")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Xyloketal derivative C53N protects against mild traumatic brain injury in mice
Authors Liang F, Su F, Wang X, Long S, Zheng Y, He X, Pang J, Pei Z
Received 22 June 2018
Accepted for publication 26 October 2018
Published 27 December 2018 Volume 2019:13 Pages 173—182
DOI https://doi.org/10.2147/DDDT.S177951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Fengyin Liang,1 Fengjuan Su,1 Xiaoxiao Wang,2 Simei Long,1 Yinglin Zheng,3 Xiaofei He,1 Jiyan Pang,3 Zhong Pei1
1Department of Neurology, Guangdong Provisional Key Laboratory for Diagnosis and Treatment of Major Neurological Diseases, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, People’s Republic of China; 2Department of Medical Oncology, Sun Yat-sen University Cancer Center, Guangzhou 510060, People’s Republic of China; 3School of Chemistry, Sun Yat-sen University, Guangzhou 510275, People’s Republic of China
Purpose: Mild traumatic brain injury (mTBI), the most common type of TBI, can result in prolonged cognitive impairment, mood disorders, and behavioral problems. Reducing oxidative stress and inflammation can rescue the neurons from mTBI-induced cell death. Xyloketal B, a natural product from mangrove fungus, has shown good antioxidative and neuroprotective effects in several disease models. Here, we investigated the potential protection afforded by a xyloketal derivative, C53N, in a closed-skull mTBI model.
Materials and methods: Skulls of mice were thinned to 20–30 µm thickness, following which they were subjected to a slight compression injury to induce mTBI. One hour after TBI, mice were intraperitoneally injected with C53N, which was solubilized in 0.5% dimethyl sulfoxide in saline. In vivo two-photon laser scanning microscopy was used to image cell death in injured parenchyma in each mouse over a 12-hour period (at 1, 3, 6, and 12 hours). Water content and oxidation index, together with pathological analysis of glial reactivity, were assessed at 24 hours to determine the effect of C53N on mTBI.
Results: Cell death, oxidative stress, and glial reactivity increased in mTBI mice compared with sham-injured mice. Treatment with 40 or 100 mg/kg C53N 1 hour after mTBI significantly attenuated oxidative stress and glial reactivity and reduced parenchymal cell death at the acute phase after mTBI.
Conclusion: The present study highlights the therapeutic potential of the xyloketal derivative C53N for pharmacological intervention in mTBI.
Keywords: xyloketal derivative, mTBI, in vivo imaging, antioxidant activity, neuroprotective activity
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability in young adults.1,2 The mild form of TBI (mTBI) is the major type and constitutes around 80% of all TBIs.3 Unlike moderate to severe forms of TBI, mTBI does not cause immediate severe symptoms. However, mTBI can result in prolonged cognitive impairment, mood disorders, and behavioral problems.4,5 Currently, there is no effective treatment for patients with mTBI. Thus, there is an urgent need for development of effective treatments. Unfortunately, the underlying molecular and biochemical mechanisms of mTBI still remain elusive. Recently, clinical and experimental studies have revealed a critical role of oxidative stress and inflammation in the pathophysiology of mTBI.6,7 mTBI induces excessive ROS and inflammation, which in turn disrupts the blood–brain barrier, causes neuronal cell death, and eventually leads to neurological dysfunction.8,9 By contrast, reducing oxidative stress and inflammation can rescue the neurons from mTBI-induced cell death.10
Marine organisms are currently receiving much attention for drug development due to their tremendous biodiversity. Xyloketal B is one of a series of novel ketal compounds isolated from the mangrove fungus Xylaria sp. from the South China Sea.11,12 Our previous studies have demonstrated that xyloketal B is a new antioxidant that has cardioprotective and neuroprotective actions.13–16 However, several weaknesses of xyloketal B limit its application. First, protection against neural cell injury was moderate despite its good in vitro antioxidative activity. Moreover, asymmetric synthesis and modification of xyloketal B are difficult because of six asymmetric carbons at the 2, 2′, 5, 5′, 6, and 6′ positions in its structure. In recent years, we have developed a series of analogs to improve xyloketal B. C53N, a derivative of xyloketal B, has demonstrated excellent antioxidative and neuroprotective effects in different disease models.14,17 In addition, C53N can be easily synthesized with excellent yield. Structures of xyloketal B and its derivatives are shown in Figure 1.
 | Figure 1 Structures of xyloketal B and its derivatives (C53N). |
In the present study, we examined the effects of the xyloketal derivative, C53N, in a closed-skull mTBI model. Using two-photon laser scanning microscopy, we assessed parenchymal cell death over 12 hours in live animals with mTBI.8,18 Xyloketal derivative C53N was administered 1 hour after mTBI at 40 or 100 mg/kg. We examined cell death at 3-hour intervals within the initial 12 hours and evaluated oxidative stress and glial reactivity 24 hours after TBI. We found that C53N significantly attenuated oxidative stress and reduced parenchymal cell death in mice with mTBI. Thus, our data suggest that C53N has great potential to be developed as an effective neuroprotectant for mTBI.
