")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Whole exome sequencing for cancer – is there evidence of clinical utility?
Authors Malhotra A, Levine S, Allingham-Hawkins D
Received 27 March 2014
Accepted for publication 13 May 2014
Published 22 August 2014 Volume 2014:4 Pages 115—128
DOI https://doi.org/10.2147/AGG.S58809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Diane Allingham-Hawkins.
Views: 348
Alka Malhotra, Susan Levine, Diane Allingham-Hawkins
Genetic Test Evaluation Program, Winifred S Hayes, Inc., Lansdale, PA, USA
Background: In recent years, whole exome sequencing (WES), which allows detection of 85% of disease-causing variants, has been used to compare tumor and normal DNA to allow the identification of variants specific to the tumor. Genetic changes in cancer are increasingly used for diagnosis and may guide treatment decisions. In this paper, we explore whether there is evidence that WES improves outcomes for patients with cancer.
Methods: Published evidence was evaluated using a methodology that combines the analytical validity, clinical validity, clinical utility and ethical, legal, and social implications (ACCE) model for genetic test evaluations with internationally accepted health technology assessment methodology. Conclusions were based on peer-reviewed published studies of >10 patients, with ≥3 studies for a given phenotype.
Results: WES has been conducted most extensively (seven studies to date) in breast cancer patients, with fewer studies of other types of cancers (eg, leukemia, prostate cancer, and ovarian cancer). Studies evaluating somatic alterations showed high intratumor and intertumor heterogeneity. In addition, both novel and previously implicated variants were identified. However, only three studies with >10 individuals have shown potential for clinical utility of WES; whereby, variants identified through WES may determine response to drug treatment.
Conclusion: Despite evidence for clinical validity of WES in cancers, clinical utility is very limited and needs to be further evaluated in large clinical studies.
Keywords: next-generation sequencing, exons, evidence-based, ACCE model, health technology assessment
Introduction
Cancers are caused by the accumulation of genetic alterations that may lead to the dysfunction of regulation of cell growth, resulting in the development of tumors.1 According to the American Cancer Society, it is estimated that >1.6 million individuals were diagnosed with cancer in the US in 2013 alone, with >580,000 resulting deaths. In the US, the most common new cases of cancers included breast cancer, prostate cancer, lung cancer, and colon cancer.2
In the past decade, there have been significant developments in next-generation technologies to sequence DNA rapidly.3,4 Massively parallel sequencing not only generates data from the entire genome in a short period of time, but it also contributes to cost reduction. Whole exome sequencing (WES) involves sequencing of all coding regions (exons) in the genome. Compared with whole genome sequencing (WGS), WES facilitates handling of data and also generates higher-quality data, since it can be performed at greater sequencing depth. The optimal sequencing depth depends on a number of factors, including, but not limited to: the region(s) being sequenced; the sample source; and the algorithm used to assemble and analyze the results. While exons represent just 1% of the genome, they account for approximately 85% of disease-causing variants, specifically for Mendelian traits;5 however, the percentage for complex traits is not known.6
In recent years, WES has been used to compare tumor DNA and normal DNA to identify variants specific to the tumor. WES of tumor DNA requires greater sequence depth than normal DNA to identify variants that are present in tumor cells only. These analyses may provide information about genes with driver variants (those with an effect on cancer development) versus passenger variants (those without an effect on cancer development).7 Through identification of potentially deleterious variants, WES may provide information about potential new avenues for diagnosis and treatment.
While clinical exome sequencing is being offered by a number of laboratories, WES specifically for cancer is currently offered by two Clinical Laboratory Improvement Amendments-certified laboratories in the US. The Baylor College of Medicine Medical Genetics Laboratories offers the Cancer Exome Sequencing test,8 and Personal Genome Diagnostics Inc., (Baltimore, MD, USA) offers the Cancer Complete™ test.9 Using proprietary methods, Personal Genome Diagnostics Inc. also conducts quality assessment and evaluation of genes and pathways of interest, identified through WES.9
This systematic review evaluates the available published evidence about the use of WES for cancer indications, with emphasis on analytical validity, clinical validity, and clinical utility.
Methods
Search strategy
Evidence evaluated for this report was obtained primarily from a search of the peer-reviewed literature in PubMed and Embase performed on June 3, 2013. Search terms included “exome” AND “sequencing” AND “cancer” AND (“diagnosis” OR “clinical” OR “utility” OR “validity”). Limits used were English language, human, and published since January 1, 1996. Additional relevant citations were also selected from the bibliographies of retrieved references.
