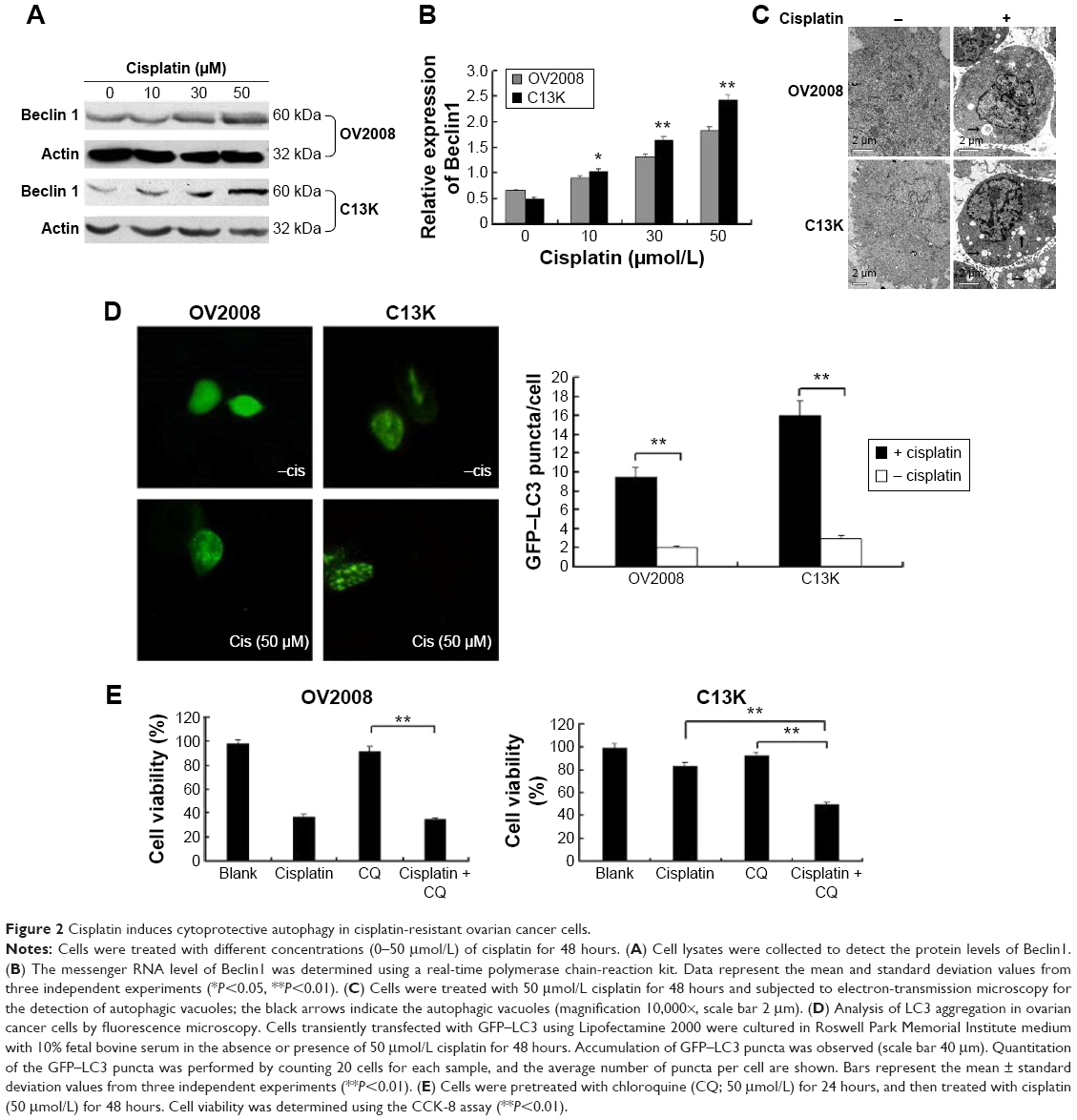 | Figure 2 Cisplatin induces cytoprotective autophagy in cisplatin-resistant ovarian cancer cells. |
Inhibition of YAP enhances the sensitivity of ovarian cancer cells to cisplatin via impairment of autophagy
C13K cells were pretransfected with control siRNA or YAP siRNA (37.5 nM) for 48 hours; this was followed by cisplatin treatment (50 μmol/L) for 48 hours. Figure 3A shows inactivation of YAP via silencing of YAP expression by siRNA in C13K cells compared to YAP expression in the cells expressing control siRNA. After transfection with YAP siRNA, the protein levels of CCND1 decreased with YAP knockdown, and the levels of CTGF and Cyr61, which are transcriptional targets of YAP, were also downregulated (Figure 3A). The cell viability of the control siRNA-plus-cisplatin group, cisplatin group, and YAP siRNA-plus-cisplatin group of C13K cells was 72.2%±10.8%, 75.6%±8.6%, and 33.26%±8.24%, respectively (Figure 3B). The fluorescence intensity of Rh-123 was increased in C13K cells treated with inhibitors of YAP siRNA in comparison with the cells treated with the control siRNA and untreated C13K cells (Figure 3C). We further demonstrated that suppression of YAP significantly augmented cisplatin-induced apoptosis, which was associated with a decrease in the levels of Atg-3 and Atg-5 and an increase in the levels of cleaved caspase 3 and PARP protein in the YAP siRNA C13K cells compared to the control siRNA C13K cells (Figure 3D and E). Furthermore, the number of autophagosomes in the YAP siRNA group was also lower than that in the control siRNA group (Figure 3F). Suppression of autophagy via inhibition of YAP in the cisplatin-treated cells suggested a regulatory role of YAP in cisplatin-resistant ovarian cancer cells.
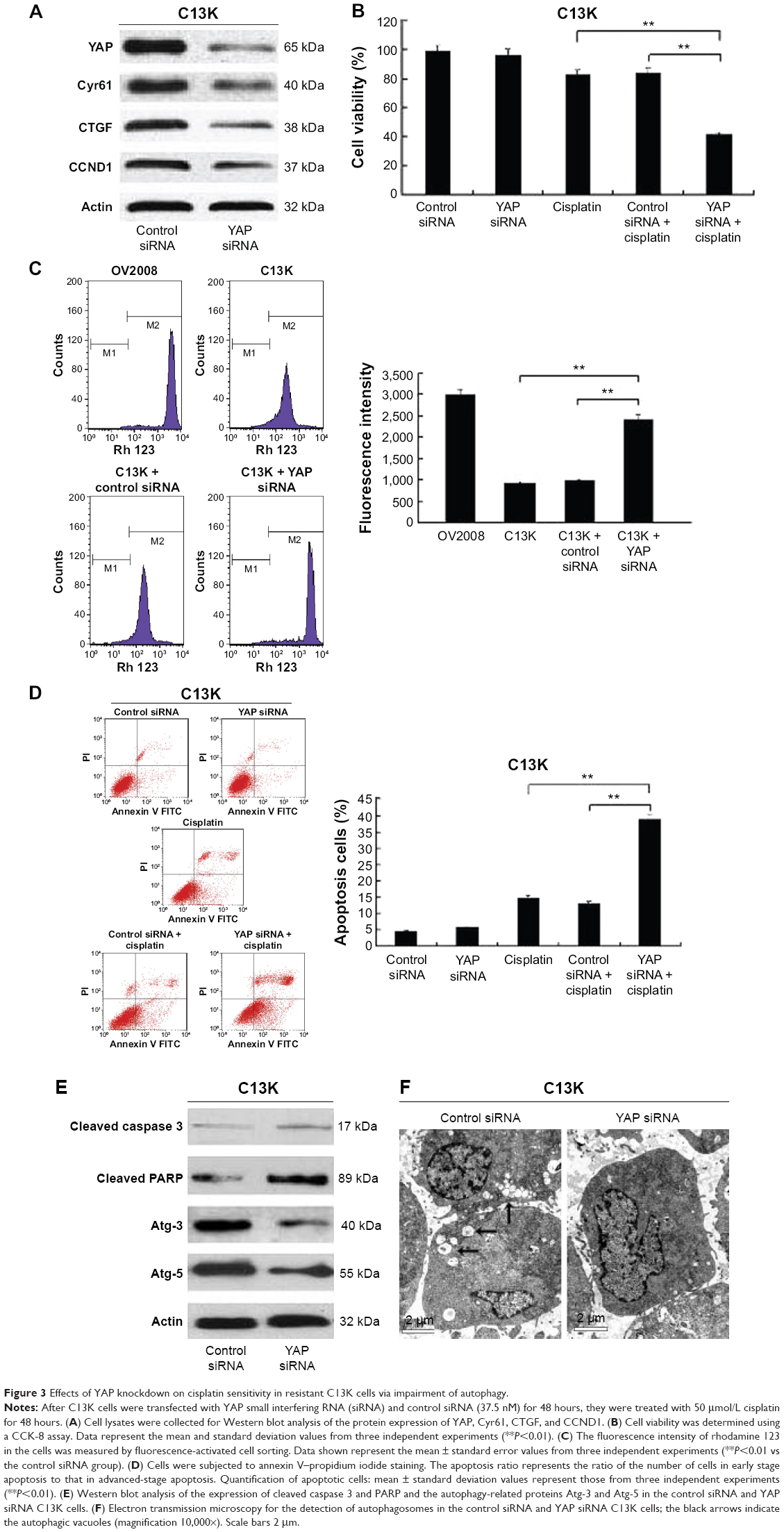 | Figure 3 Effects of YAP knockdown on cisplatin sensitivity in resistant C13K cells via impairment of autophagy. |
YAP interferes with autophagic flux by enhancing autolysosome degradation in cisplatin-resistant ovarian cancer cells
To investigate which step was targeted by YAP, chloroquine was used as an inhibitor of autophagosome formation and degradation. We found that the differences in LC3B degradation between C13K cells treated with control siRNA and C13K cells treated with YAP siRNA were attenuated in the presence of chloroquine in cells treated with cisplatin (Figure 4A). Moreover, additional treatment with rapamycin (20 μM), an inducer of autophagosome formation, did not lead to any further increase in the LC3B protein level in YAP siRNA cells compared with the control siRNA cells (Figure 4A and B). The data suggest that YAP might interfere with autophagy by targeting the degradation and not the formation step of autophagy. Lysosomal acidification is necessary for the catalytic activity of lysosomal enzymes. Therefore, we used FITC–dextran to compare the acidification of lysosomes in the different treatment groups. As expected, there were no significant differences in FITC–dextran between the different treatment groups (Figure 4C). Moreover, the activity of acid phosphatase, a representative lysosomal enzyme, was not affected by aberrant YAP protein levels either (Figure 4D). These findings suggest that the effect of YAP on autophagy is not brought about via lysosomal acidification. YAP may increase autolysosome degradation, and it does not interfere with the fusion of autophagosomes and lysosomes.