Materials and methods
Chemicals and reagents
We synthesized and purified xyloketal derivative C53N in the School of Chemistry at Sun Yat-sen University, People’s Republic of China, according to a previously reported method.14
Reduction of 3,4-dihydro-2H-pyran-5-carboxylate followed by electrophilic aromatic substitution with phloroglucinol carboxylic acid gave C53. C53 was purified by flash chromatography (Pet:EtOAc=50:1–30:1), followed by crystallization to obtain the pure compound. A solution of C53 (0.2 g, 0.6 mmol) and diethylamine (0.2 g, 6 mmol) in acetone (20 mL) was stirred at room temperature for 30 minutes. Acetone and excess diethylamine were removed under reduced pressure with a rotary evaporator, leaving the C53N compound as a white foam, with a total yield of 58%. 1H nuclear magnetic resonance (NMR) data were recorded on a 400 MB NMR spectrometer operating at 400 MHz for 1H. HPLC and NMR spectroscopy showed that the purity of C53N was >95%; 1H NMR (400 MHz, CDCl3) δ 5.31 (d, J=2.5 Hz, 1H), 5.25 (t, J=2.8 Hz, 1H), 4.13–3.90 (m, 2H), 3.76–3.54 (m, 2H), 3.00 (q, J=7.3 Hz, 4H), 2.74–2.54 (m, 4H), 2.16 (s, 1H), 2.15–2.08 (m, 2H), 1.79–1.54 (m, 8H), 1.29 (t, J=7.3 Hz, 6H).
We dissolved the compound in dimethyl sulfoxide (DMSO) and stored it at 4°C until use. We purchased all other reagents from Sigma-Aldrich (St Louis, MO, USA), unless otherwise specified.
Mice and drug administration
Male C57BL/6 mice (3 months old, weighing 20–25 g) were purchased from Guangdong Medical Laboratory Animal Center, People’s Republic of China. Animal care and experimental procedures were performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal experiments were approved by the Ethical Committee on Animal Experimentation of The First Affiliated Hospital, Sun Yat-sen University, People’s Republic of China. The animal ethics number was [2017] 060.
Animals were randomly divided into four groups (n=5–7 per group) as follows:
- Group 1: sham mice with a thinned skull, but without compression TBI that received vehicle only (0.5% DMSO in saline) by intraperitoneal injection
- Group 2: mTBI mice that received vehicle only (0.5% DMSO in saline) by intraperitoneal injection 1 hour after compression injury
- Group 3: mTBI mice that received 40 mg/kg C53N in 0.5% DMSO by intraperitoneal injection 1 hour after compression injury
- Group 4: mTBI mice that received 100 mg/kg C53N in 0.5% DMSO by intraperitoneal injection 1 hour after compression injury.
mTBI preparation
Animals were deeply anesthetized with 5% isoflurane, and anesthetization was maintained with 2% isoflurane in a mixture of 20% oxygen and 80% air. We used a regulated heating pad (Reward, Shenzen, People’s Republic of China) to maintain the rectal temperature of the animals at 37°C±0.5°C.
We placed the animals in a stereotaxic apparatus and used a sterile scalpel to cut an incision through the midline skin of the skull to expose the bone. We used a microdrill to create a 2.0×2.0 mm thinned cranial window over the left somatosensory cortex. The cranial window was located between bregma and lambda sutures. We induced compression injury according to a previous report,18 with some modifications. Briefly, we thinned the skull for 1–2 minutes to a thickness of 20–30 μm. Under stereotactic guidance, we then gently pressed the pliable skull bone downward 0.5 mm to create a constant concavity in the bone using the blunt end (~1.0 mm width) of a surgical instrument, such that the pliable skull bone compressed the surface of the brain.
Transcranial propidium iodide staining
Transcranial application of propidium iodide (PI; Sigma-Aldrich) has been used to reliably label dead cells in the meninges and parenchyma through the intact, thinned skull.8 Therefore, we used PI to assess cell death at the compression site at 1, 3, 6, and 12 hours after injury in each mouse. Briefly, PI (1.5 mM) in artificial cerebrospinal fluid was placed on the thinned skull for 30 minutes at each time point. After a single wash with artificial cerebrospinal fluid, brain cortex was imaged by two-photon microscopy.
For lesion volume detection, PI was administered by transcranial application over the compression site for 30 minutes before fixation at 24 hours. The total number of PI-positive cells in the focal lesion was calculated using the ImageJ software (NIH, Bethesda, MD, USA) “analyze particles” tool.
Labeling of blood vessels
At the same time points as the PI staining mentioned above, 0.2 mL of 2% (wt/vol) solution of 2 MDa fluorescein-dextran (FD2000S; Sigma-Aldrich) in saline was administered intravenously to each mouse to label the blood vessels fluorescent green.
In vivo two-photon imaging
To conduct in vivo two-photon imaging, we fixed the mouse in a custom-fabricated metal frame that held its head with cyanoacrylate and dental cement. The animal was then placed on the stage of a Leica DM6000 CFS (Leica Microsystems, Wetzlar, Germany). Leica LAS AF 2.5 software was used for laser scanning and data acquisition. All images were acquired using two-channel non-descanned detector detection, with emission filters of 525/50 and 585/50 nm, on a TCS SP5 MP System (Leica Microsystems). A Leica NA 0.95, 25× water immersion objective was used to visualize the vascular network and dead cells. Cells labeled by PI (exhibiting red fluorescence) were defined as dead cells and blood vessels were labeled green. Stacks of images were acquired using a step size of 2.0 μm to a depth of 300 μm.
