")
Back to Journals » International Journal of Nanomedicine » Volume 14
Would Colloidal Gold Nanocarriers Present An Effective Diagnosis Or Treatment For Ischemic Stroke?
Authors Amani H , Mostafavi E , Alebouyeh MR , Arzaghi H , Akbarzadeh A, Pazoki-Toroudi H, Webster TJ
Received 26 March 2019
Accepted for publication 17 September 2019
Published 7 October 2019 Volume 2019:14 Pages 8013—8031
DOI https://doi.org/10.2147/IJN.S210035
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Hamed Amani,1,2,* Ebrahim Mostafavi,3,* Mahmoud Reza Alebouyeh,4,* Hamidreza Arzaghi,5 Abolfazl Akbarzadeh,6 Hamidreza Pazoki-Toroudi,7 Thomas J Webster3
1Department of Medical Nanotechnology, Faculty of Advanced Technologies in Medicine, Iran University of Medical Sciences, Tehran, Iran; 2Physiology Research Center, Faculty of Medicine, Iran University of Medical Sciences, Tehran, Iran; 3Department of Chemical Engineering, Northeastern University, Boston, MA, USA; 4Anesthesia Department, Faculty of Medicine, Iran University of Medical Sciences, Tehran, Iran; 5Department of Medical Biotechnology, Faculty of Allied Medical Sciences, Iran University of Medical Sciences, Tehran, Iran; 6Department of Medical Nanotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran; 7Physiology Research Center and Department of Physiology, Faculty of Medicine, Iran University of Medical Sciences, Tehran, Iran
*These authors contributed equally to this work
Correspondence: Thomas J Webster
Department of Chemical Engineering, Northeastern University, Boston, MA, USA
Email [email protected]
Hamidreza Pazoki-Toroudi
Physiology Research Center and Department of Physiology, Iran University of Medical Sciences, P.O. Box 14515-763, Tehran, Iran
Fax +98-21-88675760
Email [email protected]
Introduction: This study was conducted to evaluate OX26-PEG-coated gold nanoparticles (GNPs) (OX26@GNPs) as a novel targeted nanoparticulate system on cell survival after ischemic stroke.
Materials and methods: Dynamic light scattering (DLS), zeta sizer, and transmission electron microscopy (TEM) were performed to characterize the OX26@GNPs. The effect of OX26@GNPs on infarct volume, neuronal loss, and necroptosis was evaluated 24 h after reperfusion using 2, 3,5-Triphenyltetrazolium chloride (TTC) staining, Nissl staining and Western blot assay, respectively.
Results: Conjugation of OX26-PEG to the surface of the 25 nm colloidal gold particles increased their size to 32±2 nm, while a zeta potential change of −40.4 to 3.40 mV remarkably increased the stability of the nanoparticles. Most importantly, OX26@GNPs significantly increased the infarcted brain tissue, while bare GNPs and PEGylated GNPs had no effect on the infarct volume. However, our results indicated an extension of necroptotic cell death, followed by cell membrane damage.
Conclusion: Collectively, our results showed that the presently formulated OX26@GNPs are not suitable nanocarriers nor contrast agents under oxidative stress for the diagnosis and treatment of ischemic stroke. Moreover, our findings suggest that the cytotoxicity of GNPs in the brain is significantly associated with their surface charge.
Keywords: targeted delivery, gold nanoparticles, necroptosis, surface charge, oxidative stress, cytotoxicity
Introduction
Stroke is a major threat to public health whose risk significantly increases with age. The global burden of stroke will keep increasing since over 1.5 billion people will be 65 years of age or older until the year 2050.1–4 Owing to rapid metabolization, high clearance from blood circulation, and poor transport across the blood-brain-barrier (BBB), targeted drug delivery to brain tissue remains a major challenge for central nervous system drug development.5,6 Other substantial challenges to delivering drugs to the brain is the great degree of complexity of molecular mechanisms involved in cell survival in the human brain.7–9 Although many of the aforementioned problems have plagued conventional drug delivery, the emergence of nanotechnology opens up many more potential avenues for researchers to solve these problems through the improvement of pharmacokinetic profiles of drugs as well as better accessibility to neurovascularity.10–12 Additionally, nanoparticles can be engineered to accumulate to the ischemic site in the brain and amplify signals for the early diagnosis of neurological diseases, such as stroke.13
Of all the available nanoparticles, gold nanoparticles with unique surface chemistry, ease of synthesis, possibility of extensive chemical modification, excellent biocompatibility as well as superior optical, electrical, and thermal properties have attracted tremendous attention as fascinating building blocks for numerous biomedical applications such as bacterial inhibition, drug and gene delivery, stem cell-based tissue engineering, photothermal therapy, biosensing and bioimaging.14–17 Gold nanoparticles (GNPs) have been found to be both oxidants and antioxidants in biological systems in a size-dependent manner. For example, Liu et al selected both 5 and 20 nm GNP sizes and investigated their antioxidant effects on focal ischemic stroke induced rats. It was found that an intraperitoneal injection of 20 nm GNPs (without being functionally targeted to the brain) remarkably exhibited neuroprotective effects in coping with oxidative stress while neuronal damage was aggravated by smaller 5 nm GNPs.18 Aside from therapeutic effects, GNPs also have the ability to bind to various biomolecules such as fluorescein-labeled hyaluronic acids to monitor reactive oxygen species (ROS) levels and subsequently identify an infarct region in ischemic stroke.19 In another study by Kim et al, fibrin-targeted GNPs were used to directly visualize cerebrovascular thrombi in a mouse embolic stroke model using a computed tomography based method.20 Additionally, Yoon et al reported in vivo multi-photon luminescence imaging using GNPs which resulted in visualization of the BBB disruption and the cerebral vasculature with high spatial resolution in a mouse model of stroke.21 Various other moieties and biomolecules can be easily attached to the surface of GNPs to obtain targeted nanoparticles for site-specific payload release.22 There is a high expression of transferrin receptor (TfR) in the brain capillary endothelium relative to other organs which makes it a desirable target for targeting delivery to the brain tissue.23 The surface of nanoparticles can be modified with antibodies against transferrin receptors (such as OX26 mAb) for their targeted delivery to the brain.24,25
Here, we designed a nanoparticulate system (OX26@GNPs) that could specifically target the brain, and went on to examine its effect to diagnose and promote neuronal cell survival under oxidative stress conditions in a rat model of middle cerebral artery occlusion (MCAO).
Methods
Supplies
Tetrachloroauric acid (HAuCl4) Immobilon®-FL PVDF membranes with 0.45 μm pore sizes, RIPA buffer (protease and phosphatase inhibitor), and polyethylene glycol were purchased from Sigma–Aldrich (Philadelphia, PA, USA). Sodium citrate and K2CO3 were supplied by Merck (Darmstadt, Germany). N-hydroxylsuccinimide (NHS) and 1-ethyl-3-(dimethylaminopropyl) carbodiimide (EDC) were obtained from the Thermo Scientific Company (Tehran, Iran).
Synthesis Of GNPs
The synthesis of GNPs was based on using the citrate reduction protocol described by Frens26. Briefly, 25 mL of 1% HAuCl4 was refluxed and heated to its boiling point using a hot plate while stirring. Then, 1 mL of 1% trisodium citrate (Na3C6H5O7) was added to this solution and the formation of GNPs was identified by observing a color change (from clear to gray and then deep pink). After 5 mins, the resulting solution was cooled quickly to room temperature.
GNP Surface Decoration With OX26 Antibody And Its Confirmation
1 mL of 1% poly (ethylene glycol)-carboxylic acid (average MW 5 kDa, Sigma-Aldrich) was added to 10 mL of the GNP solution under continuous stirring at room temperature for 24 h. Then, the resulting solution was centrifuged for 30 min at 14,000 rpm to separate the unbound constituents of C-PEGylated NPs, and again the pellet was resuspended in 0.1 M phosphate-buffered saline (pH 7.4). To this solution was added 20 µL of EDC/NHS (Sigma-Aldrich) (75 mM/30 mM, v/v, 1:1) to activate the GNP solution as a coupling agent. After washing with DI water, 5 μL of the CD71 antibody (OX26) (Santa Cruz Biotechnology) was added to the resulting solution. After continuous stirring for 5 h at room temperature, the non-bonded OX26 antibodies were removed by centrifugation at 20,000 rpm for 60 min at 4°C. Finally, the pellet was resuspended in 5 mL of 0.1 M phosphate-buffered saline (pH 7.4). FTIR spectrum of dried bare and OX26@GNPs was performed by an IR Pestige-21 Shimadzu spectrometer to confirm GNP surface decoration with an OX26 antibody. However, measurement of UV–Vis spectra of the solution at 500–600 nm was carried out using a 3–5 mm quartz cuvette on a UV–Vis Perkin Elmer Lambda 25 spectrophotometer (United Kingdom).
Stability And Morphology Of Bare And OX26@GNPs
In order to check GNP shape and stability, a droplet of the diluted pink solution containing bare GNPs or OX26@GNPs was dropped onto a formvar carbon-coated copper grid and dried in air for 2 days. Transmission electron microscopy (TEM) images of the diluted pink solution of GNPs were taken by a TEM, H-7650 at an accelerating voltage of 100 kV.
Size And Surface Charge Of Bare And OX26@GNPs
The hydrodynamic diameter of the GNPs was determined by dynamic light scattering (DLS). The surface charge or zeta potentials of both bare and OX26@GNPs was carried out using a Malvern Zetasizer Nano ZS (Malvern, Worcestershire, UK).
Ethical Approval
All procedures were performed in accordance with the guidelines contained in the guide for the care and use of laboratory animals 8th edition 2011 (the guide). All of the experimental procedures and protocols were approved by the Institutional Ethics Committee on Animal Care and Experimentation of Iran University of Medical Sciences. Eight week male Wistar rats, each weighing between 230–260g, were obtained from the animal laboratory of the Iran University of Medical Sciences. The animals were maintained under standard laboratory conditions (in a controlled temperature of 23–25°C), on a 12/12-h light/dark schedule with food and sterile water ad libitum. To maintain social interactions, five rats were kept per cage.