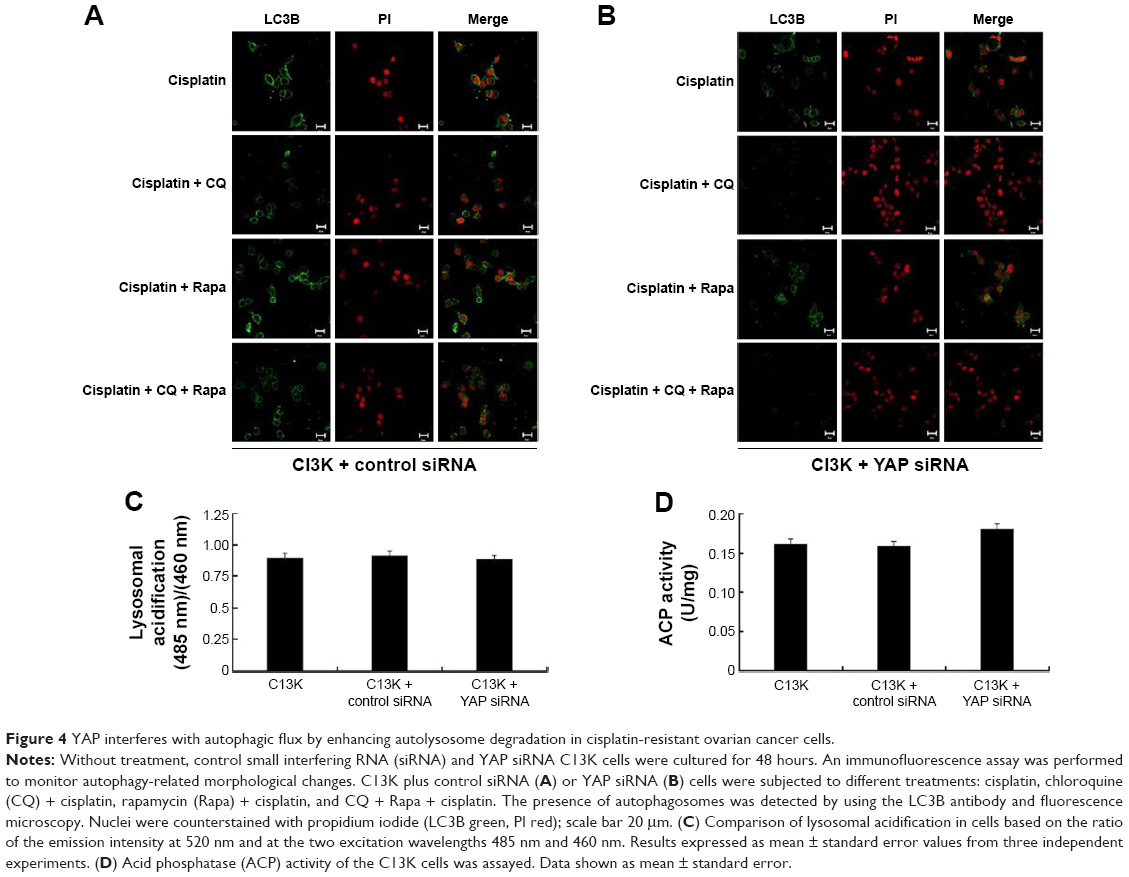 | Figure 4 YAP interferes with autophagic flux by enhancing autolysosome degradation in cisplatin-resistant ovarian cancer cells. |
Discussion
In this study, we utilized cisplatin-sensitive OV2008 cells and cisplatin-resistant C13K cells, which make an ideal pair of cell lines for studying ovarian cancer resistance, since the C13K cells are three times as resistant to cisplatin than the parental cell line OV2008. To illustrate the role of YAP in cisplatin resistance in ovarian cancer, we compared the levels of YAP, Cyr61, CTGF, and CCND1 in the two ovarian cancer cell types. CTGF and Cyr61 have been identified as direct transcriptional targets of YAP, and the role of CTGF and Cyr61 in the development of resistance to paclitaxel and cisplatin treatment has been demonstrated in osteosarcoma and ovarian cancer cells.27,28 In the current study, the basal mRNA and protein expression of YAP, Cyr61, and CTGF was high in the C13K cells but low in the OV2008 cells. Further, we found that the expression of CCND1 mRNA in the C13K cells was six fold that in the OV2008 cells. These findings suggest that the hyperactivation of YAP is involved in the development of cisplatin resistance in ovarian cancer cells.
