")
Back to Journals » Vascular Health and Risk Management » Volume 10
XANTUS: rationale and design of a noninterventional study of rivaroxaban for the prevention of stroke in patients with atrial fibrillation
Authors Camm AJ, Amarenco P, Haas S, Hess S, Kirchhof P, van Eickels M, Turpie A
Received 1 March 2014
Accepted for publication 8 May 2014
Published 17 July 2014 Volume 2014:10 Pages 425—434
DOI https://doi.org/10.2147/VHRM.S63298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
A John Camm,1 Pierre Amarenco,2 Sylvia Haas,3 Susanne Hess,4 Paulus Kirchhof,5,6 Martin van Eickels,4 Alexander GG Turpie7
1Division of Clinical Sciences, St George's, University of London, London, UK; 2Department of Neurology and Stroke Center, Paris-Diderot-Sorbonne University, Paris, France; 3Vascular Center, Munich, Germany; 4Medical Affairs, Bayer HealthCare Pharmaceuticals, Berlin, Germany; 5Centre for Cardiovascular Sciences, University of Birmingham and Sandwell & West Birmingham Hospitals NHS Trust, Birmingham, UK; 6Department of Cardiovascular Medicine, University of Münster, Münster, Germany; 7Department of Medicine, McMaster University, Hamilton, ON, Canada
Abstract: Atrial fibrillation (AF) is associated with a fivefold increase in the risk of stroke. The Phase III ROCKET AF (Rivaroxaban Once-Daily Oral Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) trial showed that rivaroxaban, an oral, direct Factor Xa inhibitor, was noninferior to warfarin for the reduction of stroke or systemic embolism in patients with AF. Compared with warfarin, rivaroxaban significantly reduced rates of intracranial and fatal hemorrhages, although not rates of bleeding overall. XANTUS (Xarelto® for Prevention of Stroke in Patients with Atrial Fibrillation) is a prospective, international, observational, postauthorization, noninterventional study designed to collect safety and efficacy data on the use of rivaroxaban for stroke prevention in AF in routine clinical practice. The key goal is to determine whether the safety profile of rivaroxaban established in ROCKET AF is also observed in routine clinical practice. XANTUS is designed as a single-arm cohort study to minimize selection bias, and will enroll approximately 6,000 patients (mostly from Europe) with nonvalvular AF prescribed rivaroxaban, irrespective of their level of stroke risk. Overall duration of follow-up will be 1 year; the first patient was enrolled in June 2012. Similar studies (XANTUS-EL [Xarelto® for Prevention of Stroke in Patients with Nonvalvular Atrial Fibrillation, Eastern Europe, Middle East, Africa and Latin America] and XANAP [Xarelto® for Prevention of Stroke in Patients with Atrial Fibrillation in Asia-Pacific]) are ongoing in Latin America and Asia-Pacific. Data from these studies will supplement those from ROCKET AF and provide practical information concerning the use of rivaroxaban for stroke prevention in AF.
Keywords: rivaroxaban, anticoagulants, atrial fibrillation, stroke, Phase IV
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia reported in clinical practice,1 and is associated with a fivefold or greater increase in the risk of stroke.2 Although effective for stroke risk reduction in AF, vitamin K antagonists (VKAs) have significant clinical limitations and are underused in practice.3,4 The past 2 years have seen the approval of three non-VKA oral anticoagulants (NOACs) for stroke prevention in AF: dabigatran, rivaroxaban, and apixaban, all of which are now recommended in guidelines.5,6
Rivaroxaban
Rivaroxaban was the first oral, direct Factor Xa inhibitor to receive approval from regulatory authorities for stroke prevention in patients with AF, based mainly on the results of the Phase III ROCKET AF trial (Rivaroxaban Once-Daily Oral Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation; NCT00403767).7 In addition, rivaroxaban has received approval in many countries, including the US8 and the countries of the European Union,9 and for use in several other different thromboembolic conditions.9
Rivaroxaban has no requirement for routine coagulation monitoring and has few drug–drug interactions.10–12 The drug undergoes renal clearance of active compound in plasma of approximately 33%.11 Pharmacokinetic analyses from two Phase II evaluations of rivaroxaban for treatment of deep-vein thrombosis and prevention of recurrent venous thromboembolism in patients with deep-vein thrombosis13,14 were adapted to provide estimates of rivaroxaban exposure in simulated patients with AF;15 these analyses indicated that 20 mg once daily would be an appropriate dosage for stroke prevention in AF. Furthermore, patients with moderate renal impairment (creatinine clearance of 30–49 mL/minute) would experience the same exposure if the dose of rivaroxaban was reduced to 15 mg once daily. Hence, 20 mg once daily (reduced to 15 mg once daily in patients with moderate renal impairment) was selected for Phase III evaluation in the ROCKET AF trial.16
ROCKET AF
In the Phase III ROCKET AF study, rivaroxaban was compared with dose-adjusted warfarin for the prevention of stroke and systemic embolism in high-risk patients with nonvalvular AF.7 Rivaroxaban was noninferior to warfarin with regard to rates of stroke or systemic embolism (intention-to-treat analysis). Overall bleeding rates for rivaroxaban versus warfarin were similar for major or nonmajor clinically relevant bleeding, although gastrointestinal bleeding was increased with rivaroxaban. However, rivaroxaban therapy resulted in significantly fewer fatal bleeding events and intracranial hemorrhages than warfarin.7 Efficacy and safety results are shown in Tables S1 and S2, respectively.