Experimental Design
Male Wistar rats were randomly assigned into six major groups (Figure 1): i) a sham-operated control group (sham, n=20): in the sham group, the animals received all procedures except middle cerebral artery occlusion (MCAO), ii) an only ischemia group (MCAO, n=20): in the MCAO group, the animals only were subjected to 45 min of ischemia and 24 h reperfusion, iii) OX26-PEG coated GNPs (OX26@GNPs, n=24): in the OX26@GNPs group, the healthy animals were divided into three subgroups and received different doses of OX26@GNPs (250, 500, and 1000 μg/mL), iv) the MCAO+OX26-PEG coated GNPs group was divided in four subgroups (MCAO+OX26@GNPs, n=48): in the MCAO+OX26-PEG coated GNPs group, the animals were subjected to 45 min ischemia, and 1 h after reperfusion received a drug at different doses (100, 250, 500, and 1000 μg/mL) intravenously, v) the MCAO+PEG coated GNPs group (MCAO+PEG@GNPs, n=8): in the MCAO+PEG coated GNPs, the animals were subjected to 45 min ischemia and 1 h after reperfusion intracerebrally received a 100 μg/mL dose of the drug, and vi) MCAO+bare GNPs (n=8): in the MCAO+bare GNPs, the animals were subjected to 45 min ischemia and 1 h after reperfusion intracerebrally received a 100 μg/mL dose of the drug. The main tested dose for the study was 500 μg/mL but in some cases, other concentrations including 250 and 1000 μg/mL were added to the groups to evaluate their influence on the stroke-induced rats.
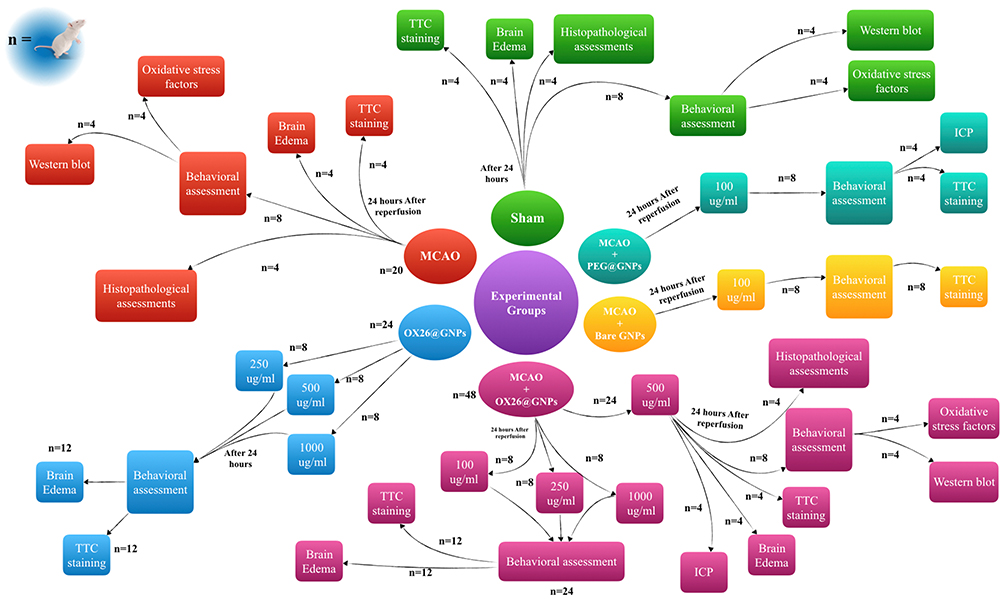 |
Figure 1 Outline scheme of the experimental design used in this study. |
Stereotaxic Surgery
Stereotaxic injection was performed according to a previous study.27 In brief, the animals were deeply anesthetized with a mixture of ketamine–xylazine and then immobilized in a stereotaxic frame (Stoelting, USA). GNPs were injected into the dorsal hippocampus (bregma: −2.5 mm; lateral: +2 mm; depth: −2.4 from the skull) using a Hamilton syringe. The total volume of the injected GNPs was 5 μl with an injection rate of 0.25 μl/min. To avoid any backflow up to the needle tract, a 2 min interval was considered after each injection.
MCAO Model
The animals were anesthetized with an intraperitoneal (IP) injection of a mixture of ketamine and xylazine and subjected to MCAO as described in a previous study (see supplementary data).28
Brain Edema Assay
At 24 h after reperfusion, four rats from each group were decapitated under deep anesthesia. Brains were removed and divided into ischemic and contralateral hemispheres. The ischemic hemisphere was weighed (wet weight) and dried at 110°C for 24 h. The degree of brain edema was determined as follows: (wet weight - dry weight)/wet weight ×100%.
Nissl Staining And Neuron Counts
At 72 h after reperfusion, four rats from each group intracardially were perfused with 0.1mol/L of phosphate-buffered saline (PBS; pH=7.4), followed by 4% paraformaldehyde in 0.1mol/L PBS. The brains were quickly dissected out, fixed in 10% paraformaldehyde for 24 h. Then, the brain tissues were dehydrated and embedded in paraffin. Finally, brains were cut into 7-μm thick sections and stained by cresyl violet to evaluate neuronal cell loss in the hippocampus area. A light microscope with OLYSIA Bio Report Soft imaging system (Muenster, Germany) was used to count cells. In the case of each section, five random fields were chosen and 100 cells were counted per field. An investigator blinded to the experimental design was employed to analyze the slides.
Neurological Deficit Score
The evaluation of a neurological deficit at 72 h of reperfusion was based on the method described by Longa et al.28 Neurological deficit scores are summarized as 0 (no observable deficits), 1 (failure to extend right forelimb), 2 (circling to the contralateral side), 3 (falling to the right), and 4 (cannot walk spontaneously and exhibit a depressed level of consciousness).
Assessment Of Cell Membrane Damage And Necrosis
Cell membrane damage was determined as the lactate dehydrogenase (LDH) release from brain slices into the medium after incubation as previously described (see supplementary data).29
Measurement Of Cerebral Infarction Volume
At the end of the neurological assessment, the animals were killed under deep anesthesia by an overdose mixture of ketamine and xylazine. The brain was rapidly removed, frozen at −20°C for 15 mins and cut into six coronal sections (2 mm thick). Sections were incubated in a solution of 2% 2,3,5-Triphenyltetrazolium chloride (TTC) at 37°C for 20 min; fixed in 10% formalin for 24 h and finally digitized. The infarcted tissue was identified as the unstained region and determined using Image J analysis software.
Western Blot Assay
At 24h after reperfusion, the animals were killed under deep anesthesia and then the brains were rapidly removed, and the ischemic ipsilateral hippocampus was dissected from the brain tissue on ice. The ischemic ipsilateral hippocampus of the brain was homogenized in cell lysis buffer with a complete protease inhibitor cocktail. After a sonication step for 5min at 4°C, tissue lysates were centrifuged at 13,300 g for 20 min. The supernatant was removed and nanodrop spectrophotometry was used to determine protein concentrations. Then, 4–20% gradient SDS/PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) was used and soluble protein (30 μg) was loaded per lane. Separated soluble proteins were transferred onto PVDF (Immobilon®-FL PVDF membrane pore size 0.45 μm-Sigma-Aldrich). PVDF membranes were stained with Sypro or Ponceau S to determine loading efficiency. A blocking solution (3% solution of BSA and 0.1% Tween-20 (TBST)) was employed to block nonspecific reaction sites. Primary antibodies used were as follows: RIP3 ((B-2): sc-374639; dilution range: 1:1000, Santa Cruz Biotechnology, USA), RIP1 Polyclonal Antibody (PA5-20811, dilution range: 1:1000, Invitrogen, USA), and mixed lineage kinase domain-like protein (MLKL) Polyclonal Antibody (PA5-43960; dilution range: 1:1000, Invitrogen, USA). After immersion in primary antibodies, PVDF membranes were incubated with a diluted secondary antibody (Anti-rabbit IgG-Peroxidase) in blocking buffer for 1 h at room temperature. β-actin (#4967, dilution range: 1:1000 Cell Signaling Technology) was applied as a protein loading control. Image J software (National Institutes of Health, Bethesda, MD, USA) was used to quantify Western blots by densitometry. In order to obtain the mean values of proteins, the ratio of expression of each protein to the expression of the β-actin in the same sample was determined based on their optical densities.
ICP-OES Analysis
To determine the Au content in different tissues of male Wistar rats at 24 h after intravenous injection of PEGylated GNPs and OX26@GNPs, we applied inductively coupled plasma optical emission spectrometry (ICP-OES) analysis. The brain, liver, and kidney tissue samples were collected, weighed and homogenized for 5 min in 3 mL of cold PBS with a tissue homogenizer. Then, homogenized tissues were digested by nitric acid and hydrogen peroxide (4:1, v/v) at 80°C for 5 min. After cooling down at room temperature, the samples were transferred to 15 mL ICP-OES tubes. Of note, ICP-OES analysis was conducted only on those rats receiving treatment with PEGylated GNPs and OX26@GNPs.
Evaluation Of Oxidative Stress Markers
The ipsilateral hippocampal tissues were dissected and homogenized in cold phosphate-buffered saline. Then, samples were centrifuged at 13,000g for 10 min at 4 °C. The supernatants were collected, and the BCA protein assay kit was used to determine protein content. MDA is the final product of cell membrane lipid peroxidation formed by oxygen-derived free radicals. To determine tissue MDA, the presence of thiobarbituric acid reactive substances in tissue homogenates was measured spectrophotometrically at 532 and 520 nm against a blank comprising all components except the tissue homogenates. Results are presented as nmol/mg tissue. GPx activity was determined in homogenates using the method described by Paglia and Valentine.30 SOD activity was determined based on the inhibition of nitroblue tetrazolium (NBT) reduction by the xanthine–xanthine oxidase system as described in a previous study.31 In this method, the xanthine–xanthine oxidase system acts as a superoxide producer. SOD activity is expressed as U/mg protein. Evaluation of catalase activity was based on the determination of H2O2 decomposition or the rate constant (k, s−1) according to the method described by Aebi.32,33 CAT activity is presented as U/g protein. The amount of total glutathione was evaluated by the formation of 5-thio-2-nitrobenzoic acid (TBA) converted from 5,5-dithiobis (2-nitrobenzoic acid) (DTNB).
Statistical Analysis
Results are expressed as the mean ±SD or mean ±SEM. Data analysis was carried out using Prism software, version 5. Statistical comparisons were made using a one-way ANOVA followed by a Tukey’s test for post hoc analysis or two-way ANOVA. p < 0.05 was considered statistically significant.
Results
Size And Zeta Potential Of OX26@GNPs
In the present study, the hydrodynamic diameter (± S.D.) of over 90% of bare GNPs was 25.27 ±2. The particle size of OX26@GNPs was about 32±4 nm (Figure 2A). As depicted in Figure 2B, the zeta potential of bare GNPs was −40.4 which increased to +3.40 mV after conjugation with PEG-OX26 mAb. Moreover, the zeta potential of PEGylated GNPs was measured as −21.7 mV (Figure S1).