Evidence evaluation
The evidence analysis used for this review is based on the ACCE model (http://www.cdc.gov/genomics/gtesting/ACCE/index.htm) that was developed by the Centers for Disease Control and Prevention.10,11 The ACCE model takes its name from an abbreviation of its main components: analytical validity; clinical validity; clinical utility; and ethical, legal, and social implications. The model is widely used to allow the performance of rapid evaluations of genetic tests.12
For this paper, the analytical validity is defined as the ability of WES technology to measure accurately and reliably the sequences of interest. Clinical validity is defined as the ability of WES to detect or predict the associated disorder or phenotype, while clinical utility focuses on what needs to be considered when evaluating the risks and benefits of introducing a genetic test into routine practice, and it is based on studies designed to investigate whether there are improvements in health outcomes as a result of using the genetic test in clinical practice. In this review, we focus on these components of the ACCE model, with less emphasis on the ethical, legal, and social implications.
Results/evidence overview
Analytical validity
Commercial laboratories offering WES mainly use the Agilent SureSelect All Exome (Agilent Technologies, Santa Clara, CA, USA) or NimbleGen Sequence Capture (SeqCap) Human Exome (Hoffmann-La Roche Ltd, Basel, Switzerland) platforms for exome capture. The analytical validity of these methods has been assessed by four studies comparing various platforms to detect variants through WES. Three studies utilized data from healthy individuals and one study evaluated data from cancer patients; however, none of the information provided was specific to cancer. In the analyses, various parameters were evaluated, including the following:
- Enrichment efficiency – proportion of base pair reads falling in the target region to the total base pair reads in any region of the genome.
- Genotype sensitivity – probability to accurately determine the genotypes in the target regions.