Following the treatment of the cells with different concentrations of cisplatin for 48 hours, we found that cisplatin treatment preferentially induced autophagy in C13K cells but not in OV2008 cells, as evidenced by the increase in LC3 aggregation and the number of LC3 autophagic vacuoles, and the upregulation of Beclin1 protein. Recent studies have consistently indicated that autophagy plays a protective role in cisplatin resistance.29,30 Similarly, we have demonstrated here that cisplatin-induced autophagy protects ovarian cancer cells from the cytotoxic effects of cisplatin. Taken together, these findings indicate that inhibition of autophagy contributed to the reversal of cisplatin resistance in ovarian cancer cells.
There is an increasing amount of evidence to confirm that activation of autophagy plays a role in chemoresistance in cancer cells, and downregulation of autophagy sensitizes cancer cells to therapeutic drugs. However, few reports have studied the cross talk between the YAP signaling pathway and autophagy in cisplatin resistance in ovarian cancer. In this study, we report a previously unrecognized role of YAP in regulating autophagy in ovarian cancer cells. First, we demonstrated that transfection of C13K cells with YAP siRNA results in a reduction in the protein expression of YAP, CTGF, Cyr61, and CCND1; an increase in the fluorescence intensity of Rh-123; and enhancement of the sensitivity of C13K cells to cisplatin. Second, we confirmed that silencing of YAP significantly augmented cisplatin-induced apoptosis, as indicated by the increase in the intensity of annexin V staining and in the amount of cleaved caspase 3 and PARP present. However, the expression of the autophagy-related proteins Atg-3 and Atg-5 was downregulated as the expression of YAP decreased. Furthermore, the amount of autophagosomes in the YAP siRNA-transfection group was lower than that in the control siRNA group. These data suggest that autophagy mediated by YAP may play a key role in cisplatin resistance here. Recently, it was shown that verteporfin, an autophagy inhibitor, may have a direct inhibitory effect on the growth of cancer cells without light activation but via disruption of the YAP-TEAD complex and prevention of YAP-induced oncogenic growth.31,32 All the findings so far indicate that disruption of the function of YAP suppresses autophagy, which makes YAP a potential target in ovarian cancer treatment.
We also show that under cisplatin exposure, YAP induced an increased autophagy via enhancement of autolysosome degradation. When we blocked autophagy with chloroquine or the mTOR-inhibitor rapamycin, an increase in the amount of LC3B protein was detected in the C13K cells transfected with YAP siRNA than in those transfected with control siRNA after treatment with cisplatin. Besides, we noticed that YAP did not exert a direct effect on lysosome acidification or the fusion of autophagosomes with lysosomes in ovarian cancer cells. These data indicate that YAP might interfere with autophagy by targeting the degradation and not the formation step of autophagy. The precise contribution of YAP to ovarian cancer may be complex, and the detailed molecular mechanism underlying the regulation of autophagy via YAP needs to be further investigated.
In summary, our study identified YAP as a novel regulator of autophagy and demonstrated its involvement in the modulation of cisplatin sensitivity. Inhibition of YAP can enhance the sensitivity of ovarian cancer cells to this chemotherapeutic drug. YAP might therefore be a novel therapeutic target for the prevention of chemotherapy resistance in tumor cells and for the reinforcement of the efficacy of cisplatin against malignant tumors.