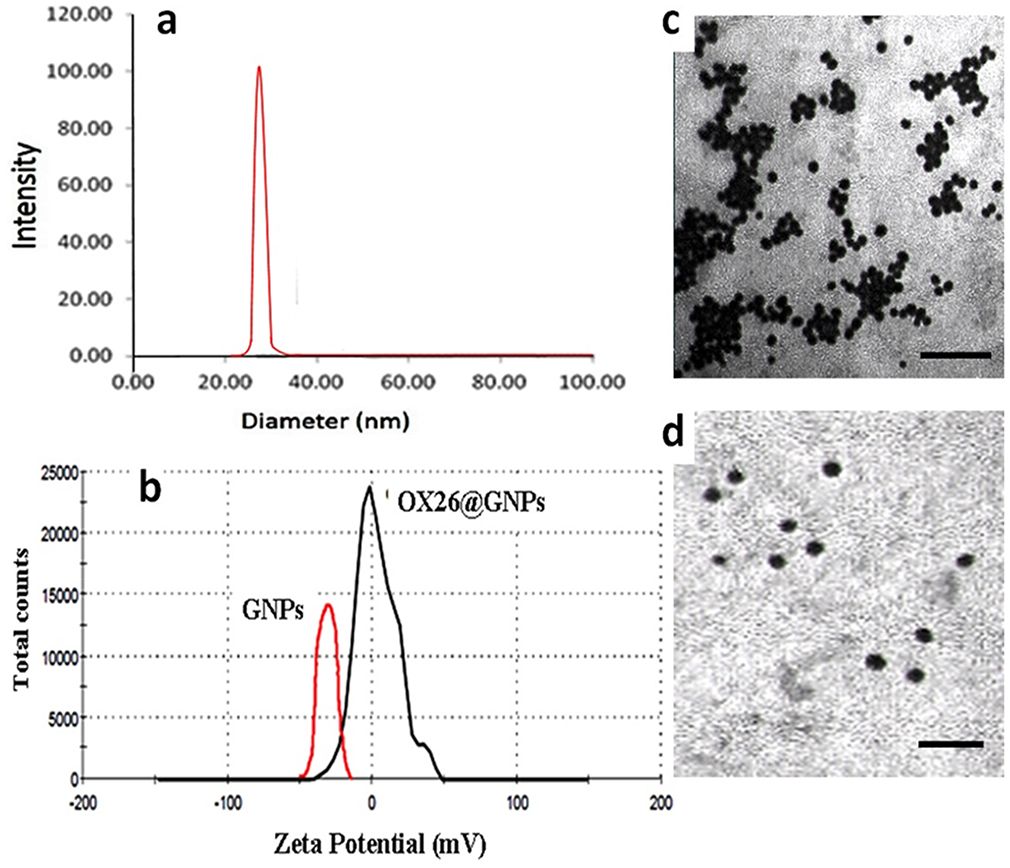 |
Figure 2 Physiochemical characterization of GNPs. (A) Particle size analysis of OX26@GNPs using dynamic light scattering (DLS), (B) Zeta potential distribution of GNPs along with OX26@GNPs. TEM images of (C) GNPs (scale bar 200 nm) and (D) OX26@GNPs (scale bar 100 nm). |
Morphology And Stability Of OX26@GNPs
As shown in Figure 2C and D, bare GNPs were aggregated owing to their high surface energy, while the PEGylation and attachment of OX26 mAb onto the surface resulted in a monodispersed and homogeneous spherical structure of particles with high stability.
Additionally, the colloidal stability of bare GNPs and OX26@GNPs were determined under in vitro and in vivo conditions (see Tables 1 and 2). For the case of the OX26@GNPs, the variations of size and zeta potential were lower compared to bare nanoparticles.
 |
Table 1 Hydrodynamic Size (nm) Of Bare And OX26@GNPs In Different Aqueous Media |
 |
Table 2 The Zeta Potential (mv) Of Bare And OX26@GNPs In Different Aqueous Media |
Confirmation Of OX26 mAb Onto The Surface Of GNPs
In order to prove that chemical bonds were formed between OX26 mAb and GNPs, gold distribution was determined by ICP-OES analysis in the brain, liver and kidney for PEGylated GNPs and OX26@GNPs 24 h after reperfusion. As shown in Figure 3, PEGylated GNPs were present in the liver and kidney whereas the distribution of OX26@GNPs was obviously different. For the case of PEGylated GNPs, besides the high liver uptake of the particles, the kidney had significant uptake compared to the brain. The gold concentration in the brain significantly increased relative to the PEGylated GNPs after conjugation with antibodies, confirming the presence of OX26 mAb on the surface of the GNPs. The results revealed a significant increase in the absorbance and intensity of the particles with an extinction peak at 527 cm−1 after conjugation. For the PEGylated GNPs, the peaks at 1100 cm−1 (C-O-C), 1342 cm−1 (C-H bending) and 2885 cm−1 (CH2 stretch) confirmed the presence of PEG on the surface of the GNPs. For the OX26-PEG-GNPs, the peaks at 1594 cm−1 (C=O in amide) and 1705 cm−1 (N=H in amide) confirmed the presence of the OX26 antibody on the surface of the GNPs (Figure S2), in keeping with previous reports.34
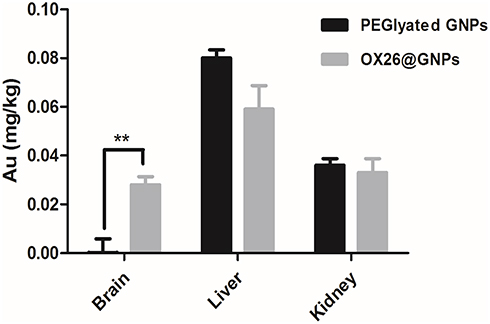 |
Figure 3 The Au content in the different tissues of male Wistar rats at 24 h after intravenous injection of 500 μg/mL PEGylated GNPs and OX26@GNPs (**P<0.01 between indicated groups). |
Moreover, immobilization of the OX26 antibody onto the surface of GNPs was quantified using the Bradford assay (Table S1). Our results showed that the OX26 antibody at a concentration of 5 μg/mL exhibited the highest immobilization on the surface of the GNPs compared to the other tested concentrations. Additionally, the number of GNPs/mL for each concentration was calculated and is presented as Table S2. As shown, there was a direct correlation between the number of GNPs/mL and increasing concentration.
The Effects On The Ischemic Brain
The ischemia resulted in a significant infarct volume compared with the sham. Intracerebral injection of bare and PEGylated GNPs at the 100μg/mL concentration in the ipsilateral hippocampal area did not affect the size of the infarcted region compared with the MCAO group.
Notably, rats treated with OX26@GNPs exhibited a significantly higher increase in infarct volume compared with the sham, MCAO group, and MCAO+ bare GNPs group, suggesting the extent of neuronal loss in brain tissue (Figure 4A and S3).
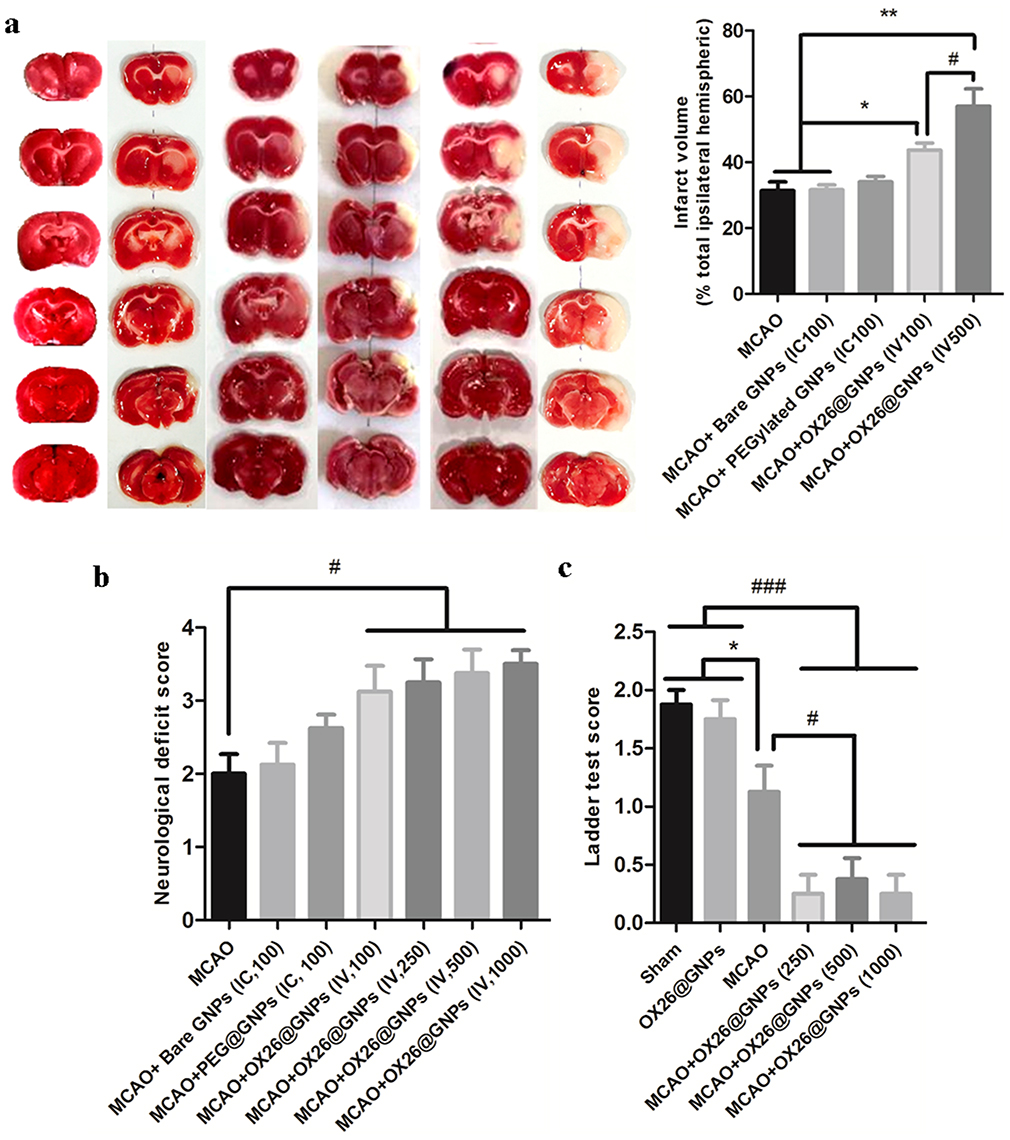 |
Figure 4 The effect of OX26@GNPs on infarct volume and neurological deficits. a) Representative images of infarct size at 72 h post-MCAO by TTC staining in the sham, MCAO and OX26@GNPs+MCAO (500 μg/mL, i.v. dose of GNPs) (**P<0.01 between indicated groups, n=5, #P<0.05 between indicated groups). b) neurological deficit score at 72 h post-MCAO (#P<0.05 between indicated groups), and c) ladder test (*P<0.05, #P<0.05, ###P<0.001 between indicated groups, data presented as means ± SEMs). |
Parallel to infarct exacerbation, we also found exacerbation of locomotor functional outcomes in MCAO + OX26@GNPs groups by the assessment of the neurological deficit scores and the ladder test (Figure 4B and C). Surprisingly, intracerebral injection of bare GNPs at a 100μg/mL concentration in the ipsilateral hippocampal area did not affect the locomotor functional outcomes compared with MCAO. Locomotor functional outcomes were exacerbated after intracerebral injection of PEGylated GNPs at the 100μg/mL concentration, although the differences did not reach a significant level. To further confirm the findings on the increase of the infarct volume when using OX26@GNPs, we also investigated whether the targeted delivery of OX26@GNPs to brain tissue can affect the neuronal vulnerability to ischemia in the hippocampus region or not. Nissl staining results revealed that the targeted delivery of OX26@GNPs to the brain tissue exhibited higher necrotic cell death relative to the sham and MCAO in the sites of ROS stress (Figure 5A–C).