- Genotype concordance – accuracy of overlap of genotype calls between the current analysis and previously identified genotype information.
- Coverage – overlap with existing publicly available databases with protein-coding variant information, as well as detection percentage depending on the depth of sequencing.
Table 1 presents comparisons between the different exome enrichment platforms, some that are currently being used by commercial laboratories in the US. Two studies were conducted in individuals of Asian descent,13,14 and two studies were of white individuals.15,16 For all studies, a high genotype concordance (>98%) was estimated.13–16 However, overall coverage rates varied between studies and platforms, ranging from >80% in two studies15,16 to 59% (for Agilent SureSelect [Agilent Technologies]) in one study.13 In addition, where available, enrichment efficiency also varied among studies, ranging from 56.4%–89.6% for the Agilent SureSelect (Agilent Technologies) platform.13,15,16 This variation may be explained by the different depths of coverage used by each study.
 | Table 1 Comparison of exome enrichment platforms |
Clinical validity
The use of WES has been reported for a number of different cancers, with a majority of studies being either case reports or having sample sizes < ten individuals, which is too small to draw meaningful conclusions. This section describes clinical validity data obtained from at least three studies of the same or similar cancer type, with each of the studies including > ten individuals.
Breast cancer
Seven studies with > ten individuals evaluated the clinical validity of WES in breast cancer (summarized in Table 2). The overall proportion of missense variants in unrelated individuals was similar in different studies, ranging from 61.2%–65.4%.17–21 In addition, studies identified varying proportions of variants and candidate genes depending on the subtype; for example, the tumor protein p53 (TP53) gene had a high proportion of variants in luminal B individuals (P=0.04), while the mitogen-activated protein kinase kinase kinase 1 (MAP3K1) gene had a high proportion of variants in luminal A individuals (P=0.02).19 In all studies, both previously identified genes – as well as those not previously detected in breast cancer studies – were identified with a larger number of variants than expected by chance.17,19–23