Postauthorization studies
Phase III clinical trials are generally registration trials that support marketing approval by the regulatory authorities. Such trials must meet strict design requirements to ensure well-defined inclusion and exclusion criteria, strict protocol adherence, appropriate clinical endpoints, and statistical validity. These requirements mean that event rates with the experimental drug and the patient characteristics in the clinical trial population may or may not fully reflect those observed in the broad range of patients seen in routine care. Therefore, postauthorization studies are needed to explore the efficacy and safety of a new agent in routine clinical practice. This is particularly true for oral anticoagulants, for which factors that could potentially influence outcomes (eg, adherence, persistence, comorbidities) might vary between the strict environment of a clinical trial and that of real-world therapy in routine practice.
XANTUS
Objectives and rationale
XANTUS (Xarelto® for Prevention of Stroke in Patients with Atrial Fibrillation; NCT01606995) is a prospective, international, postauthorization, observational, noninterventional Phase IV study in patients with nonvalvular AF prescribed rivaroxaban for prevention of stroke in routine practice. This paper describes the objectives, design, and protocol for the XANTUS study, as well as key limitations.
Because the XANTUS study is intended to assess the use of rivaroxaban in routine, real-world clinical practice, study procedures will not interfere with the clinical management of patients or with the prescribing behaviors of attending physicians. In contrast to ROCKET AF, patients with nonvalvular AF may be included in XANTUS regardless of their risk of stroke, although it is presumed that they will be prescribed rivaroxaban in accordance with the appropriate prescribing information. Data collected will include risk assessment, bleeding events, thromboembolic events, and adverse events (AEs) that occur in patients with nonvalvular AF receiving rivaroxaban in routine care according to the local marketing authorization. Information on the clinical management of these events will also be collected.
The key goal for XANTUS is to confirm the safety profile of rivaroxaban across the broad range of patient risk profiles encountered in the setting of routine clinical practice. This will include assessment of rivaroxaban’s safety in patients at increased risk of thromboembolic or bleeding events. It is also expected that the data collected during the course of the study will provide a comprehensive overview of the outcomes associated with rivaroxaban therapy and factors influencing its use in the clinical management of stroke prophylaxis in AF, across different geographic regions, care settings, patient populations, and health care systems.
XANTUS study program
The XANTUS study described below is the prototype for a series of four separate Phase IV studies designed to cover different global regions (Table 1), and which collectively comprise the XANTUS program. It is intended that all four of these studies will use the same protocol and will contribute results to the same central database. A pooled analysis is also planned.
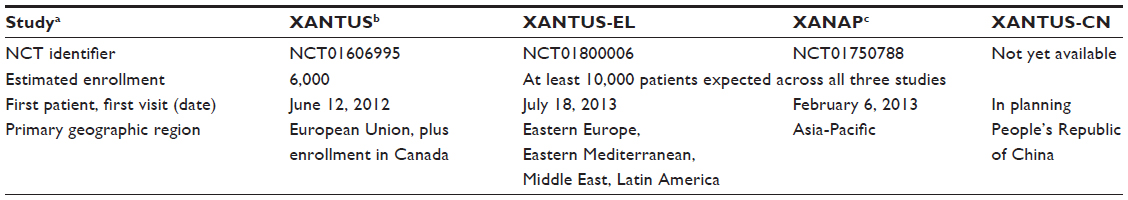 | Table 1 Clinical studies comprising the XANTUS program |
Design and protocol
XANTUS is designed as a single-arm observational, noninterventional cohort study (Figure 1). Overall duration of follow-up for each patient will be 1 year, with intermediate follow-up visits at approximately 3-month intervals.