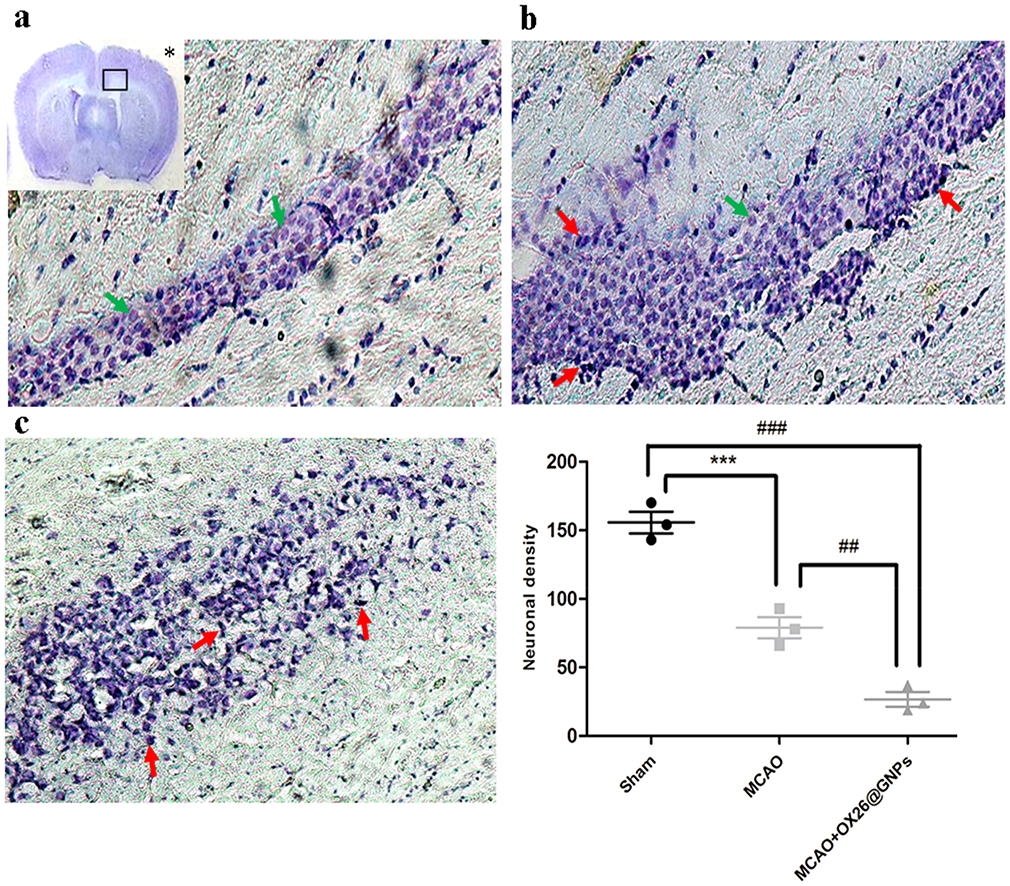 |
Figure 5 Nissl-stained brain sections of the CA1subregion of the hippocampus 72 h post-MCAO at a 500 μg/mL dose of OX26@GNPs (magnification 200×). *: macro picture demonstrates the region where the assessments were done. (A) Sham, (B) MCAO, and (C) MCAO+ OX26@GNPs. The green arrows demonstrate intact cells and red arrows demonstrate necrotic cells. The graph demonstrates the density of intact neuronal cells in the hippocampal CA1subregion (***P<0.001, ##P<0.01, ###P<0.001 between indicated groups). |
We also examined whether the targeted delivery of OX26@GNPs to brain tissue alters brain function through myelin damage, neutrophil recruitment and axonal loss (see Figures S4A–C and S5A–C).
Moreover, our results showed higher brain water content in rats subjected to MCAO and treated with OX26@GNPs relative to only OX26@GNPs, sham, and MCAO (Figure 6A). In order to check the exerted damage to the cell membrane, we measured tissue LDH activity at different doses. At 24 h after reperfusion, LDH activity was significantly higher in brain slices of rats subjected to MCAO and treated with OX26@GNPs relative to only OX26@GNPs, sham, and MCAO. These results suggested that GNPs may strongly interact with membranes and increase their vulnerability to ischemia. Our results showed that although LDH activity and brain edema were significantly increased in rats subjected to MCAO and treated with OX26@GNPs, no significant difference was observed between different doses of nanoparticles (Figure 6B).
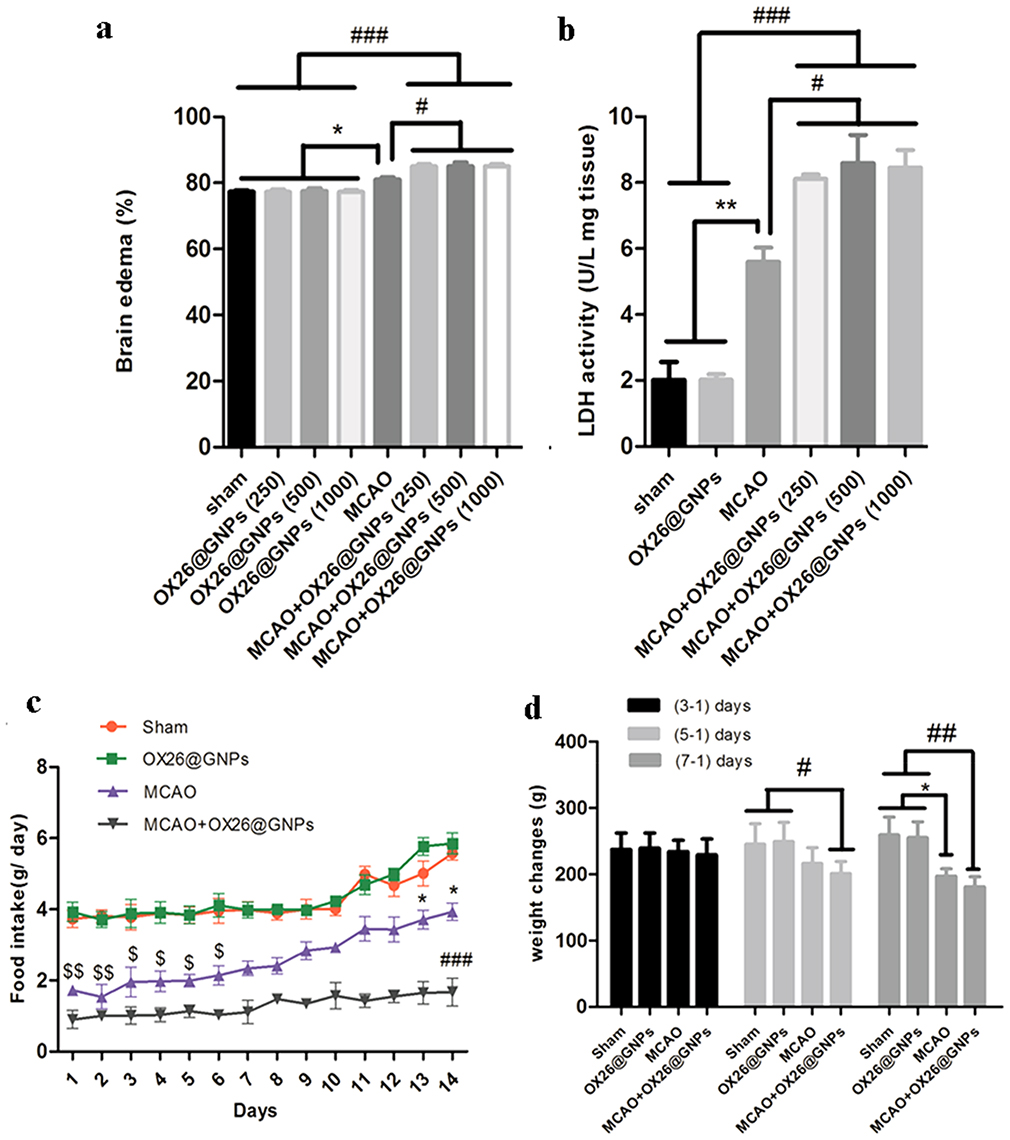 |
Figure 6 The effects of OX26@GNPs on: (A) Brain edema (*P<0.05, #P<0.05, ###P<0.001 between indicated groups), (B) brain LDH activity (**P<0.01, #P<0.05, ###P<0.001 between indicated groups), (C) food intake ($$P<0.01 in 1 and 2 days, $ P<0.05 in 3–6 days vs. sham and OX26@GNPs groups; ###P<0.001 in 1–14 days vs. sham and OX26@GNPs groups; *P<0.05 in 13 and 14 days vs. MCAO+OX26@GNPs group, 500 μg/mL, i.v.), D) body weight changes (# P<0.05 in 3–1 days; *P<0.05 and ## P<0.01 in 7–1 days between indicated groups). |
Systemic Toxicity And Side Effects On Other Organs
We examined the effect of intravenously administered OX26@GNPs on food intake (500μg/mL; 1–14 days post-injection) and body weight (500μg/mL; 1–7 days post-injection). We did not see any change in daily food intake behavior and body weight between the sham and healthy rats treated with an intravenous administration of OX26@GNPs. Significant variations in daily food intake behavior were found in the MCAO group (1–6 days) and rats subjected to MCAO and treated with OX26@GNPs relative to other groups (1–14 days). However, significant body weight loss was observed in MCAO (1–7 days) and MCAO+ OX26@GNPs (1–5 days, and 1–7 days) (Figure 6C and D).