Lesion volume calculation
Lesion volumes were calculated by PI staining according to published literature.19 Briefly, we sliced fixed brains into 50 μm thick sections, captured images of the lesion site by confocal microscopy, and measured the lesion volume using ImageJ software. The total infarct volume was defined as the sum of the PI staining volumes.
Brain edema measurement
The percentage brain water content was determined using the wet–dry method. The percentage water content (% H2O) was calculated for each hemisphere as follows: % H2O=[(wet weight − dry weight)/wet weight] ×100%.
ROS test and glutathione (GSH) measurement
We used commercial kits to assess ROS (Hydrogen Peroxide Assay Kit S0038; Beyotime) and GSH (Cellular Glutathione Peroxidase Assay Kit S0056; Beyotime) levels based on the protocols provided by the manufacturer. Briefly, we perfused the injured cortex with 0.9% saline (100 μL/10 mg tissue) and extracted brain tissue in a tissue homogenizer instrument (SANYO, MSS.150-CX15) at 24 hours postinjury. We then mixed the tissue by vortexing, followed by centrifugation at 14,000 rpm for 5 minutes. We collected the supernatant and diluted with tissue lysates. The concentrations of ROS and GSH were evaluated by measurement of the absorbance at 560 nm for ROS and 420 nm for GSH.
Histological analysis
The brain regions under the thinned skull window from each brain were dissected into 10 μm thick serial coronal slices. The following cell types were labeled: neurons (anti-NeuN; 1:400; EMD Millipore, Billerica, MA, USA), astrocytes (anti-GFAP; 1:400; Sigma-Aldrich), and microglia (anti-Iba1; 1:400; Wako, Osaka, Japan). Two-dimensional images were captured from the stained frozen sections using a Leica SP5 confocal microscope equipped with a 25× objective. Images were collected using sequential scanning with the 405, 488, and 561 nm lasers to produce combined overlays.
Statistical analysis
SPSS version 22.0 (IBM Corporation, Armonk, NY, USA) was used for statistical analysis. Shapiro–Wilk test was used to check data distribution. Parametric tests (Student’s t-test and ANOVA) were applied as data were found to be normally distributed. P-values below 0.05 were considered statistically significant.
Results
Synthesis of xyloketal derivative C53N
Xyloketal derivative C53N was synthesized according to a previously reported method.14 Reduction of 3,4-dihydro-2H-pyran-5-carboxylate was followed by electrophilic aromatic substitution with phloroglucinol carboxylic acid to give C53. C53N is a diethylamine salt of C53, which has improved water solubility. The scheme for synthesis of C53N is shown in Figure 2.
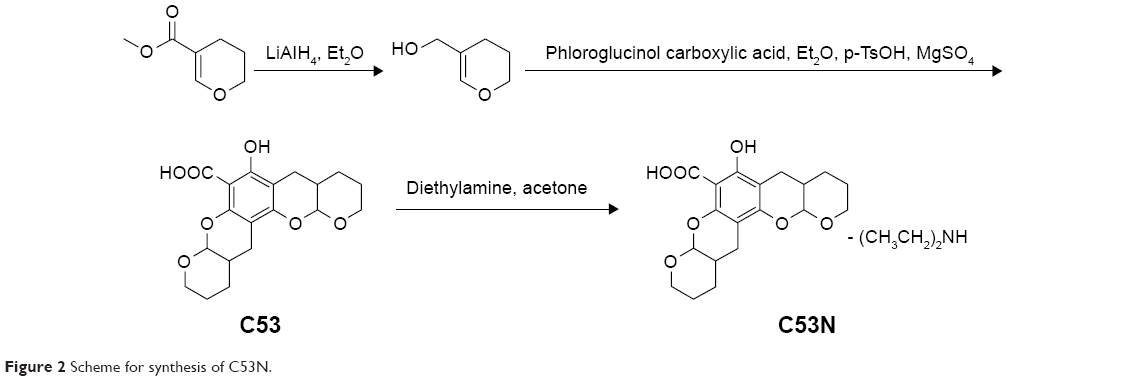 | Figure 2 Scheme for synthesis of C53N. |
C53 was purified by flash chromatography (Pet:EtOAc=50:1–30:1), followed by crystallization to obtain the pure compound. A solution of C53 (0.2 g, 0.6 mmol) and diethylamine (0.2 g, 6 mmol) in acetone (20 mL) was stirred at room temperature for 30 minutes. Acetone and excess diethylamine were removed under reduced pressure with a rotary evaporator to leave the C53N compound as a white foam, with a total yield of 58%.
Xyloketal derivative C53N reduced cell death after mTBI
We evaluated the effects of C53N in a closed-skull mTBI model. PI was used to label dead cells at 1, 3, 6, and 12 hours after compression brain injury. Leica software (LAS AF Lite) was used to reconstruct stacks of images into three-dimensional vasculature images (Figure 3A). PI-positive dead cells were evident in the parenchyma in all the animals with compression injury, while there were no dead cells in the brain parenchyma of sham mice. Figure 3B shows images of the plane 100 μm below the meninges at each time point, which correspond to the images in Figure 3A. Compared with vehicle-treated animals, the number of dead cells was significantly lower at 6 or 12 hours following mTBI in animals receiving 40 or 100 mg/kg xyloketal derivative C53N (P<0.05; Figure 3C). Our data suggest that C53N reduces brain cell death during acute brain injury.