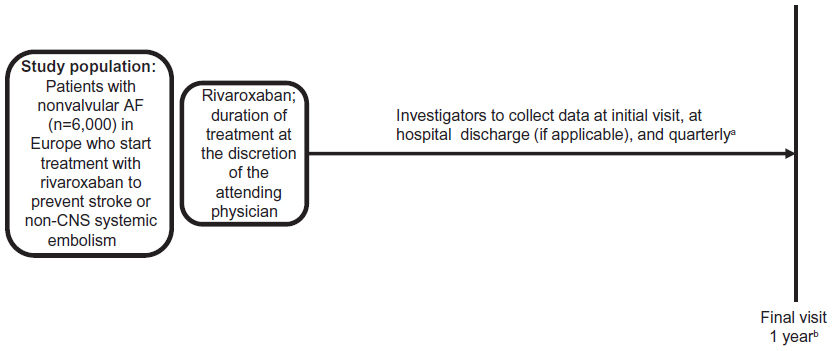 | Figure 1 XANTUS study design, an observational, single-arm cohort study. The objective is to collect real-life data on adverse events, bleeding, thromboembolic events, and mortality in patients with nonvalvular AF treated with rivaroxaban. The same basic design will be used for all studies in the XANTUS program. |
Study protocol
Patient population, enrollment, and follow-up
Eligible patients comprise men and women aged 18 years or older with nonvalvular AF who start rivaroxaban therapy to reduce the risk of stroke or systemic embolism and who have provided written informed consent. To reduce selection bias, all patients at least 18 years old presenting with nonvalvular AF will be screened sequentially and documented in an anonymous log file independent of therapy. Patients prescribed rivaroxaban will be eligible for the study, and all such patients must be enrolled consecutively. If a patient is not eligible for any reason (eg, refusal to provide consent), then the reason must be documented in the log file. Contraindications to rivaroxaban therapy should be considered according to local marketing authorizations. For XANTUS, enrollment will take place at up to 300 sites, each of which should enroll a minimum of 20 and a maximum of 60 patients. Sites must be able to capture data electronically and provide long-term follow-up for patients.
After the initial screening visit, follow-up visits will occur at the time of hospital discharge, if applicable, and approximately every 3 months thereafter. The overall follow-up period will be 1 year, and will conclude with a final patient visit. For patients who discontinue therapy before the end of the 1-year follow-up, the observation period will end approximately 30 days after the last dose of rivaroxaban (Figure 1 and Supplementary material).
Drug administration
The decision to prescribe rivaroxaban is at the sole discretion of the treating physician, including the dose and duration of rivaroxaban therapy. Any prior use of an anticoagulant will be documented in the case report form (CRF) and all concomitant medications or other therapies will also be documented. For patients withdrawing from the study, the reasons and any follow-on therapy are to be documented in the CRF. Temporary interruptions of rivaroxaban therapy will also be documented.
Data and information to be collected
Baseline data
Baseline demographics will be collected at the initial visit and will include:
- age, sex, ethnicity, smoking history, and alcohol use
- previous therapies or interventions to treat AF, including cardioversion and ablation
- prior medication (medication taken before study commencement), including any prior use of anticoagulants
- concomitant medications (ie, medications taken in addition to rivaroxaban at time of commencing rivaroxaban therapy)
- vital signs and laboratory tests, if performed as part of routine care
- other medical history at baseline, such as bleeding diathesis or risk factors for stroke in AF (eg, hypertension, diabetes, heart failure, prior stroke)
- stroke and bleeding risk profiles; risk profiles will be based on such risk scores as CHADS2 (Congestive heart failure, Hypertension, Age, Diabetes mellitus, Stroke)18 CHA2DS2-VASc (Vascular disease, Age, Sex category),19 or HAS-BLED (Hypertension, Abnormal liver/renal function, Stroke history, Bleeding predisposition, Labile international normalized ratios, Elderly, Drug/alcohol usage).20
Study outcomes to be captured
Primary outcomes
The primary outcomes to be collected are those that can assess and estimate the safety of rivaroxaban in routine practice. These will be recorded as AEs or serious AEs, and include major bleeding, defined as in ROCKET AF,7 all-cause mortality, and any other AEs and serious AEs. The sites of major bleeding events will also be captured. Outcome definitions are provided in the Supplementary material.