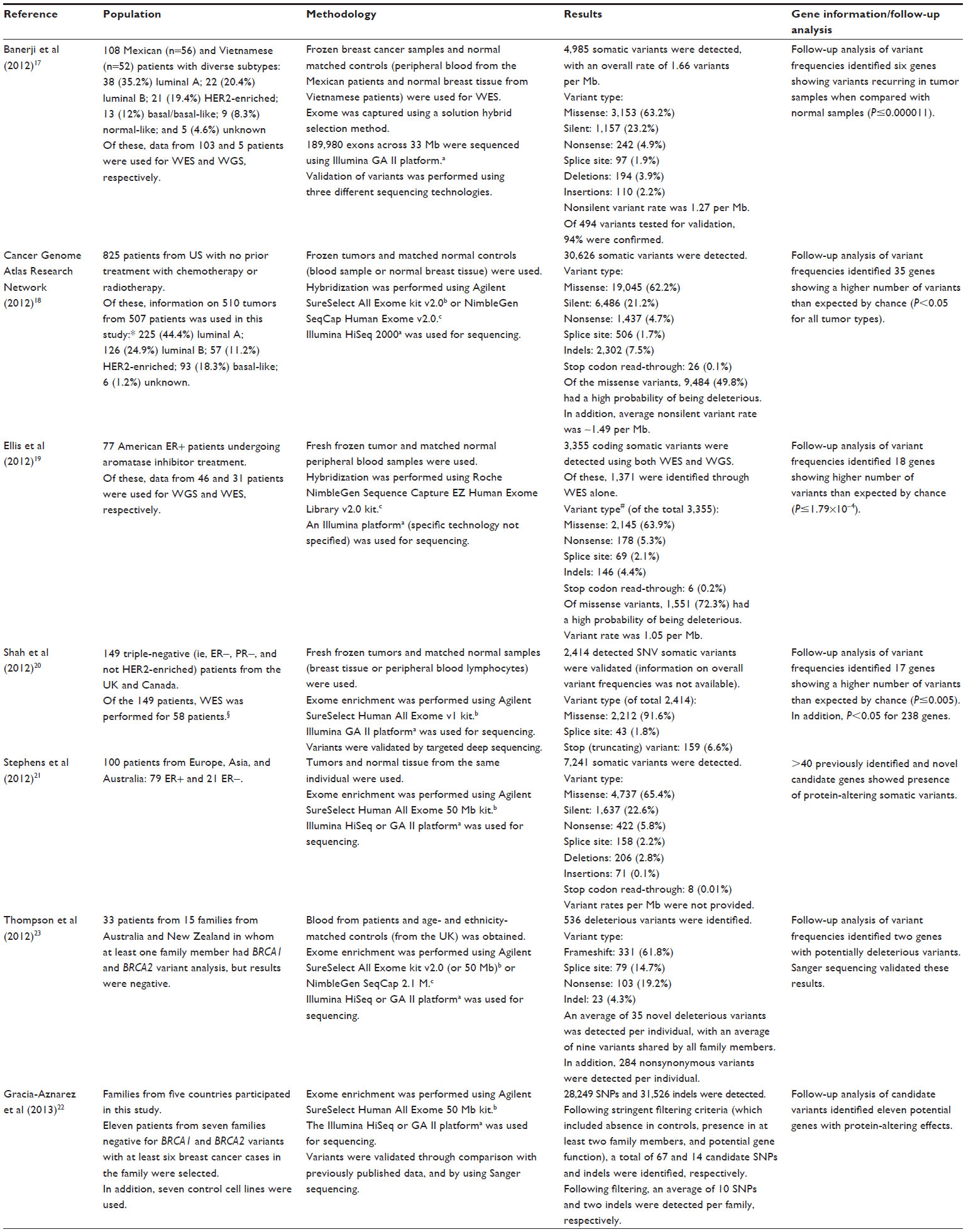 | Table 2 WES in breast cancer patients |
Two studies were specifically conducted in families that were negative for the breast cancer 1, early-onset (BRCA1) gene and the breast cancer 2, early-onset (BRCA2) gene.22,23 One study identified the Bloom syndrome, RecQ helicase-like (BLM) and Fanconi anemia, complementation group C (FANCC) genes with higher frequency of variants that may have an effect on breast cancer development;23 whereas, the second study identified eleven candidate genes.22
Leukemia and lymphoma
Table 3 presents results of WES in two types of leukemia – two studies for chronic lymphocytic leukemia (CLL)24,25 and one study for acute myeloid leukemia (AML).26 While all three studies identified a large proportion of genes with variants with potential deleterious effects, these genes did not overlap between AML and CLL.24–26 In the CLL studies, the splicing factor 3b, subunit 1 (SF3B1) gene was identified as having a higher rate of protein-altering variants. Specifically, CLL patients with variants in this gene showed a shorter time to disease progression (P=0.0001) and lower 10-year survival rate (P=0.002).24
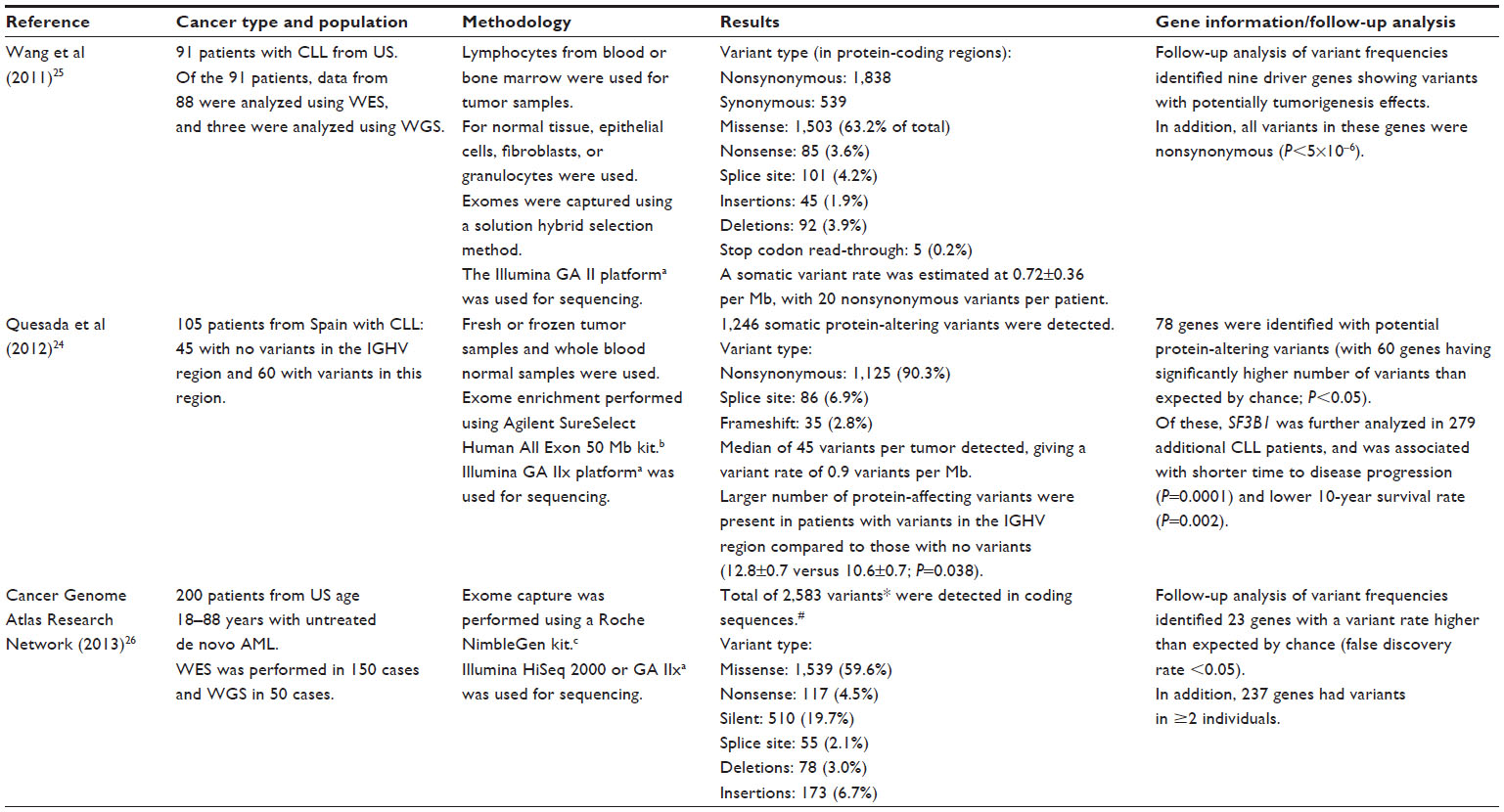 | Table 3 WES in leukemia patients |
In addition to the previously mentioned leukemia studies, two WES studies have also been conducted in a related cancer, diffuse large B-cell lymphoma (DLBCL), which affects lymph nodes.27,28
- A DLBCL WES study was conducted by Lohr et al27 (2012) using 55 frozen tumor samples and matched normal DNA from 55 US patients. Exomes were captured using a solution hybrid selection method followed by sequencing using the Illumina HiSeq platform (Illumina Inc., San Diego, CA, USA). Of the 55 tumors, six had very low variant rates and were, therefore, excluded