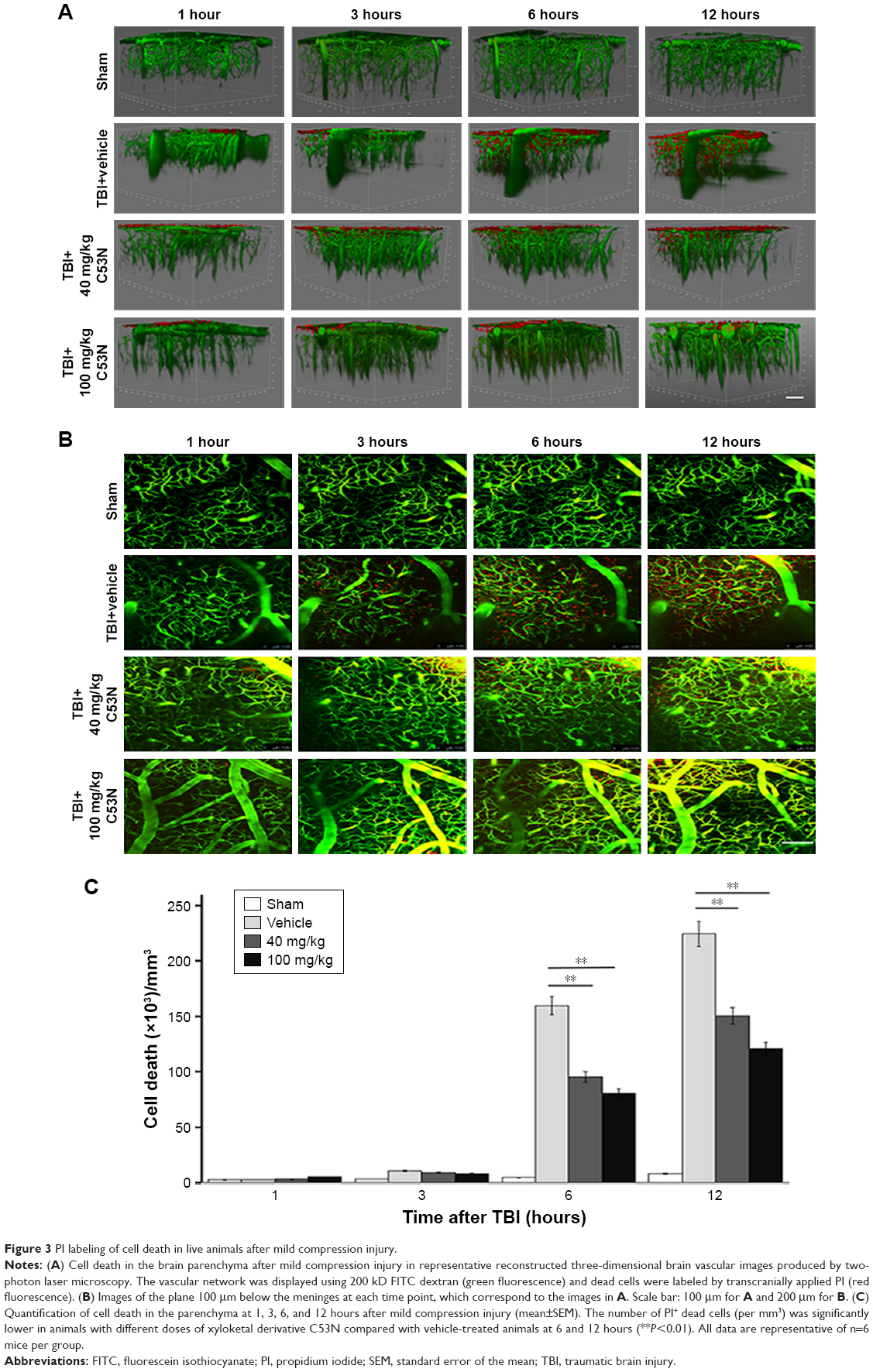 | Figure 3 PI labeling of cell death in live animals after mild compression injury. |
Xyloketal derivative C53N reduced lesion size post-mTBI
Lesion volume was evaluated according to cell death by PI staining 24 hours post-mTBI. As shown in Figure 4A, dead cells and neuronal loss were observed in the injury site in all TBI mice, while there were no PI-positive cells in sham mice. Mice treated with xyloketal derivative C53N showed significantly reduced cell death and neuronal loss in the cortex compared with vehicle-treated mice after mTBI (Figure 4B).