Secondary outcomes
- Symptomatic thromboembolic events
- Nonmajor bleeding events, defined as any bleeding event that does not meet the criteria for a major hemorrhage
- Rates of AEs and serious AEs across patients with different baseline risk profiles for stroke or bleeding (eg, CHADS2, CHA2DS2-VASc, or HAS-BLED).
Rates of AEs and serious AEs will also be evaluated within other important medical subgroups (defined at baseline). These include, among others:
- age, sex, and ethnicity
- body mass index
- prior use of oral anticoagulants
- concomitant medications
- creatinine clearance
- baseline type of AF
- prior stroke, transient ischemic attack (TIA), or non-systemic embolism.
Other outcomes
These include the use of concomitant medications initiated any time after start of rivaroxaban therapy. Over-the-counter medications such as acetylsalicylic acid should also be included. Treatment satisfaction is to be reported by patients using standardized questionnaires completed at quarterly intervals. Patients who received a VKA in the 4 weeks prior to enrolling in the study will also be asked to complete a questionnaire at the initial visit. The questionnaires used will be translated, and validated with versions of either the Anti-clot Treatment Scale or the Treatment Satisfaction Questionnaire for Medication. Also included are health care-resource use, including health care visits, length of stay for hospitalizations, and nurse visits in the community (eg, for administration of a parenteral anticoagulant), details of interventions and how they were managed, and reasons for any switch from or interruption of rivaroxaban therapy, including assessment of persistence with therapy
Reporting of adverse events
AEs and serious AEs will be recorded on the AE report form that is appended to the CRF. The AE report form will document the seriousness, duration, actions taken, outcomes, and relation to rivaroxaban therapy for each AE. For patients who discontinue rivaroxaban early, documentation concerning AEs will also cover the 30-day period after cessation of therapy.
Additionally, the manner in which bleeding and thromboembolic events are managed in the routine practice setting will be recorded. This includes any use of thrombolytics or surgical intervention after ischemia or use of prohemostatic agents (eg, prothrombin-complex concentrates) for major bleeding.
These cumulative data will provide a comprehensive overview of the use of rivaroxaban in routine practice and of how clinical events arising during stroke prophylaxis in AF are managed in this setting.
Statistical considerations
Overview
The main goal of the XANTUS study is to develop estimates of the rates of treatment-emergent AEs arising in routine clinical practice. AEs are considered treatment-emergent when they occur from the day of the first dose of rivaroxaban, and up to 2 days after the last dose in the event of discontinuation.
Statistical analyses will be descriptive, exploratory, and generally limited to frequency tables or summary statistics (eg, means ± standard deviation or medians ± quartiles), eg, for demographic data. The primary analysis population will be the full safety population, defined as all patients who have taken at least one dose of rivaroxaban, although analyses will be performed not only for the full study sample but also for each participating country where numbers are sufficient for statistical analysis. Each of the four studies in the XANTUS program will report separately, but there will also be a prespecified pooled analysis. In addition, results will be derived for medically relevant subgroups, eg, elderly patients, patients with renal impairment or prior stroke/TIA, and patients with different stroke-risk profiles, eg, CHADS2 or CHA2DS2-VASc categories. The combined data set is also expected to permit the determination of regional differences in the use of rivaroxaban and to allow the investigators to identify any differences in treatment approaches for different ethnicities.
Presentation of events of interest
Both raw incidence proportions (patients with events/number of treated patients) and incidence rates (eg, patients with events per 100 patient-years) will be presented. Each such estimate will be presented with corresponding 95% confidence intervals (CIs). In addition, Kaplan–Meier plots will be generated to show the time course up to the first event of interest.
Rationale for sample size
The overall incidence of major bleeding in ROCKET AF was 5.6% (395 of 7,111) over a maximum follow-up period of 1,263 days. Over the first year, 3.5% (250 of 7,111) of patients experienced a major bleeding event, and it is assumed that this figure is representative of the rate of major bleeding in patients receiving rivaroxaban in a real-world setting. A sample size of 5,000 patients would yield a 95% CI of 3.008–4.047, assuming a rate of major bleeding of 3.5%. In other words, assuming an underlying event rate of 3.5%, the CI would be ≤1.1% with a probability of 95%. A lower event rate with the same sample size would result in a narrower CI. This is considered sufficiently precise for the purposes of the study. However, because it was also assumed that 20% of patients would be unevaluable, target enrollment was increased to 6,000 patients.