from further analysis. In the remaining 49 samples, 6,233 total somatic variants were detected (missense, 4,093 [65.6%]; nonsense, 326 [5.2%]; silent, 1,549 [24.9%]; splice site, 123 [2.0%]; deletion, 98 [1.6%]; and insertion, 39 [0.6%]). A mean nonsynonymous variant rate was estimated at 3.2 per Mb (range, 0.6–7.8). Based on the WES results, 58 genes were identified as having a higher number of variants than expected (defined as a false discovery rate Q value <0.1; whereby, the analyses were corrected for multiple comparisons. In this study, the Q value indicated that <10% of significant tests were expected to result in a false-positive).27
- In a second study, as part of the Hematologic Malignancies Research Consortium, Zhang et al28 (2013) analyzed a total of 73 frozen biopsies from US patients with DLBCL. These were divided into a discovery set (34 tumors and 34 matched unaffected bone marrow samples) and a validation set (the remaining 39 tumors). In addition, exomes from 21 DLBCL cell lines were sequenced. Exome enrichment was performed using the Agilent SureSelect All Exome kit (Agilent Technologies), and the Illumina HiSeq platform (Illumina Inc.) was used for sequencing. Following a first step of filtering, a total of 5,884 somatic variants were detected in the discovery set. Somatic variant frequencies were: 53.8%, missense; 1.1%, nonsense; 2.4%, frameshift; and 42.7%, silent. Upon further filtering, which included the removal of silent variants, 2,589 (discovery set) and 4,928 variants (discovery and validation sets combined) were identified. Validation of variant detection was performed using three methods, resulting in 99% concordance in all cases. The follow-up of the WES results identified 322 strong candidate genes with recurrent variants in the DLBCL patients.28
Endometrial and ovarian cancer
Table 4 presents findings of four WES studies in endometrial and ovarian cancers, with the largest study performed by the Cancer Genome Atlas Research Network using 316 tumor samples. Three of the four studies showed the presence of different types of variants, including the missense, nonsense, splice site, deletions, and insertions.29–31