Then, we examined the overall toxicity of OX26@GNPs in animals subjected to MCAO at different times (500μg/mL; 1, 7, and 14 days post-injection). There were no significant statistical differences in terms of red blood cell (RBC) count, and white blood cell (WBC) count between experimental groups at different times (Figure 7A and B). As for the possible toxicity of OX26@GNPs on liver and kidney tissues of animals subjected to MCAO, it was found that the intravenous administration of nanoparticles did not cause any noticeable changes in the levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine, and urea (Figure 7C–F), suggesting no damage to the liver and kidney.
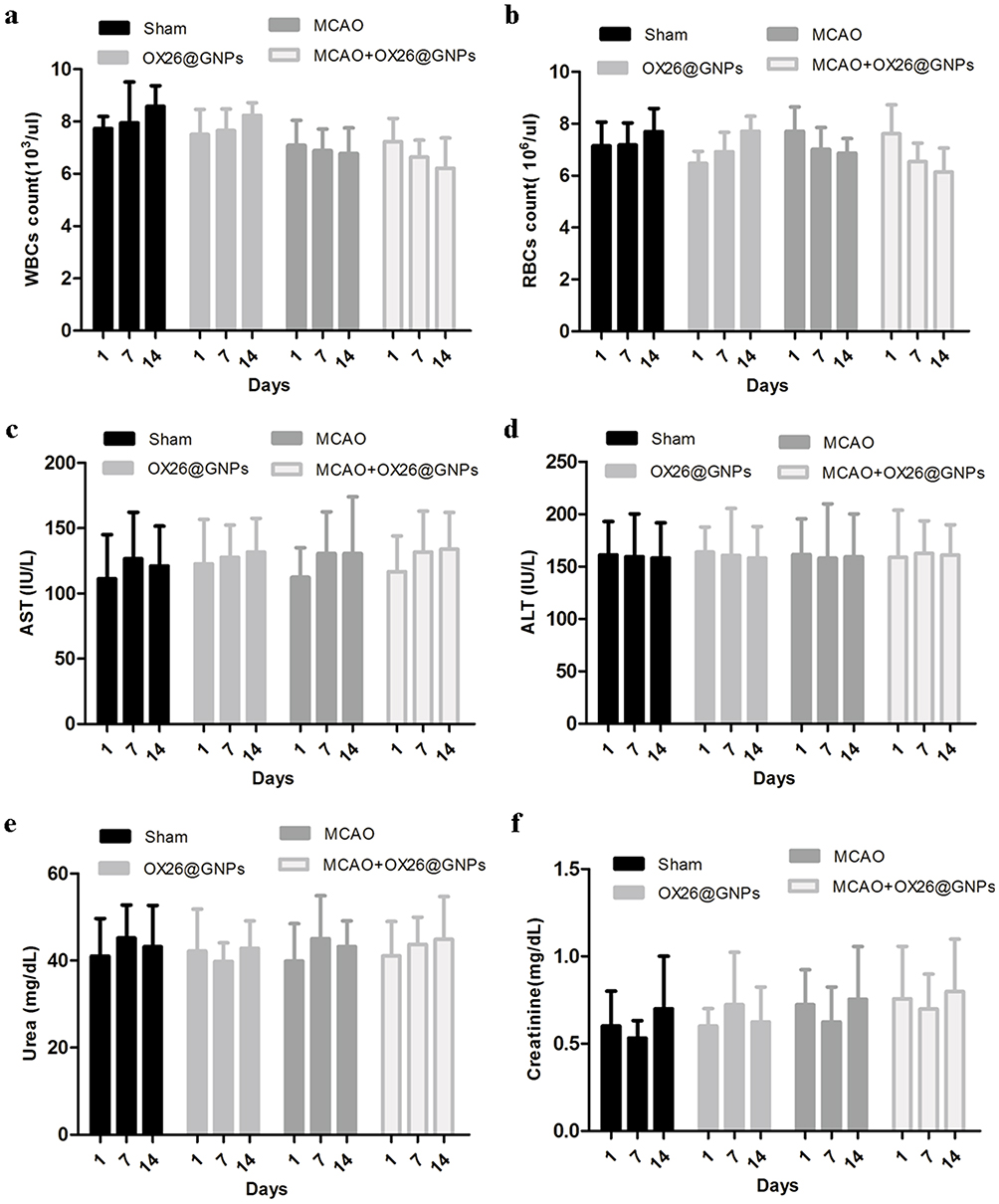 |
Figure 7 The effects of OX26@GNPs administration (500 μg/mL, i.v.) on: (A) WBC count, (B) RBC count, (C) AST, (D) ALT, (E) urea, and (F) creatinine. Data presented as mean ± SD. |
However, to examine whether intravenously administered OX26@GNPs can endow biological toxicity in other organs, we investigated tissue damage in the liver, kidney, spleen, lung, heart, and testis (500 μg/mL; 7 days post-injection). There were no signs of damage, inflammation and alveolar obstruction (Figure 8A–F).
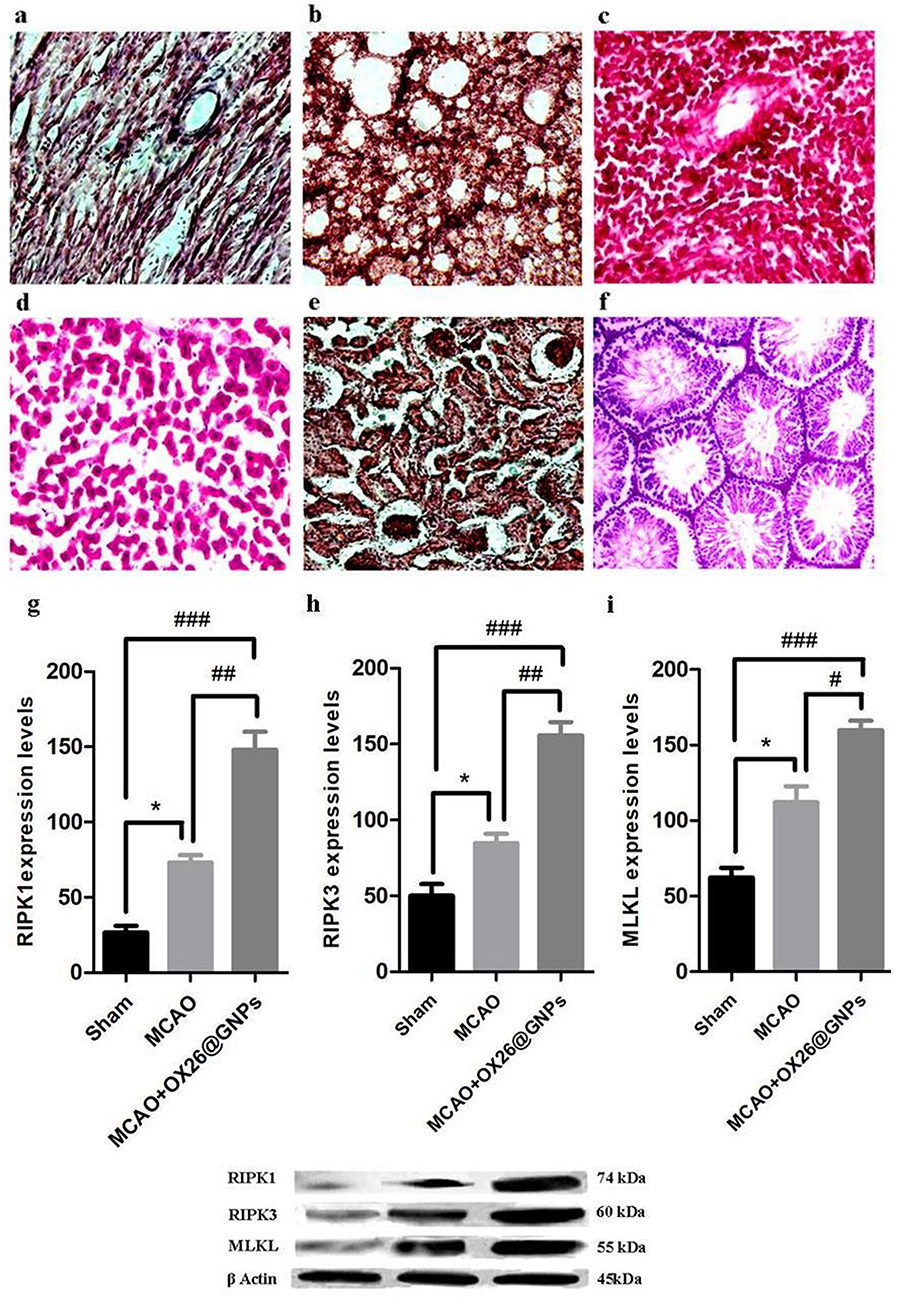 |
Figure 8 The effects of OX26@GNPs (500 μg/mL, i.v.) on the (A) heart, (B) lung, (C) spleen, (D) liver, (E) kidney and (F) testis 7 days post-injection (magnification 200×). The effects of OX26@GNPs (500 μg/mL, i.v.) 24 h post-MCAO in experimental groups on the expression levels of (G) RIPK1, (H) RIPK3, and (I) MLKL (*P<0.05, ##P<0.01, # P<0.05, and ###P<0.001 between indicated groups). Data presented as mean ±SEM. |
Molecular Targets Of OX26@GNPs Under Oxidative Stress
A significantly increased expression of receptor-interacting protein kinase 1(RIPK1) was found in MCAO relative to the sham. Intravenously administered OX26@GNPs significantly elevated RIP1 levels relative to the sham and MCAO groups, suggesting that OX26@GNPs caused significant neuronal loss in the hippocampus region of the ipsilateral hippocampus through TNF-induced necroptosis (Figure 8G). To confirm the findings on necroptosis targeting by OX26@GNPs, we measured receptor-interacting protein kinase 3 (RIPK3) protein by a Western blot assay (Figure 8H). Necroptosis cell death induced by MCAO was markedly increased when OX26@GNPs were intravenously administered 1h after reperfusion. We then asked if OX26@GNPs triggered necroptosis in neuronal cells of the hippocampal region by targeting the RIPK1-RIPK3-MLKL axis. MLKL protein expression was robustly induced by MCAO. Higher levels of the MLKL protein was observed after intravenous administration of OX26@GNPs (Figure 8I).