Data integrity and quality
All outcome variables and covariates will be recorded on a standardized CRF. Reporting bias will be minimized by onsite quality reviews to verify source data from at least 10% of the sites. Furthermore, 50% of the sites will receive random visits to monitor study conduct. Selection bias will be minimized by requiring investigators to document consecutive patients prescribed rivaroxaban, with no omissions.
Data management and administrative organization
The parent XANTUS study is a postauthorization safety study funded by Bayer HealthCare. Each of the four studies in the program will have its own steering committee, and the program as a whole will be overseen by an executive steering committee. Major events, specifically major bleeding, stroke, systemic embolism, TIA, and myocardial infarction, will be adjudicated centrally by the same adjudication committee for all four studies, blinded to individual patient data. Data verification at study sites should facilitate identification of any relevant outcomes not submitted for adjudication. The study will use one centralized database to receive results, and results from all four studies in the program will be stored in the same database.
Discussion
Registration clinical trials, such as ROCKET AF, are required to mandate strict inclusion and exclusion criteria and to enforce tight control of both the intervention therapy and the comparator. In the case of ROCKET AF (and other trials), this was further complicated by the double-blind, double-dummy protocol. The advantage of this approach is that the efficacy and safety of rivaroxaban relative to warfarin are now well established.7 Conversely, there are limited data concerning the clinical utility of rivaroxaban for long-term use, in contrast to the strictly controlled environment of a Phase III clinical trial.
Noninterventional studies and registries have a pivotal role in confirming the efficacy and safety of newly approved interventions across the wide range of patients (demographics, comorbidities, health care access, etc) treated in routine practice, an environment in which control of patient therapy is often not as strict as in a clinical trial. For example, the recently completed XAMOS (XArelto in the prophylaxis of post-surgical venous thromboembolism after elective Major Orthopaedic Surgery of hip or knee) study21 was a noninterventional study in which the use of rivaroxaban in routine practice for the prevention of venous thromboembolism after elective hip or knee replacement was evaluated, although local standard of care was included as a comparator. The results confirmed the superior efficacy and similar safety of rivaroxaban, relative to the standard of care, reported in the Phase III RECORD (REgulation of Coagulation in ORthopedic surgery to prevent Deep venous thrombosis and pulmonary embolism) program.22
Noninterventional studies such as XAMOS or XANTUS and registries such as GARFIELD (Global Anticoagulant Registry in the FIELD)-AF,23 ORBIT-AF (Outcomes Registry for Better Informed Treatment of Atrial Fibrillation),24 and the Dresden NOAC registry (NCT01588119) can go a long way toward meeting the need for real-world data on the efficacy and safety of rivaroxaban, and in particular confirming the safety of rivaroxaban in the routine practice setting. Postauthorization studies for other NOACs are also in process (eg, NCT01774370 [SPARK {Safety Study of Pradaxa in Atrial Fibrillation Patients by Regulatory Requirement of Korea}] and NCT01491178 for dabigatran).
Postauthorization studies, such as XANTUS, can also contribute important additional information on various aspects of patient management in routine care. For rivaroxaban, this might include the following:
- When bleeding events or strokes do occur in patients receiving rivaroxaban, how are they managed, particularly in the first few days after a stroke?
- What are the most common reasons for temporary interruptions of rivaroxaban therapy?
- How are cardioversion and ablation procedures managed?
- How is switching to a parenteral anticoagulant managed in practice (where relevant), and how is reinitiation of rivaroxaban therapy managed?
- How are conversions from/to conventional VKA treatment to/from rivaroxaban performed in routine practice? This may be of particular importance in light of the transient increase in events observed at the end of the ROCKET AF study period in patients transitioning from blinded rivaroxaban to open-label warfarin.
- Are there signals that either efficacy or safety of rivaroxaban in this real-world cohort differ markedly from the published experience in controlled clinical trials?
XANTUS will also provide useful information on factors that can affect patient compliance with therapy, potentially an important determinant of the effectiveness of stroke prophylaxis. The study may also contribute to optimizing management of patients at increased risk of both ischemic and hemorrhagic events, such as the use of various comedications, patients with renal impairment, and those who have experienced a prior stroke.