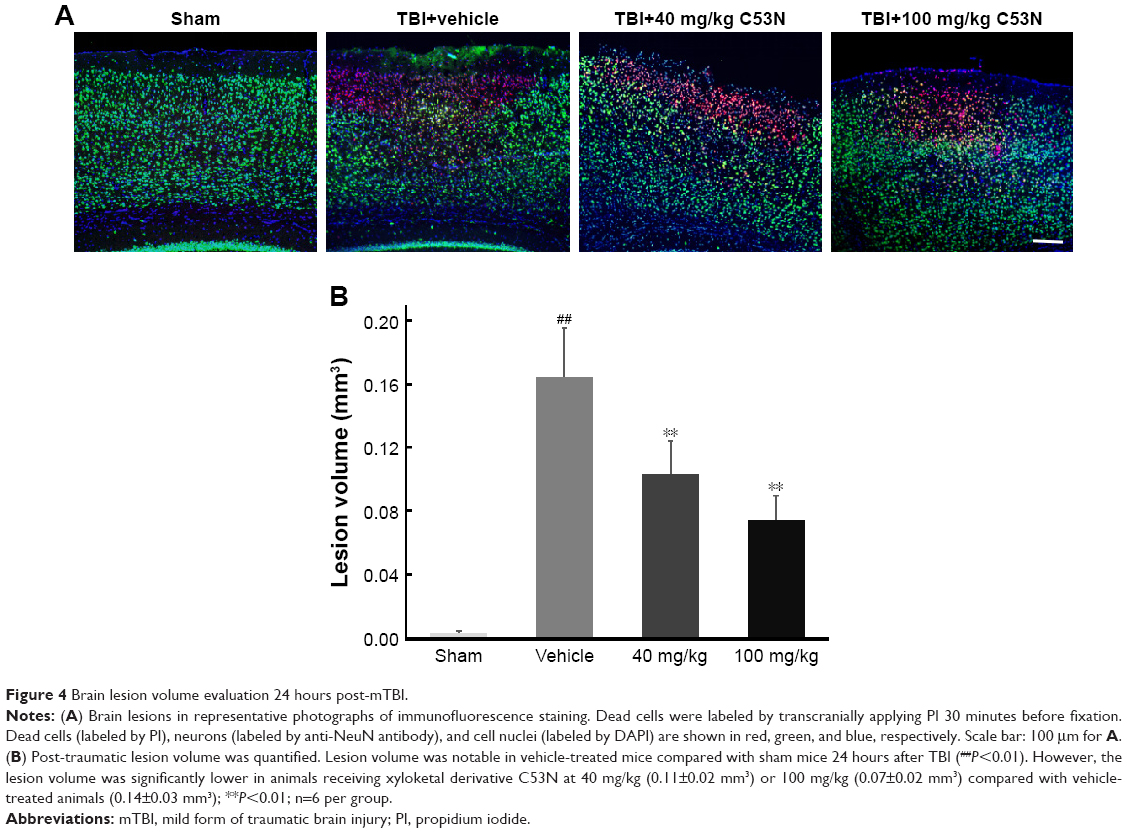 | Figure 4 Brain lesion volume evaluation 24 hours post-mTBI. |
Xyloketal derivative C53N decreases edema 24 hours post-mTBI
As shown in Figure 5, following mTBI, the brain injury site had a higher percentage of water content compared with sham mice. In mice treated with 40 mg/kg C53N, the water content was significantly lower than in mice treated with vehicle. The water content was also significantly lower in mice treated with 100 mg/kg C53N compared with mice treated with vehicle. These data imply that the xyloketal derivative C53N decreases edema 24 hours post-mTBI.
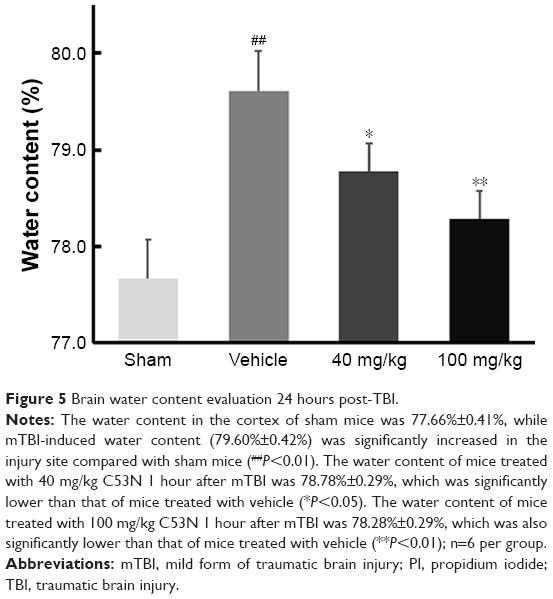 | Figure 5 Brain water content evaluation 24 hours post-TBI. |
Xyloketal derivative C53N reduces ROS and increases GSH post-mTBI
We used commercial kits to measure the levels of H2O2, a potent ROS species, and GSH in injured cortex at 24 hours postinjury. We found that ROS, as evidenced by H2O2, was >2.5 times higher in mTBI mice than in sham mice (P<0.01), but animals that received different doses of xyloketal derivative C53N had decreased H2O2 in the injury site compared with vehicle-treated mice (Figure 6A). In addition, as shown in Figure 6B, GSH in mTBI mice was 68.4% of that in sham mice. There was no difference in the concentration of GSH in mice treated with 40 mg/kg xyloketal derivative C53N, but mice that received 100 mg/kg xyloketal derivative C53N had an increased concentration of GSH compared with vehicle-treated mice (*P<0.01). Thus, the present study indicates that xyloketal derivative C53N protects against mTBI at least partly through its antioxidant action.