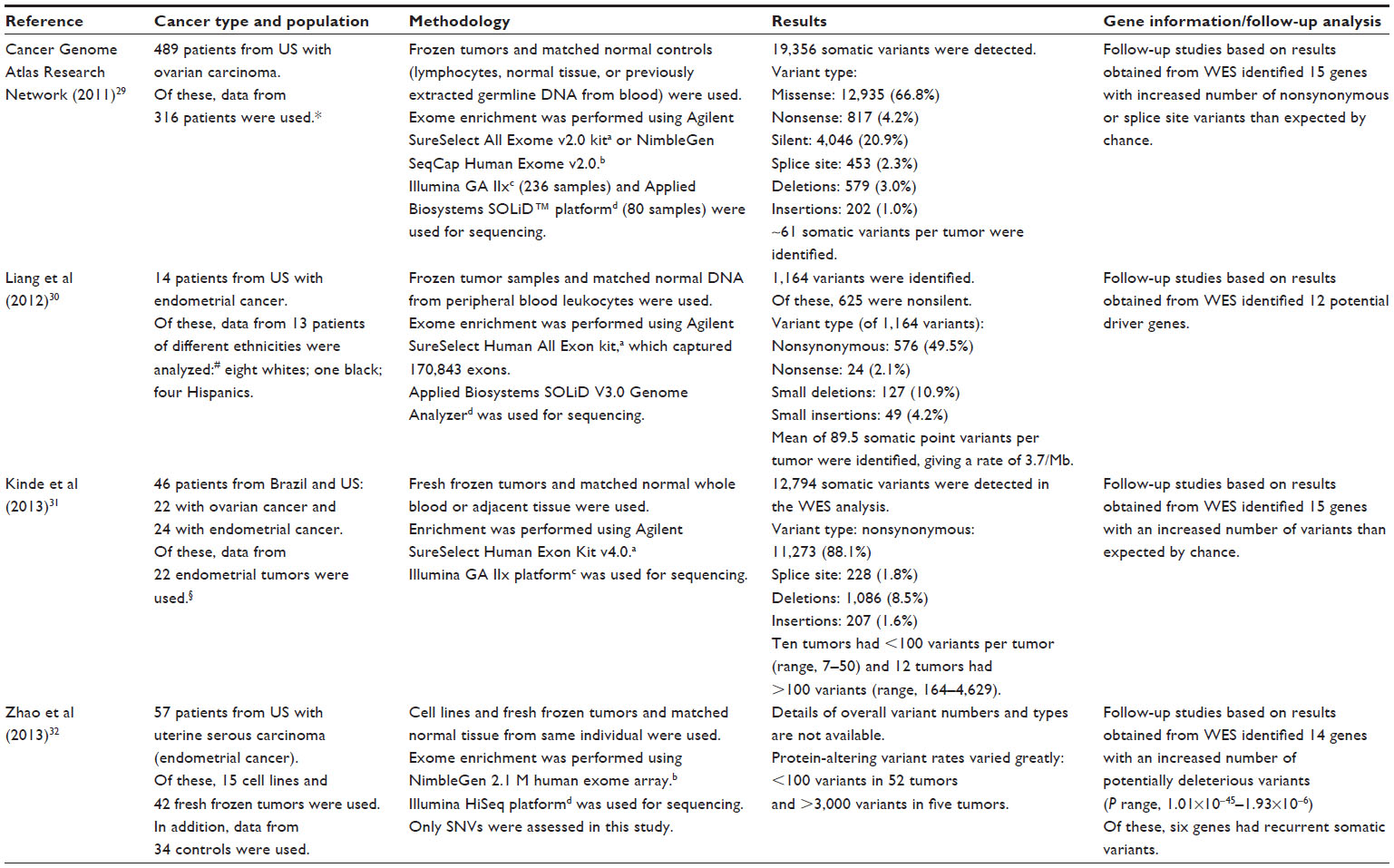 | Table 4 WES in endometrial or ovarian cancer patients |
The number of variants per tumor varied greatly (some tumors with <100 variants and others having >3,000 variants), showing the high degree of genetic heterogeneity both within and among tumors.31,32 In addition, the WES analyses resulted in the identification of nine to 15 genes per study, with more variants (including potentially deleterious variants in some studies) than would be expected by chance.29–32
Lung-related cancers
WES studies in lung cancers have been conducted for non-small-cell lung carcinoma (two studies)33,34 and lung adenocarcinoma (one study).35 These studies are described in Table 5. All the studies presented in this table used the Agilent SureSelect platform for exome enrichment, followed by the use of Illumina platforms (either Genome Analyzer II or HiSeq [Illumina Inc.]) for sequencing. The two non-small cell-lung carcinoma studies showed differences in the variant type, based on the subtype of the cancer;33,34 for example, the ratio of nonsynonymous to synonymous variants was nominally statistically significantly higher in adenocarcinoma patients compared with squamous cell carcinoma patients (3.6 versus 2.8; P=0.05).33 Follow-up of potential candidate genes identified >15 genes in all studies,33–35 with some genes having a highly significant proportion of variants (P range; 2×10−14–7×10−4).33
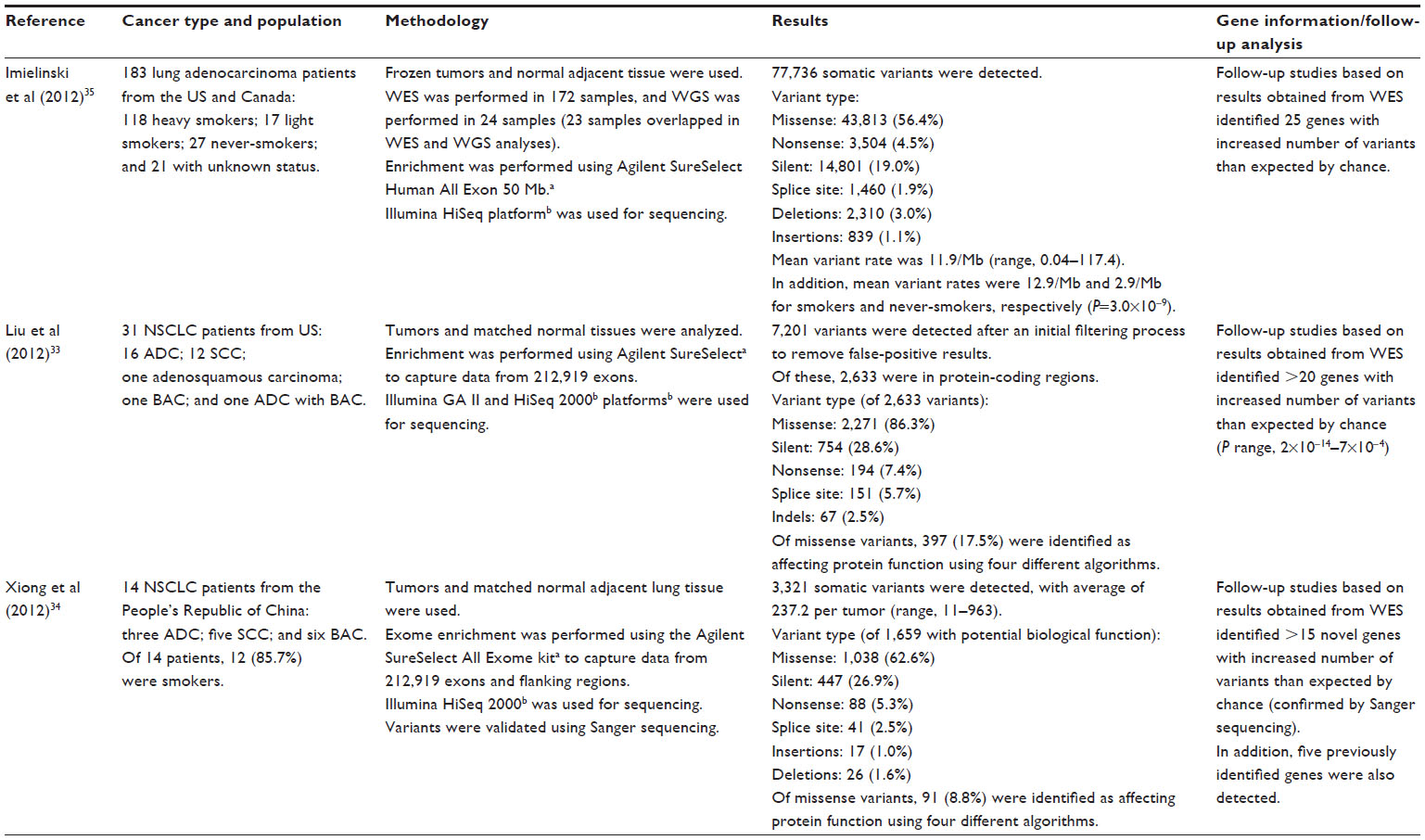 | Table 5 WES in lung-related cancer patients |
WES in other types of cancer
WES studies have been conducted in other types of cancers, including colon and colorectal cancer,36,37 prostate cancer,38,39 and head and neck squamous cell carcinoma.40,41 Since the methodology and criteria used to evaluate the results are similar to those described previously (ie, include assessment of the genotype landscape and, in some cases, identification of potential candidate genes to be further evaluated), these studies are not described in this review.