Uptake of NOACs into routine practice is expected to occur gradually after marketing approval is received. However, if a study such as XANTUS was to include a comparator arm (eg, warfarin therapy), then the noninterventional aspects of such a nonrandomized study might result in patients most likely to experience difficulties with warfarin therapy (eg, those at high risk of bleeding or poor compliance) being those most likely to be prescribed rivaroxaban. Conversely, patients most likely to do well on therapy might be more likely to receive a VKA. This would inevitably result in a considerable degree of selection bias, and for this reason a single-arm cohort study was considered the most appropriate design. A limitation of this design is that any relative risk estimates are not feasible. In addition, the strictly limited data available for patients screened but not enrolled preclude any comparison of treatment-selection rationales. A further limitation is that because XANTUS is strictly an observational study, estimates of event rates will not be corrected or normalized for baseline characteristics or for different durations of rivaroxaban therapy. However, the prospective definition of events and outcomes to be captured is expected to enhance the accuracy with which events are recorded. Event rates within predefined baseline subgroups (eg, stroke-risk categories) will also be determined.
There are several practical questions concerning the management of certain events, such as ischemic stroke, in a patient receiving an NOAC. For example, should thrombolytic therapy be used, and if so, what factors should be considered? Similarly, understanding the way in which bleeding events are managed will also be of use, including factors determining if and when such agents as prothrombin-complex concentrates are used or when rivaroxaban can be restarted after an intracranial hemorrhage. XANTUS is expected to make a substantial contribution to answering such questions, and should show how clinicians are approaching this and other everyday clinical challenges.
Conclusion
XANTUS will provide a comprehensive data set to support the safety and clinical utility of rivaroxaban for stroke-risk reduction in patients with AF in routine practice (ie, beyond the strict confines of the randomized clinical trial environment). The study will also provide important insights into the management of AEs occurring in patients receiving rivaroxaban therapy, as well as factors that contribute to maximizing patient satisfaction and optimizing real-world outcomes.
Acknowledgments
The authors would like to acknowledge Seamus McMillan, who provided editorial support with funding from Bayer HealthCare Pharmaceuticals, and Janssen Scientific Affairs LLC.
Disclosure
AJC has been a consultant for AstraZeneca, Bayer HealthCare, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, Pfizer, Sanofi, Aryx, and Johnson & Johnson. PA has been a consultant for Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Pfizer, Boehringer Ingelheim, Daiichi Sankyo, AstraZeneca, Sanofi, Boston Scientific, Edwards, Lundbeck, Merck, and Kowa Pharmaceutical. S Haas has been a consultant for Bayer HealthCare Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, Pfizer, and Sanofi. PK has received consulting fees and honoraria from 3M Medica, MEDA Pharma, AstraZeneca, Bayer HealthCare, Biosense Webster, Boehringer Ingelheim, Daiichi Sankyo, the German Cardiac Society, Medtronic, Merck, MSD, Otsuka Pharma, Pfizer/Bristol-Myers Squibb, Sanofi, Servier, Siemens, and Takeda. MVE and S Hess are employees of Bayer HealthCare Pharmaceuticals. AGGT has been a consultant for Bayer HealthCare, Janssen Pharmaceutical Research & Development, Astellas, Portola, and Takeda. The authors have no other conflicts of interest.