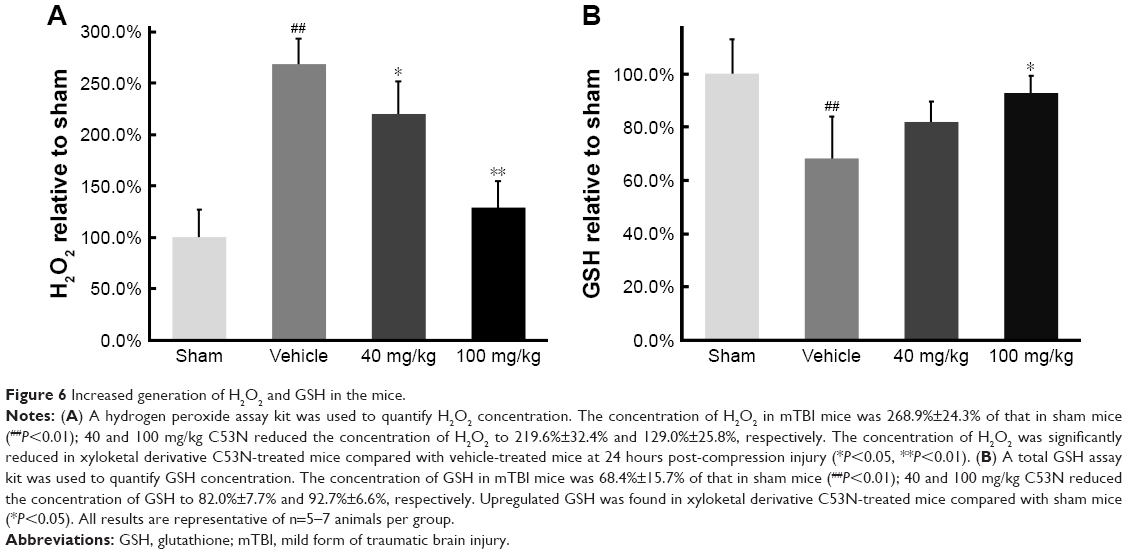 | Figure 6 Increased generation of H2O2 and GSH in the mice. |
Xyloketal derivative C53N decreases glial activity post-mTBI
To investigate whether the xyloketal derivative C53N can act on neuroinflammation after TBI, we examined reactive astrocytes and microglia using anti-GFAP and anti-IBA1 antibodies, respectively. As shown in Figure 7A and B, few GFAP-positive astrocytes were detected in sham mice. However, GFAP-positive astrocytes were evident at 24 hours post-trauma. By contrast, the xyloketal derivative C53N at 40 or 100 mg/kg significantly decreased the number of astrocytes at 24 hours compared with vehicle treatment (P<0.05). As shown in Figure 7C and D, microglia were scattered in the cortex of sham mice. These microglia exhibited the features of quiescent microglia, including small cell bodies and fine, ramified processes (Figure 7E). However, Iba1-positive microglia were evident at 24 hours post-trauma. The morphology of microglia in the injured cortex resembled microglial reactive-like phenotypes, such as an amoeboid shape with large cell bodies and thickened, short processes (Figure 7E). Administration of xyloketal derivative C53N at 100 or 40 mg/kg significantly inhibited Iba1-positive microglia post-trauma (P<0.05; Figure 7D) and attenuated the microglial reactive-like phenotype to a lesser degree. Thus, the present data suggest that the xyloketal derivative C53N significantly decreases glial activity post-mTBI.
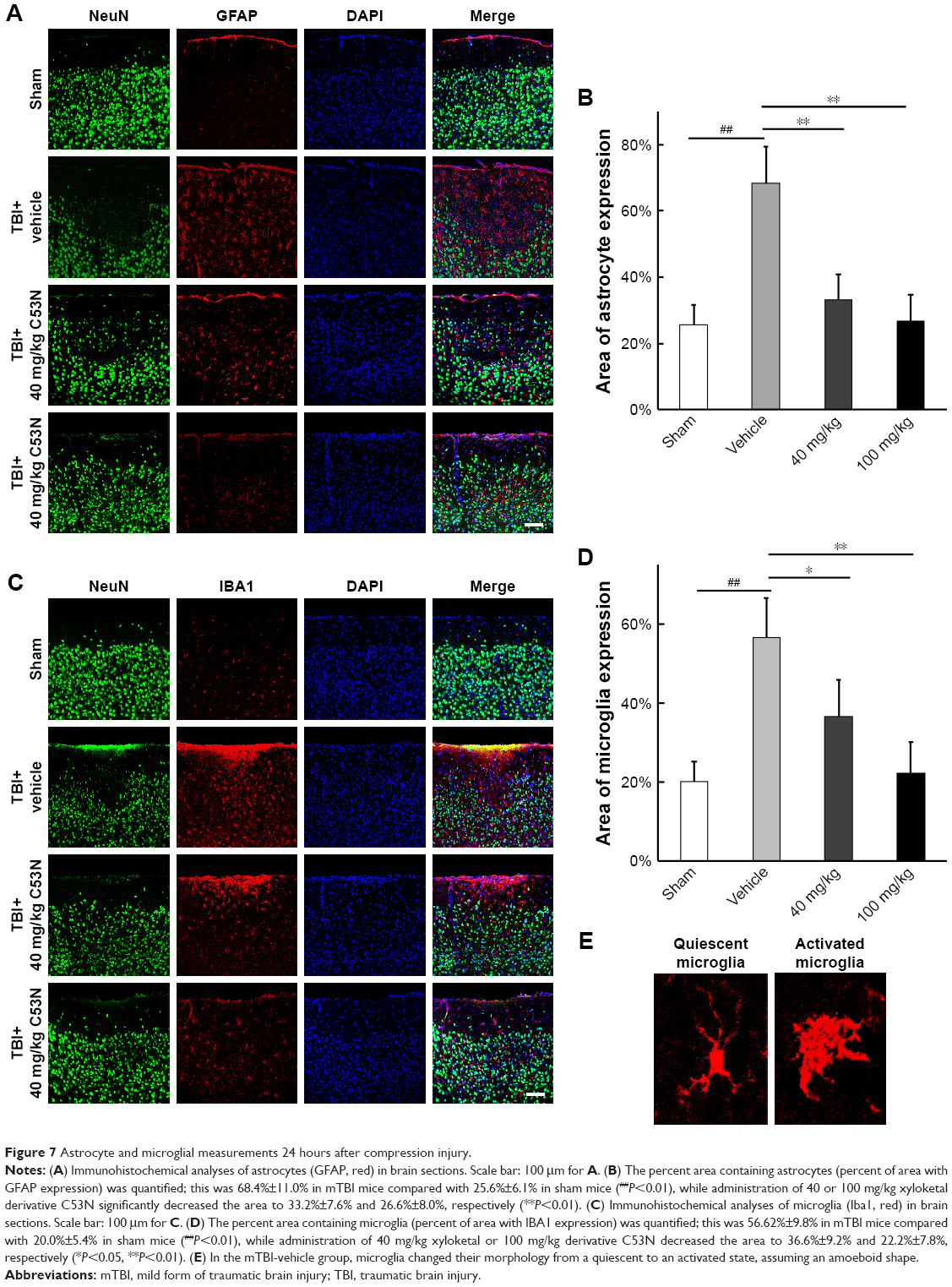 | Figure 7 Astrocyte and microglial measurements 24 hours after compression injury. |
Discussion
The mTBI is frequently underestimated as the immediate physical symptoms decrease rapidly and conventional neuroimaging studies often do not show visible evidence of brain lesions. In the present study, we used a balance beam and Rotarod to test the functional outcome of mice at 24 hours after mTBI; however, there were no differences between mTBI and sham mice (data not shown). This is probably because it was such a small TBI that functional damage would not be evident within a short space of time. A more sophisticated functional test may be needed to score mice when using this model.
TBI induces a complex reaction that can result in permanent damage and neurological dysfunction. Cell death is one of the most common outcomes of brain injury. The nucleic acid stain, PI, has been widely used as a simple and reliable technique for real-time monitoring of dead cells8 and pathological assessment of infarct size. In the present study, we used PI staining to monitor the injury in the compressed site in vivo within 12 hours of mTBI, visualized with a two-photon microscope. We found that mTBI gradually increased parenchymal cell death over the 12-hour period. Consistently, a large number of neurons were lost in the PI-stained area 24 hours after TBI, but mice treated with 40 or 100 mg/kg of xyloketal derivative C53N showed a significantly reduced amount of parenchymal neuron death in the acute stage of TBI.