To investigate whether necroptosis might be induced by oxidative stress in the CA1 hippocampal region, we measured the oxidative stress markers in the experimental groups. A significantly decreased content of glutathione, decreased activity of antioxidant enzymes (such as superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT), as well as increased levels of malondialdehyde (MDA)), were found as early as 24 hrs after reperfusion, which was enhanced by intravenously administered OX26@GNPs, indicating that the nanoparticles increased neuronal demise by oxidative stress-induced necroptosis (Figure 9A–E).
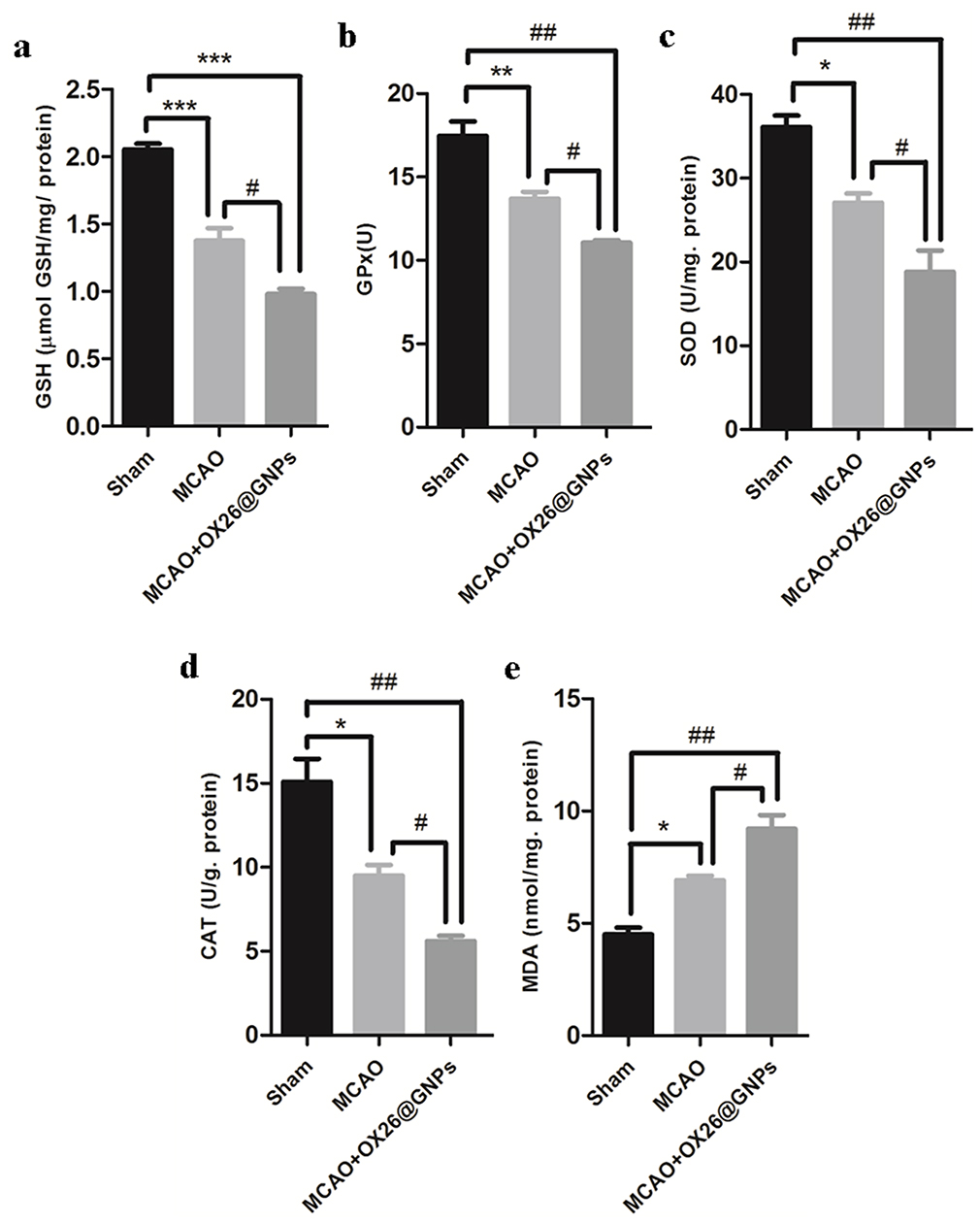 |
Figure 9 The effects of OX26@GNPs (500 μg/mL, i.v.) 24 h post-MCAO in experimental groups on: (A) GSH content, (B) GPx, (C) SOD, (D) CAT and (E) MDA in brain tissue under oxidative stress (***P<0.001, **P<0.01, *P<0.05, #P<0.05, ###P<0.001 and ## P<0.01 between indicated groups). |
Discussion
The preservation of neuronal survival during an ischemic stroke has emerged as one of the greatest medical challenges in recent years.35,36 Currently, many therapeutic agents have been examined to treat degenerative disorders and other diseases.3,37–40 It was found that GNPs may have antioxidant effects in the range of 20–40 nm while smaller sizes (1–5 nm) trigger necrosis cell death by the impairment of the mitochondrial electron transport chain, endogenous ROS generation and depletion of the intracellular antioxidant pool.41 A fundamental question is whether the targeted delivery of GNPs in the size range of 20–40 nm to the site of ROS stress might contribute to cell survival. In this study, we designed a nanoparticulate system to examine the effects of the targeted delivery of GNPs to the brain on neuronal fate under oxidative stress. We also investigated the effects of surface charges of GNPs on neuronal survival under oxidative stress.
The morphology and stability of bare and OX26@GNPs were determined using various spectroscopic and microscopic methods. Our results showed an increase in the hydrodynamic diameter of the GNPs after conjugation that may be due to the increasing layer thickness of the nanoparticles after conjugation.
Moreover, an increase in zeta potential was found after conjugation, suggesting a higher uptake of OX26@GNPs by neuronal cells. In keeping with our findings, a report by Cho et al indicated that surface charge plays a pivotal role in the internalization of gold nanoparticles. They found that positively charged functional groups facilitate interactions between GNPs and negatively charged cell membranes.42 Our finding revealed that surface charges of GNPs strongly affect their toxicity under oxidative stress. We found that positively charged functional groups enhanced neuronal loss while negative charge surfaces did not affect neuronal loss. This could be attributed to the stronger interactions of GNPs with a positive surface charge with the cell membrane and resulting membrane damage. In a comprehensive study by our group, we reported that surface properties of bionanomaterials, such as surface chemistry and surface charge, significantly affect cellular behaviors.43 A study by Karakocak et al demonstrated that surface modification of GNPs and changes in surface chemistry strongly affect cell uptake and subsequently cellular behavior.44 They found that the native 50 and 100 nm GNPs could not enter the cell whereas that GNPs coated with end-thiolated hyaluronate facilitated their entry in the cells through CD44 receptors that in turn resulted in a greater payload of the drug to cells.44
Likewise, in accordance to our findings, a previous study also reported that cationic NPs can cause a more pronounced disruption of plasma-membrane integrity and consequently mitochondrial and lysosomal damage.45 Moreover, Yang et al showed that chemical composition, particle size, and shape of the nanoparticles are strongly linked to their toxicity. They mentioned that oxidative stress was a key route in inducing the cytotoxicity of nanoparticles.46 Therefore, there is a close association between the stability of nanomaterials and their future applications.47 Our results demonstrated that the variation of size and zeta potential of OX26@GNPs in different aqueous media was lower compared to bare nanoparticles, which was probably due to the hydrated PEG chains resulting in steric repulsion and subsequently a reduction in protein adsorption onto the surface of the nanoparticles. To explore the role of OX26@GNPs in cell fate during focal cerebral ischemia, we measured the infarcted region in different groups.
Our results showed that the intravenous injection of OX26@GNPs markedly enhanced infarct volume and neurological deficit scores through an increase in neuronal and axonal loss, neutrophil recruitment, myelin, and membrane damage. On the other hand, previous studies indicated that the chief cause of mortality following ischemic stroke is neuronal swelling and subsequent cytotoxic brain edema. The aberrant entry of sodium and chloride ions causes an osmotic imbalance, which is consistent with water entry and cytotoxic edema.48 A central question is whether the targeted delivery of OX26@GNPs to brain tissue after ischemia may affect neuronal swelling because of their detrimental effects on myelin sheaths and cell membranes.