References
You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):e531S–e575S. | |
Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22(8):983–988. | |
Nieuwlaat R, Olsson SB, Lip GY, et al. Guideline-adherent antithrombotic treatment is associated with improved outcomes compared with undertreatment in high-risk patients with atrial fibrillation. The Euro Heart Survey on Atrial Fibrillation. Am Heart J. 2007;153(6):1006–1012. | |
Ansell J, Hollowell J, Pengo V, Martinez-Brotons F, Caro J, Drouet L. Descriptive analysis of the process and quality of oral anticoagulation management in real-life practice in patients with chronic non-valvular atrial fibrillation: the international study of anticoagulation management (ISAM). J Thromb Thrombolysis. 2007;23(2):83–91. | |
Camm AJ, Lip GY, De Caterina R, et al. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719–2747. | |
Furie KL, Goldstein LB, Albers GW, et al. Oral antithrombotic agents for the prevention of stroke in nonvalvular atrial fibrillation: a science advisory for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43(12):3442–3453. | |
Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–891. | |
Janssen Pharmaceuticals. Xarelto (rivaroxaban) [prescribing Information]. 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202439s008lbl.pdf. Accessed January 27, 2014. | |
Bayer Pharma. Xarelto (rivaroxaban) [summary of product characteristics]. 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000944/WC500057108.pdf. Accessed April 23, 2014. | |
Mueck W, Becka M, Kubitza D, Voith B, Zuehlsdorf M. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct factor Xa inhibitor – in healthy subjects. Int J Clin Pharmacol Ther. 2007;45(6):335–344. | |
Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939 – an oral, direct factor Xa inhibitor – after multiple dosing in healthy male subjects. Eur J Clin Pharmacol. 2005;61(12):873–880. | |
Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat Rev Drug Discov. 2011;10(1):61–75. | |
Buller HR, Lensing AW, Prins MH, et al. A dose-ranging study evaluating once-daily oral administration of the factor Xa inhibitor rivaroxaban in the treatment of patients with acute symptomatic deep vein thrombosis: the Einstein-DVT Dose-Ranging Study. Blood. 2008;112(6):2242–2247. | |
Agnelli G, Gallus A, Goldhaber SZ, et al. Treatment of proximal deep-vein thrombosis with the oral direct factor Xa inhibitor rivaroxaban (BAY 59-7939): the ODIXa-DVT (Oral Direct Factor Xa Inhibitor BAY 59-7939 in Patients with Acute Symptomatic Deep-Vein Thrombosis) study. Circulation. 2007;116(2):180–187. | |
Mueck W, Lensing AW, Agnelli G, Décousus H, Prandoni P, Misselwitz F. Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep-vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50(10):675–686. | |
ROCKET AF Study Investigators. Rivaroxaban-once daily, oral, direct factor Xa inhibition compared with vitamin K antagonism for prevention of stroke and embolism trial in atrial fibrillation: rationale and design of the ROCKET AF study. Am Heart J. 2010;159(3):340–347. | |
Wong KS, Hu DY, Oomman A, et al. Rivaroxaban for stroke prevention in East Asian patients from the ROCKET AF trial. Stroke. Epub April 24, 2014. | |
Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285(22):2864–2870. | |
Lip GYH, Nieuwlaat R, Pisters R, Lane D, Crijns H. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor based approach: the Euro Heart Survey on Atrial Fibrillation. Chest. 2010;137(2):263–272. | |
Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093–1100. | |
Turpie AG, Schmidt AC, Kreutz R, et al. Rationale and design of XAMOS: noninterventional study of rivaroxaban for prophylaxis of venous thromboembolism after major hip and knee surgery. Vasc Health Risk Manag. 2012;8:363–370. | |
Turpie AG, Haas S, Kreutz R, et al. A non-interventional comparison of rivaroxaban with standard of care for thromboprophylaxis after major orthopaedic surgery in 17,701 patients with propensity score adjustment. Thromb Haemost. 2014;111(1):94–102. | |
Kakkar AK, Mueller I, Bassand JP, et al. International longitudinal registry of patients with atrial fibrillation at risk of stroke: Global Anticoagulant Registry in the FIELD (GARFIELD). Am Heart J. 2012;163(1):13–19. | |
Piccini JP, Fraulo ES, Ansell JE, et al. Outcomes registry for better informed treatment of atrial fibrillation: rationale and design of ORBIT-AF. Am Heart J. 2011;162(4):606–612. |
Supplementary material
Outcome definitions
Stroke
Stroke is defined as a new, sudden, focal neurologic deficit resulting from a presumed cerebrovascular cause that is not reversible within 24 hours and not due to a readily identifiable cause, such as a tumor or seizure.
Whenever possible, computed tomography scanning or magnetic resonance imaging should be used to assist in the classification. Stroke will be classified as:
- primary hemorrhagic – stroke with focal collections of intracerebral blood; hemorrhagic stroke will be recorded as bleeding and as stroke; note that subarachnoid, subdural, and epidural hemorrhages will be recorded as bleeding events
- primary ischemic – stroke without focal collections of intracranial blood; hemorrhagic conversion of a primary ischemic infarction will be recorded
- uncertain – no imaging or autopsy data available.
Death within 30 days of stroke onset will be considered a fatal stroke.