Oxidative stress plays a critical role in the pathophysiology of TBI.20,21 The severity of TBI-induced brain damage has been directly linked to the degree of oxidative stress, whereas inhibition of oxidative stress can significantly reduce brain damage and improve the outcomes of TBI.22,23 Oxidative stress may result from an imbalance between pro-oxidant and antioxidant systems caused by either excessive production of ROS or depletion of GSH.24 GSH is a major cellular non-enzymatic antioxidant that prevents mammalian cells from oxidative damage, while depletion of GSH is an important mechanism for the induction of apoptosis.25 Our previous studies demonstrated that xyloketal B and its derivative have an excellent antioxidant effect. In the present study, xyloketal derivative C53N significantly reduced the concentration of ROS, but increased the concentration of GSH, indicating that the C53N derivative still maintained good antioxidant activity to protect against mTBI.
Glial cells are the most abundant brain cells and account for over 70% of the brain tissue. Physiologically, glial cells maintain homeostasis of the brain by providing support and recycling waste products. However, glial cells can be harmful under pathological conditions. For example, astrocytes, the most common brain glial cells, have been reported to be involved in different neurological diseases. Furthermore, microglia, the key cells in the brain’s innate immune system, can mediate immune responses by releasing proinflammatory cytokines.26 It has been reported that microglia and astrocytes are the major glial cells involved in immune reactivity and inflammation in the brain.27 The inflammatory response, including activated astrocyte infiltration (astrogliosis) and microglial activation (microgliosis) at the site of injury, has been well documented in TBI.8 Following TBI, reactive astrocytes participate in the inflammatory response by activation of inflammatory pathways, which leads to secretion of different chemokines/cytokines.28 Similarly, TBI activates the M1 subtype of non-phagocytic microglia during the acute stage to induce neuronal apoptosis.29 In the present study, activated astrocytes and microglia increased at 24 hours after mTBI. However, treatment with the xyloketal derivative C53N 1 hour after TBI evidently decreased the induction of activated astrocytes and microglia.
In the present study, we investigated the effectiveness of different concentrations of C53N on mTBI only at one intervention time point (1 hour after TBI), which indicated a protective effect in the acute stage of TBI. To be more practical for clinical use, it will be interesting and essential to study the time window of C53N and its long-term protective effects.