To explore this question, we assessed brain edema by measuring brain water content for three different doses of OX26@GNPs (250, 500, and 1000 µg/mL). Analysis of this data demonstrated that the targeted delivery of OX26@GNPs to brain tissue seemed to increase the vulnerability of neurons to cell death following ischemia by damage to the membrane and alteration of Na+ and Cl− influx. It is worth noting that GNPs can be distributed to other organs such as the lungs, liver, and spleen. They have the ability to induce toxicity via mitochondrial damage and excessive production of intracellular ROS.49 Currently, numerous reports have shown that the behavior of inorganic NPs in biological systems, as well as their toxicity, is closely associated with their physicochemical properties including surface chemistry, chemical compositions, size, charge, surface coating, and functional moieties. Moreover, exposure routes and doses of inorganic NPs can strongly influence their behavior in biological systems. A previous study has reported that GNPs accumulate in the liver and their long-term administration might create noticeable changes in genes related to oxidative stress.50 No signs of damage were observed in other organs. We further went on to clarify the mechanism behind the increased neuronal loss and infarct size by OX26@GNPs via investigation of necroptosis cell death in the CA1 hippocampal region. After ischemia or reperfusion, glutamate release from astrocytes or dying neurons initiates a cascade of events including disruption of intracellular Ca2+ homeostasis, excessive generation of ROS, and mitochondrial cytochrome-c release which, in turn, promote activation of caspases.51,52
Aside from the above mentioned events, dead or injured neuronal cells activate microglia that indirectly leads to activation of the Fas/TNFR family of death-domain receptors (DRs).53 Generally, after injury or ischemia, DRs have the ability to trigger a pathological cascade and subsequently caspase-independent cell death in a RIPK1 related mechanism.54 Indeed, excessive generation of mitochondrial ROS and their sensing by three crucial cysteines in RIPK1 lead to autophosphorylation of this protein on the serine residue 161 (S161).55 To ascertain whether OX26@GNPs can exert detrimental effects via initiation of necroptosis, we measured the expression levels of RIPK1 in the hippocampus region of the ipsilateral hippocampus. Western blot analyses demonstrated that the expression of this protein was higher than the MCAO group following intravenous administration of OX26@GNPs. On the other hand, previous studies have shown that the assembly of RIPK1 and RIPK3 is a crucial step of necroptosis which, in turn, results in necrosome complex formation and recruitment of several molecules to facilitate the process.56 Indeed, RIPK1phosphorylation facilitates the recruitment of RIPK3 to RIPK1 and subsequent necrosome formation. After cerebral ischemia, apoptosis-inducing factor (AIF) is released from mitochondria and interacts with RIPK3 to create a RIPK3-AIF complex. The RIPK3-AIF complex could be translocated into the nucleus and initiate programmed necrosis of neuronal cells via chromatin condensation and DNA degradation.57 Alternatively, Ca2+-calmodulin-dependent protein kinase (CaMKII) can be activated by RIPK3 and trigger necroptosis via opening of the mitochondrial permeability transition pore.58 Western blot analyses also demonstrated that the expression of RIPK3 was higher than the MCAO group following intravenous administration of OX26@GNPs. MLKL is a molecule that can be recruited by the necrosome and its phosphorylation and trimerization which by RIPK3 results in its translocation to the plasma membrane and disruption of plasma membrane integrity in a transient receptor potential melastatin related 7 (TRPM7)- dependent mechanism.59
To confirm that OX26@GNPs trigger necroptosis in neuronal cells of the hippocampal region by targeting the RIPK1-RIPK3-MLKL axis, we also measured the expression of this protein. As mentioned above, our results showed a higher expression of this protein in the MCAO + OX26@GNPs group compared with other groups. Alternatively, a recent study demonstrated that the necroptosis by RIPK3 activation occurs following a cascade of events including agitation of the endoplasmic reticulum (ER) stress, increased levels of intracellular Ca2+ ions, overexpression of xanthine oxidase (XO), ROS overproduction, and subsequently the mitochondrial permeability transition pore (mPTP) opening through voltage-dependent anion channel-1(VDAC1) oligomerization and mitochondrial ATP-sensitive potassium channels.60,61
Persistent generation of ROS can overwhelm the endogenous antioxidant defense system via inactivation of antioxidant enzymes (such as SOD) and depletion of low-molecular-weight antioxidants.62 During oxidative stress, NO interacts with increased free radicals (such as the superoxide anion (O−2)) and subsequently causes peroxynitrite, a reactive species.63 After peroxynitrite formation, a cascade of events is commonly observed involving lipid peroxidation which is accompanied with increased MDA levels, protein oxidation, and an acute inflammatory response.64 To investigate whether necroptosis might be induced by oxidative stress in the CA1 hippocampal region, we measured oxidative stress markers in the experimental groups. Our results confirmed depletion of the intracellular antioxidant pool and increases in lipid peroxidation following intravenous administration of OX26@GNPs. Generally, our results demonstrated that the targeted delivery of GNPs to the brain under oxidative stress increased neuronal loss in a RIPK1-RIPK3-MLKL dependent mechanism (Figure 10).
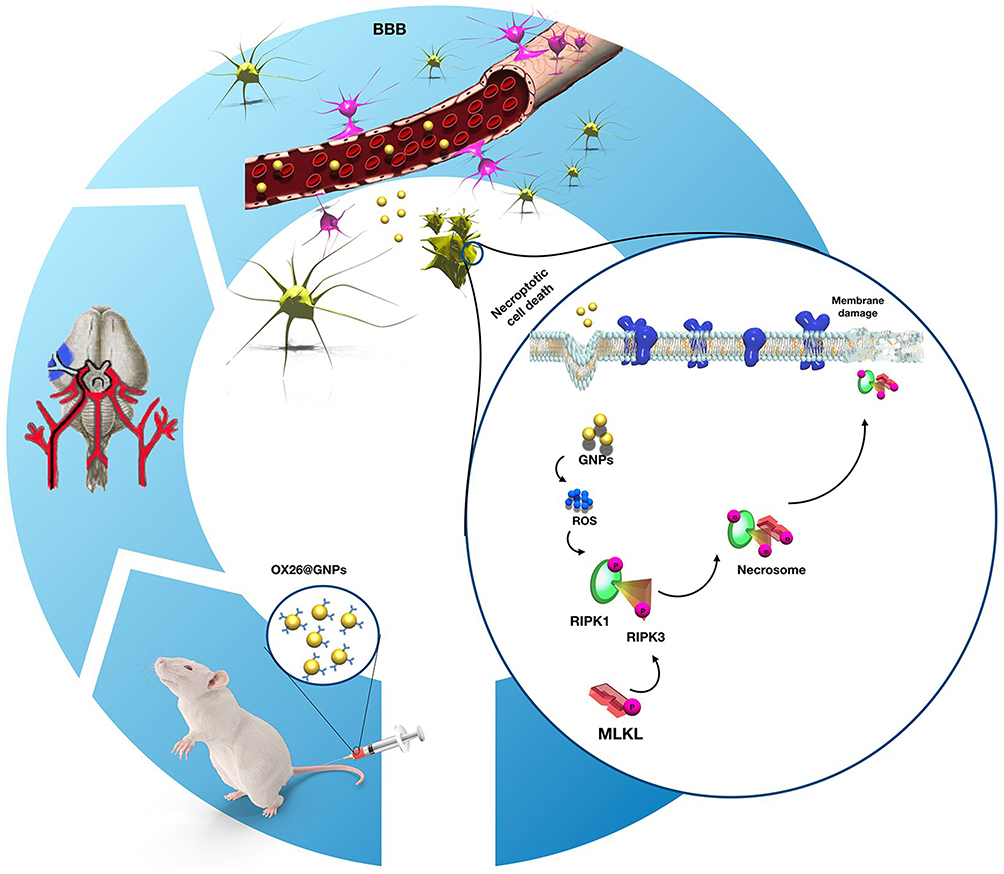 |
Figure 10 Proposed signaling pathways involved in OX26@GNPs-induced toxicity in a rat model of MCAO. |
Collectively, the results of this study suggested that the biological impacts of GNPs on neuronal cells under oxidative stress can be strongly linked to their surface charge. The GNPs as formulated here are not suitable nanocarriers for targeted drug delivery to the brain under oxidative stress conditions. Moreover, OX26@GNPs are not suitable contrast agents in X-ray imaging and computed tomography for enhanced detection of oxidative stress in the brain because they increase injury and neuronal loss through enhancement of necroptotic cell death and depletion of antioxidant defenses.
Acknowledgments
The authors would like to thank Dr. Amin Shiralizadeh Dezfuli for his help in the synthesis and conjugation of nanoparticles. This research was supported by a research grant from Iran University of Medical Sciences, Tehran, Iran (Number: 94-04-30-26504).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Chamorro Á, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016;15:869–881. doi:10.1016/S1474-4422(16)00114-9
2. Amani H, Habibey R, Shokri F, et al. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci Rep. 2019;9:6044. doi:10.1038/s41598-019-42633-9
3. Ajami M, Eghtesadi S, Razaz JM, et al. Expression of Bcl-2 and Bax after hippocampal ischemia in DHA + EPA treated rats. Neurol Sci. 2011;32:811. doi:10.1007/s10072-011-0621-5
4. Amani H, Kazerooni H, Hassanpoor H, Akbarzadeh A, Pazoki-Toroudi H. Tailoring synthetic polymeric biomaterials towards nerve tissue engineering: a review. Artif Cells Nanomed Biotechnol. 2019;47:3524–3539. doi:10.1080/21691401.2019.1639723
5. Gaudin A, Yemisci M, Eroglu H, et al. Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat Nanotechnol. 2014;9:1054. doi:10.1038/nnano.2014.274
6. Amani H, Mostafavi E, Arzaghi H, et al. Three-dimensional graphene foams: synthesis, properties, biocompatibility, biodegradability, and applications in tissue engineering. ACS Biomater Sci Eng. 2018. doi:10.1021/acsbiomaterials.8b00658
7. Zarch AV, Toroudi HP, Soleimani M, Bakhtiarian A, Katebi M, Djahanguiri B. Neuroprotective effects of diazoxide and its antagonism by glibenclamide in pyramidal neurons of rat hippocampus subjected to ischemia-reperfusion-induced injury. Int J Neurosci. 2009;119:1346–1361. doi:10.1080/00207450802338721
8. Rahgozar M, Pazokitoroudi H, Bakhtiarian A, Djahanguiri B. Diazoxide, a K(ATP) opener, accelerates restitution of ethanol or indomethacin-induced gastric ulceration in rats independent of polyamines. J Gastroenterol Hepatol. 2001;16:290–296. doi:10.1046/j.1440-1746.2001.02433.x
9. Gorjipour F, Dehaki MG, Totonchi Z, et al. Inflammatory cytokine response and cardiac troponin I changes in cardiopulmonary bypass using two cardioplegia solutions; del Nido and modified St. Thomas’: a randomized controlled trial. Perfusion. 2017;32:394–402. doi:10.1177/0267659117691119
10. Saraiva C, Praça C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: overcoming blood-brain barrier to treat neurodegenerative diseases. J Control Release. 2016;235:34–47. doi:10.1016/j.jconrel.2016.05.044
11. Moon SU, Kim J, Bokara KK, et al. Preparation of an antitumor and antivirus agent: chemical modification of α-MMC and MAP30 from Momordica Charantia L. with covalent conjugation of polyethyelene glycol. Int J Nanomedicine. 2012;7:2751. doi:10.2147/IJN.S30631
12. Mostafavi E, Soltantabar P, Webster TJ. Biomaterials in Translational Medicine. Elsevier; 2019:191–212.
13. Amani H, Habibey R, Hajmiresmail S, Latifi S, Pazoki-Toroudi H, Akhavan O. Antioxidant nanomaterials in advanced diagnoses and treatments of ischemia reperfusion injuries. J Mater Chem B. 2017;5:9452–9476. doi:10.1039/C7TB01689A
14. Jazayeri M, Amani H, Pourfatollah A, Avan A, Ferns G, Pazoki-Toroudi H. Enhanced detection sensitivity of prostate-specific antigen via PSA-conjugated gold nanoparticles based on localized surface plasmon resonance: GNP-coated anti-PSA/LSPR as a novel approach for the identification of prostate anomalies. Cancer Gene Ther. 2016;23:365. doi:10.1038/cgt.2016.42
15. Daraee H, Eatemadi A, Abbasi E, Fekri Aval S, Kouhi M, Akbarzadeh A. Application of gold nanoparticles in biomedical and drug delivery. Artif Cells Nanomed Biotechnol. 2016;44:410–422. doi:10.3109/21691401.2014.955107
16. Encabo-Berzosa MM, Sancho-Albero M, Crespo A, et al. The effect of PEGylated hollow gold nanoparticles on stem cell migration: potential application in tissue regeneration. Nanoscale. 2017;9(28):9848–9858.