Transient ischemic attack
An event matching the definition for stroke but lasting less than 24 hours will be considered a transient ischemic attack (TIA). The duration of symptoms for a TIA will be recorded, as will the results of any imaging procedures. However, TIA events with neuroimaging documentation of a cerebral infarction in the appropriate location for the clinical syndrome will be recorded as an ischemic stroke.
Non-central nervous system systemic embolism
Non-central nervous system systemic embolism is defined as abrupt vascular insufficiency associated with clinical or radiologic evidence of arterial occlusion in the absence of other likely mechanisms (eg, trauma, atherosclerosis, instrumentation). In the presence of atherosclerotic peripheral vascular disease, diagnosis of embolism to the lower extremities requires angiographic demonstration of abrupt arterial occlusion.
Myocardial infarction
Diagnosis of myocardial infarction will be considered relative to positive coronary angiography. In the absence of coronary angiography, myocardial infarction is defined as: clinical symptoms consistent with myocardial ischemia and cardiac biomarker elevation (troponin I or T, creatine-kinase muscle and brain subunit) greater than the site’s upper limit of normal; development of new pathologic Q waves in at least two contiguous leads on the electrocardiogram; or autopsy confirmation.
Major bleeding
This is defined as overt bleeding associated with:
- a fall in hemoglobin of ≥2 g/dL
- a transfusion of two or more units of packed red blood cells or whole blood
- occurrence at a critical site (intracranial, intraspinal, intraocular, pericardial, intra-articular, intramuscular with compartment syndrome, retroperitoneal)
- death.
Withdrawal from study
Patients may withdraw from the study at any time, and are not required to provide a reason. No patient will be disadvantaged by withdrawing.
However, because this is a noninterventional study, withdrawal from the study is independent of the patient’s particular therapy. Where a patient discontinues therapy prematurely, follow-up will be documented until 30 days after the discontinuation of treatment.
Every effort will be made to ascertain the reason for withdrawal from the study or for premature discontinuation of therapy; this will be recorded in the case report form.
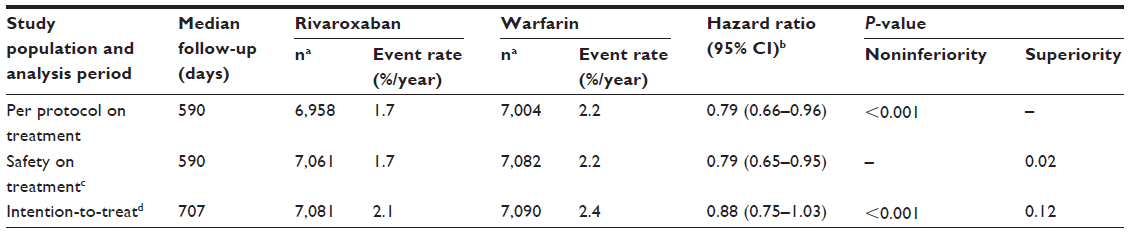 | Table S1 Primary efficacy results for ROCKET AF |
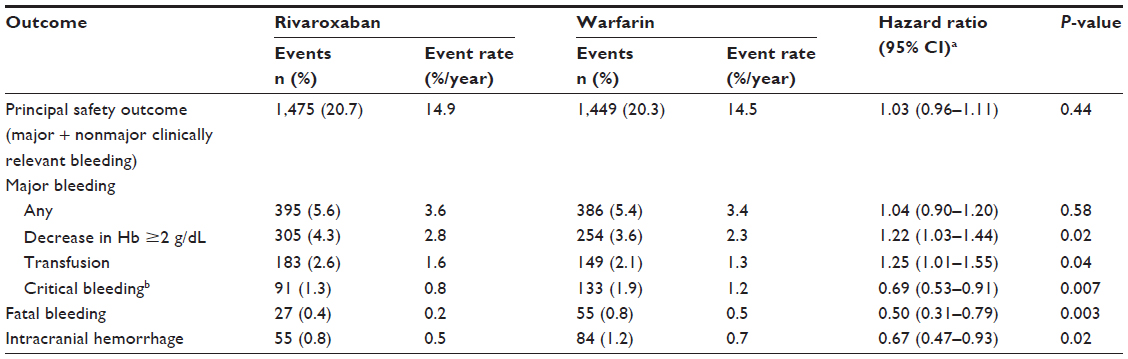 | Table S2 Rates of bleeding outcomes in ROCKET AF |
Reference
Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–891. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.