Clinical utility
Three studies evaluating treatment outcomes based on the results obtained through WES have suggested potential for clinical utility.19,42,43
- In one of the breast cancer studies described in Table 2, Ellis et al19 (2012) evaluated the correlation between variants in specific genes (identified through WES and WGS) and response to aromatase inhibitors in patients with estrogen receptor-positive breast cancer. The protein Ki-67 is a marker of tumor proliferation, and high levels of this protein are associated with a poor prognosis even after aromatase inhibitor treatment. Genetic variants were analyzed at baseline and postsurgery in relation to Ki-67 levels. One gene, GATA-binding protein 3 (GATA3), showed no correlation at baseline, but showed enrichment of variants in samples showing a decline in Ki-67 levels (P=0.01) postsurgery. These results suggest that the presence of GATA3 variants may be a predictive marker to identify individuals who will respond to treatment with aromatase inhibitors.19
- A second study by Sohn et al42 (2012) evaluated WES data from 174 patients with ovarian cancer who were part of the Cancer Genome Atlas study described in the “Clinical validity” section. In patients with high-grade serous ovarian cancer, the standard treatment is surgery followed by platinum-based chemotherapy. However, platinum resistance is observed in approximately 30% patients. Details of the study are:42
- All patients had platinum response data and WES results available. From the WES results, individuals were classified into two groups, based on the number of variants detected: hypervariant (>61.9 variants per patient); or hypovariant (<61.9 per patient). In addition, platinum status after receiving prior platinum treatment was determined: platinum-sensitive (no platinum treatment for ≥6 months with no disease progression or recurrence); or platinum-resistant (<6 months to disease progression or recurrence).
- Of the 174 patients, 122 (70.1%) and 52 (29.9%) were classified as platinum-sensitive and platinum-resistant, respectively. The number of variants was statistically significantly different between the two groups (mean number of variants of 67.2±38.9 and 49.4±25.2 for the sensitive and resistant groups, respectively; P<0.001). In addition, a multivariate logistic regression analysis identified somatic hypervariant as an independent risk factor for determining platinum response (odds ratio =3.616; 95% CI, 1.595–8.200; P=0.002).
- Hypervariant status was also statistically significantly associated with longer overall survival (P<0.001) and progression-free survival (P=0.001).
- Finally, a third study evaluated WES in patients with acute lymphoblastic leukemia (ALL). ALL patients who experience remission following aggressive chemotherapy, but then relapse, are generally treated with chemotherapy again. However, 40% of these patients do not benefit from this subsequent chemotherapy treatment. Tzoneva et al43 (2013) performed WES in five pediatric patients who had prior chemotherapy, remission, and relapsed ALL. Details of the study are:43
- Variants were detected using the Agilent SureSelect kit (Agilent Technologies) for exome capture, and Illumina HiSeq 2000 (Illumina Inc.) for sequencing.
- The WES analysis identified variants in the 5′-nucleotidase, cytosolic II (NT5C2) gene. Follow-up analysis of this gene in 133 individuals with relapsed ALL identified 23 additional variants in this gene. Nine individuals with available DNA at time of diagnosis showed the absence of NT5C2 variants at diagnosis, but these were acquired during relapse. In addition, individuals with NT5C2 showed association with relapse even when receiving treatment (P=0.002), and early relapse compared to late relapse (P<0.05).
- The presence of NT5C2 variants in relapsed patients suggests that this may be a potential marker to identify individuals at increased risk for relapse, despite chemotherapy treatment.
Discussion and conclusion
An increasing number of WES studies have been performed in cancer patients in recent years, comparing tumor tissue to normal tissues. WES allows simultaneous detection of variants in protein-coding regions across the genome with a single assay instead of performing a large number of single tests to identify potential genetic contributors to the cancer being analyzed. WES also allows exploration of the heterogeneous nature of cancers. In this review, we evaluated the analytical validity, clinical validity, and clinical utility of WES for cancer indications.