Conclusion
In summary, we tested protection by the xyloketal derivative C53N in a closed-skull mTBI model and showed that the treatment significantly decreased the water content, oxidative stress, and glial reactivity, thereby attenuating parenchymal cell death in the acute phase after compression injury. Thus, the present study highlights the therapeutic potential of xyloketal derivative C53N as a pharmacological intervention for mTBI.
Acknowledgments
This work was supported by grants from the National Key Clinical Department and Key Discipline of Neurology, Guangdong Provincial Key Laboratory for Diagnosis and Treatment of Major Neurological Diseases (No 2017B030314103), the Southern China International Cooperation Base for Early Intervention and Functional Rehabilitation of Neurological Diseases (No 2015B050501003), Guangdong Provincial Engineering Center for Major Neurological Disease Treatment, Guangdong Provincial Science and Technology Plan Project (No 2016B030230002; No 2017A040406007), the Science and Technology Planning Project of Guangzhou (No 201605030012; No 201604020010), and the Natural Science Foundation of Guangdong Province, People’s Republic of China (No 2017A030313064). We also thank Ann Turnley, PhD, from Liwen Bianji, Edanz Group China, for editing the English text of a draft of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380(9847):1088–1098. | ||
Corrigan JD, Hammond FM. Traumatic brain injury as a chronic health condition. Arch Phys Med Rehabil. 2013;94(6):1199–1201. | ||
Wible EF, Laskowitz DT. Statins in traumatic brain injury. Neurotherapeutics. 2010;7(1):62–73. | ||
Plassman BL, Havlik RJ, Steffens DC, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55(8):1158–1166. | ||
Fehily B, Fitzgerald M. Repeated mild traumatic brain injury: potential mechanisms of damage. Cell Transplant. 2017;26(7):1131–1155. | ||
Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2015;51(3):966–979. | ||
Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, Mcintosh TK. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol. 2001;24(1–3):169–181. | ||
Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, Mcgavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505(7482):223–228. | ||
Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14(2):128–142. | ||
Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2010;12(1):125–169. | ||
Lin Y, Wu X, Feng S, et al. Five unique compounds: xyloketals from mangrove fungus Xylaria sp. from the South China Sea coast. J Org Chem. 2001;66(19):6252–6256. | ||
Pettigrew JD, Wilson PD. Synthesis of xyloketal A, B, C, D, and G analogues. J Org Chem. 2006;71(4):1620–1625. | ||
Chen WL, Qian Y, Meng WF, et al. A novel marine compound xyloketal B protects against oxidized LDL-induced cell injury in vitro. Biochem Pharmacol. 2009;78(8):941–950. | ||
Li S, Shen C, Guo W, et al. Synthesis and neuroprotective action of xyloketal derivatives in Parkinson’s disease models. Mar Drugs. 2013;11(12):5159–5189. | ||
Liu S, Luo R, Xiang Q, Xu X, Qiu L, Pang J. Design and synthesis of novel xyloketal derivatives and their protective activities against H2O2-induced HUVEC injury. Mar Drugs. 2015;13(2):948–973. | ||
Zeng Y, Guo W, Xu G, et al. Xyloketal-derived small molecules show protective effect by decreasing mutant Huntingtin protein aggregates in Caenorhabditis elegans model of Huntington’s disease. Drug Des Devel Ther. 2016;10:1443–1451. | ||
Li ZX, Chen JW, Yuan F, et al. Xyloketal B exhibits its antioxidant activity through induction of HO-1 in vascular endothelial cells and zebrafish. Mar Drugs. 2013;11(2):504–522. | ||
Liang F, Luo C, Xu G, et al. Deletion of aquaporin-4 is neuroprotective during the acute stage of micro traumatic brain injury in mice. Neurosci Lett. 2015;598:29–35. | ||
Wolff RA, Chien GL, van Winkle DM. Propidium iodide compares favorably with histology and triphenyl tetrazolium chloride in the assessment of experimentally-induced infarct size. J Mol Cell Cardiol. 2000;32(2):225–232. | ||
Bayir H, Kochanek PM, Kagan VE. Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci. 2006;28(4–5):420–431. | ||
Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7(1):51–61. | ||
Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012;1822(5):675–684. | ||
Mustafa AG, Wang JA, Carrico KM, Hall ED. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem. 2011;117(3):579–588. | ||
Aquilano K, Baldelli S, Cardaci S, Rotilio G, Ciriolo MR. Nitric oxide is the primary mediator of cytotoxicity induced by GSH depletion in neuronal cells. J Cell Sci. 2011;124(Pt 7):1043–1054. | ||
Fan S, Yu Y, Qi M, et al. P53-mediated GSH depletion enhanced the cytotoxicity of NO in silibinin-treated human cervical carcinoma HeLa cells. Free Radic Res. 2012;46(9):1082–1092. | ||
Gyoneva S, Ransohoff RM. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell–cell communication by chemokines. Trends Pharmacol Sci. 2015;36(7):471–480. | ||
Witcher KG, Eiferman DS, Godbout JP. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. 2015;38(10):609–620. | ||
Laird MD, Vender JR, Dhandapani KM. Opposing roles for reactive astrocytes following traumatic brain injury. Neurosignals. 2008;16(2–3):154–164. | ||
Yang ST, Lin JW, Chiu BY, Hsu YC, Chang CP, Chang CK. Astragaloside improves outcomes of traumatic brain injury in rats by reducing microglia activation. Am J Chin Med. 2014;42(6):1357–1370. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.