17. Mehranfar S, Abdi Rad I, Mostafavi E, Akbarzadeh A. The use of stromal vascular fraction (SVF), platelet-rich plasma (PRP) and stem cells in the treatment of osteoarthritis: an overview of clinical trials. Artif Cells Nanomed Biotechnol. 2019;47:882–890. doi:10.1080/21691401.2019.1576710
18. Liu Z, Shen Y, Wu Y, et al. An intrinsic therapy of gold nanoparticles in focal cerebral ischemia-reperfusion injury in rats. J Biomed Nanotechnol. 2013;9:1017–1028.
19. Hyun H, Lee K, Min KH, et al. Ischemic brain imaging using fluorescent gold nanoprobes sensitive to reactive oxygen species. J Control Release. 2013;170:352–357. doi:10.1016/j.jconrel.2013.06.002
20. Kim J-Y, Ryu JH, Schellingerhout D, et al. Direct imaging of cerebral thromboemboli using computed tomography and fibrin-targeted gold nanoparticles. Theranostics. 2015;5:1098. doi:10.7150/thno.11679
21. Yoon H-J, Lee E-S, Kang M, Jeong Y, Park J-H. In vivo multi-photon luminescence imaging of cerebral vasculature and blood–brain barrier integrity using gold nanoparticles. J Mater Chem B. 2015;3:2935–2938. doi:10.1039/C4TB01759E
22. Jazayeri MH, Amani H, Pourfatollah AA, Pazoki-Toroudi H, Sedighimoghaddam B. Various methods of gold nanoparticles (GNPs) conjugation to antibodies. Sens Biosensing Res. 2016;9:17–22. doi:10.1016/j.sbsr.2016.04.002
23. Loureiro JA, Gomes B, Coelho MA, Carmo Pereira MD, Rocha S. Targeting nanoparticles across the blood-brain barrier with monoclonal antibodies. Nanomedicine. 2014;9:709–722. doi:10.2217/nnm.14.27
24. Singh D, Kapahi H, Rashid M, Prakash A, Majeed ABA, Mishra N. Recent prospective of surface engineered nanoparticles in the management of neurodegenerative disorders. Artif Cells Nanomed Biotechnol. 2016;44:780–791. doi:10.3109/21691401.2015.1029622
25. Monsalve Y, Tosi G, Ruozi B, et al. PEG-g-chitosan nanoparticles functionalized with the monoclonal antibody OX26 for brain drug targeting. Nanomedicine. 2015;10:1735–1750. doi:10.2217/nnm.15.29
26. Frens G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature. 1973;241:20–22.
27. He Z, Guo JL, McBride JD, et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. 2018;24:29. doi:10.1038/nm.4443
28. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi:10.1161/01.str.20.1.84
29. Chan FK-M, Moriwaki K, De Rosa MJ. Immune Homeostasis. Springer; 2013:65–70.
30. Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–169.
31. Sun Y, Oberley LW, Li Y. A simple method for clinical assay of superoxide dismutase. Clin Chem. 1988;34:497–500.
32. Aebi H.Methods of Enzymatic Analysis.
33. Gharegozloo S, Ataie A, Abdizadeh H, Mostafavi E, Parnian M, Khodadadi A. High performance Ni–cNTs catalyst: synthesis and characterization. RSC Adv. 2016;6:47072–47082. doi:10.1039/C6RA03052A
34. Farooq MU, Novosad V, Rozhkova EA, et al. Gold Nanoparticles-enabled efficient dual delivery of anticancer therapeutics to hela cells. Sci Rep. 2018;8:2907. doi:10.1038/s41598-018-21331-y
35. Duan X, Lu L, Wang Y, et al. The long-term fate of mesenchymal stem cells labeled with magnetic resonance imaging-visible polymersomes in cerebral ischemia. Int J Nanomedicine. 2017;12:6705. doi:10.2147/IJN
36. Shen J, Zhao Z, Shang W, et al. Ginsenoside Rg1 nanoparticle penetrating the blood-brain barrier to improve the cerebral function of diabetic rats complicated with cerebral infarction. Int J Nanomedicine. 2017;12:6477. doi:10.2147/IJN
37. Javedan G, Shidfar F, Davoodi SH, et al. Conjugated linoleic acid rat pretreatment reduces renal damage in ischemia/reperfusion injury: unraveling antiapoptotic mechanisms and regulation of phosphorylated mammalian target of rapamycin. Mol Nutr Food Res. 2016;60:2665–2677. doi:10.1002/mnfr.201600112
38. Mehrjerdi FZ, Aboutaleb N, Pazoki-Toroudi H, et al. The protective effect of remote renal preconditioning against hippocampal ischemia reperfusion injury: role of KATP channels. J Mol Neurosci. 2015;57:554–560. doi:10.1007/s12031-015-0636-0
39. Amani H, Ajami M, Maleki SN, et al. Targeting signal transducers and activators of transcription (STAT) in human cancer by dietary polyphenolic antioxidants. Biochimie. 2017;142:63–79. doi:10.1016/j.biochi.2017.08.007
40. Tejada S, Manayi A, Daglia M, et al. Wound healing effects of curcumin: a short review. Curr Pharm Biotechnol. 2016;17:1002–1007. doi:10.2174/1389201017666160721123109
41. Pan Y, Leifert A, Ruau D, et al. Gold nanoparticles of diameter 1.4 nm trigger necrosis by oxidative stress and mitochondrial damage. Small. 2009;5:2067–2076. doi:10.1002/smll.200900466
42. Cho EC, Xie J, Wurm PA, Xia Y. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009;9:1080–1084. doi:10.1021/nl803487r
43. Amani H, Arzaghi H, Bayandori M, et al. Controlling cell behavior through the design of biomaterial surfaces: a focus on surface modification techniques. Adv Mater Interfaces. 2019;6:1900572. doi:10.1002/admi.v6.13
44. Karakocak BB, Liang J, Biswas P, Ravi N. Controlling cell behavior through the design of biomaterial surfaces: a focus on surface modification techniques. Carbohydr Polym. 2018;186:243–251. doi:10.1016/j.carbpol.2018.01.046
45. Fröhlich EJIJON. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. doi:10.2147/IJN.S36111.
46. Yang H, Liu C, Yang D, Zhang H, Xi Z. Comparative study of cytotoxicity, oxidative stress and genotoxicity induced by four typical nanomaterials: the role of particle size, shape and composition. J Appl Toxicol. 2009;29:69–78. doi:10.1002/jat.1385
47. Soenen SJ, Parak WJ, Rejman J, Manshian B. (Intra)cellular stability of inorganic nanoparticles: effects on cytotoxicity, particle functionality, and biomedical applications. Chem Rev. 2015;115:2109–2135. doi:10.1021/cr400714j
48. Rungta RL, Choi HB, Tyson JR, et al. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. 2015;161:610–621. doi:10.1016/j.cell.2015.03.029
49. Wan J, Wang J-H, Liu T, Xie Z, Yu X-F, Li W. Surface chemistry but not aspect ratio mediates the biological toxicity of gold nanorods in vitro and in vivo. Sci Rep. 2015;5:srep11398. doi:10.1038/srep11398
50. Yang L, Kuang H, Zhang W, Aguilar ZP, Wei H, Xu H. Comparisons of the biodistribution and toxicological examinations after repeated intravenous administration of silver and gold nanoparticles in mice. Sci Rep. 2017;7:3303. doi:10.1038/s41598-017-03015-1
51. Ghadernezhad N, Khalaj L, Pazoki-Toroudi H, Mirmasoumi M, Ashabi G. Metformin pretreatment enhanced learning and memory in cerebral forebrain ischaemia: the role of the AMPK/BDNF/P70SK signalling pathway. Pharm Biol. 2016;54:2211–2219. doi:10.3109/13880209.2016.1150306
52. Pazoki‐Toroudi HR, Hesami A, Vahidi S, Sahebjam F, Seifi B, Djahanguiri B. The preventive effect of captopril or enalapril on reperfusion injury of the kidney of rats is independent of angiotensin II AT1 receptors. Fundam Clin Pharmacol. 2003;17:595–598. doi:10.1046/j.1472-8206.2003.00188.x
53. Cruz SA, Qin Z, Stewart AF, Chen -H-H. Natural polyphenols effects on protein aggregates in Alzheimer’s and Parkinson’s prion-like diseases. Neural Regen Res. 2018;13:252. doi:10.4103/1673-5374.233432
54. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311. doi:10.1038/nature14191
55. Zhang Y, Su SS, Zhao S, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi:10.1038/ncomms14329
56. Tian Z, Huang L, Pei X, et al. Electrochemical synthesis of three-dimensional porous reduced graphene oxide film: preparation and in vitro osteogenic activity evaluation. Colloids Surf B Biointerfaces. 2017;155:150–158. doi:10.1016/j.colsurfb.2017.04.012
57. Xu Y, Wang J, Song X, et al. RIP3 induces ischemic neuronal DNA degradation and programmed necrosis in rat via AIF. Sci Rep. 2016;6:29362. doi:10.1038/srep29362
58. Zhang T, Zhang Y, Cui M, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress–induced myocardial necroptosis. Nat Med. 2016;22:175–182. doi:10.1038/nm.4017
59. Cai Z, Jitkaew S, Zhao J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55. doi:10.1038/ncb2883
60. Zhou H, Zhang Y, Hu S, et al. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017;63(1). doi:10.1111/jpi.12413.
61. Zhu P, Hu S, Jin Q, et al. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi:10.1016/j.redox.2018.02.019
62. Münzel T, Gori T, Keaney JF
63. Habibey R, Pazoki‐Toroudi H. Morphine dependence protects rat kidney against ischaemia-reperfusion injury. Clin Exp Pharmacol Physiol. 2008;35:1209–1214. doi:10.1111/j.1440-1681.2008.04986.x
64. Lawson M, Jomova K, Poprac P, Kuča K, Musílek K, Valko M. Nutritional Antioxidant Therapies: Treatments and Perspectives. Springer; 2017:283–305.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.