Acknowledgments
We thank Dr Wen-Cheng Ding (Cancer Biology Research Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology) for supplying the cell lines. This study was funded by grant 1508085MH193 from the National Natural Science Foundation of Anhui Province, grant 20133420120007 from the Specialized Research Fund for the Doctoral Program of Higher Education (Young Scholar), and grant 2011KJ02 from the National Natural Science Foundation (Young Scholar Cultivation Plan) of the First Affiliated Hospital, Anhui Medical University.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. | ||
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. | ||
Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–539. | ||
Hu YL, Jahangiri A, Delay M, Aghi MK. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012;72:4294–4299. | ||
Carew JS, Nawrocki ST, Cleveland JL. Modulating autophagy for therapeutic benefit. Autophagy. 2007;3:464–467. | ||
Wu WK, Coffelt SB, Cho CH, et al. The autophagic paradox in cancer therapy. Oncogene. 2012;31:939–953. | ||
Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289:17163–17173. | ||
Pennati M, Lopergolo A, Profumo V, et al. miR-205 impairs the autophagic flux and enhances cisplatin cytotoxicity in castration-resistant prostate cancer cells. Biochem Pharmacol. 2014;87:579–597. | ||
Yu L, Gu C, Zhong D, et al. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014;355:34–45. | ||
Bao L, Jaramillo MC, Zhang Z, et al. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol Med Rep. 2015;11:91–98. | ||
Zhang Y, Cheng Y, Ren X, et al. NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene. 2012;31:1055–1064. | ||
Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. | ||
Zender L, Spector MS, Xue W, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. | ||
Xu MZ, Yao TJ, Lee NP, et al. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer. 2009;115:4576–4585. | ||
Avruch J, Zhou D, Bardeesy N. YAP oncogene overexpression supercharges colon cancer proliferation. Cell Cycle. 2012;11:1090–1096. | ||
Muramatsu T, Imoto I, Matsui T, et al. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis. 2011;32:389–398. | ||
Wang Y, Dong Q, Zhang Q, Li Z, Wang E, Qiu X. Overexpression of Yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010;101:1279–1285. | ||
Zhao Y, Yang X. The Hippo pathway in chemotherapeutic drug resistance. Int J Cancer. 2015;137:2767–2773. | ||
Huo X, Zhang Q, Liu AM, et al. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar [sic] carcinoma. Oncol Rep. 2013;29:840–846. | ||
Touil Y, Igoudjil W, Corvaisier M, et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res. 2014;20:837–846. | ||
Jiang N, Hjorth-Jensen K, Hekmat O, et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene. 2015;34:2764–2776. | ||
Jeong W, Kim SB, Sohn BH, et al. Activation of YAP1 is associated with poor prognosis and response to taxanes in ovarian cancer. Anticancer Res. 2014;34:811–817. | ||
Zhang X, George J, Deb S, et al. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene. 2011;30:2810–2822. | ||
Maejima Y, Kyoi S, Zhai P, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488. | ||
Lu HY, Chang YJ, Fan NC, et al. Synergism through combination of chemotherapy and oxidative stress-induced autophagy in A549 lung cancer cells using redox-responsive nanohybrids: a new strategy for cancer therapy. Biomaterials. 2015;42:30–41. | ||
Altan N, Chen Y, Schindler M, Simon SM. Defective acidification in human breast tumor cells and implications for chemotherapy. J Exp Med. 1998;187:1583–1598. | ||
Tsai HC, Huang CY, Su HL, Tang CH. CTGF increases drug resistance to paclitaxel by upregulating survivin expression in human osteosarcoma cells. Biochim Biophys Acta. 2014;1843:846–854. | ||
Rho SB, Woo JS, Chun T, Park SY. Cysteine-rich 61 (CYR61) inhibits cisplatin-induced apoptosis in ovarian carcinoma cells. Biotechnol Lett. 2009;31:23–28. | ||
Fukuda T, Oda K, Wada-Hiraike O, et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol Oncol. 2015;137:538–545. | ||
Qiao S, Tao S, de la Vega MR, et al. The antimalarial amodiaquine causes autophagic-lysosomal and proliferative blockade sensitizing human melanoma cells to starvation- and chemotherapy-induced cell death. Autophagy. 2013;9:2087–2102. | ||
Liu-Chittenden Y, Huang B, Shim JS, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. | ||
Brodowska K, Al-Moujahed A, Marmalidou A, et al. The clinically used photosensitizer verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res. 2014;124:67–73. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.