Analytical validity evidence was provided by four studies – three studies in healthy individuals and one in cancer patients.13–16 A number of limitations are noted. Two studies used data from only one individual, which limits statistical power.13,15 In addition, only one study analyzed data collected from cancer patients; however, cancer status was not specifically accounted for in the analyses.14 The tissue types evaluated in cancer may be limited with respect to the quantity, preparation (for example, fresh frozen), and heterogeneity of clinically significant variants. In addition, the very wide spectrum of variants present in cancers may increase the complexity of data analysis required. Therefore, more studies that specifically address the analytical issues of WES in cancer specimens need to be conducted.
Furthermore, the exome comprises only about 1% of the genome and variants in noncoding regions, and other DNA alterations (including genomic rearrangements and copy number variations) will not be detected. Therefore, there is a possibility of missing clinically important variants. In addition, WES does not provide functional information about variants; consequently, results need to be followed up to determine the biological function of identified variants. Finally, WES relies on bioinformatic algorithms to infer variant information and the selection of genes for further analysis. Given that such algorithms are still being developed, current methods may be limited with respect to the optimal variant (and gene) selection.
Clinical validity studies have been mostly limited to assessment of genetic landscape information; whereby, details about the types of variants (including missense, nonsense, silent, insertions, and deletions) are provided. Follow-up of the WES results to identify genes with potential roles in cancer development have rarely been conducted, with no further assessment in most cases. Additional studies using adequate sample sizes and with follow-up of identified variants to assess their functional roles are therefore needed. Such analyses may lead to a better understanding of diagnostic and prognostic factors, and potential treatment avenues for cancer patients.
While no study provided direct evidence of clinical utility, three studies suggest the potential for clinical utility. Follow-up analyses showed enrichment of GATA3 variants (identified by WES) in samples showing a decline in Ki-67 levels, which is a marker for response to aromatase inhibitor treatment. This association suggests that presence of GATA3 variants may be a predictive marker to identify individuals who will respond to treatment with aromatase inhibitors.19 A second study showed that somatic hypervariation detected through WES not only was a predictive factor for determining platinum-based chemotherapy response in ovarian cancer treatment, but it was also statistically significantly associated with longer overall survival and progression-free survival.42 Lastly, a WES study identified NT5C2 variants that were associated with ALL relapse even when patients were receiving treatment, suggesting that NT5C2 may be a potential marker to identify individuals who may experience ALL relapse despite chemotherapy treatment.43 While these three studies suggest potential for clinical utility for WES in cancer, there are no studies that specifically demonstrate improvements in patient outcomes.
To assess the potential impact of studies published after June 2013, an updated search of the literature was conducted in March 2014. Although additional studies have been published since June 2013, these represented further genetic landscape evaluation with no additional evidence for clinical utility.
WES results may have ethical implications. One ethical issue that may arise with WES studies is the identification of an incidental finding with clinical significance, ie, a variant with a known disease-causing effect, but for a different condition than that being studied. This information may affect not only a patient, but it may have implications for the patient’s family members. The methods of providing such information to patients are not entirely clear, although some recommendations have recently been made by the American College of Medical Genetics and Genomics.44 In addition, given the limited number of follow-up studies (and small sample sizes) addressing treatment outcomes, there are potential safety concerns if a treatment decision is based solely on WES results.
In conclusion, while there is evidence for clinical validity, mainly for breast cancer, clinical utility evidence for WES is lacking for all types of cancer. Therefore, WES has not yet been proven to improve outcomes for patients with cancer.
Acknowledgments
The authors would like to acknowledge the contributions of the members of the Hayes Genetic Test Evaluation team, particularly Lisa Spock, Linnie Wieselquist, and Charlotte Kuo-Benitez. Funding for the Health Technology Assessment that informed this work was provided by Winifred S Hayes, Inc.
Disclosure
The authors are employees at Winifred S Hayes, Inc., an independent health technology research and consulting company. None of the employees at this company has any financial or personal interest in any of the technologies reviewed by Winifred S Hayes, Inc. No input on report content or conclusions is permitted by manufacturers. The authors report no other conflicts of interest in this work.
References
Shyr D, Liu Q. Next generation sequencing in cancer research and clinical application. Biol Proced Online. 2013;15(1):4. | |
Cancer.org [homepage on the Internet]. Cancer Facts and Figures 2013. Atlanta: American Cancer Society; 2014. Available from: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-036845.pdf. Accessed March 19, 2014. | |
Bick D, Dimmock D. Whole exome and whole genome sequencing. Curr Opin Pediatr. 2011;23(6):594–600. | |
Yu Y, Wu BL, Wu J, Shen Y. Exome and whole-genome sequencing as clinical tests: a transformative practice in molecular diagnostics. Clin Chem. 2012;58(11):1507–1509. | |
Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–755. | |
Chakravarti A. Population genetics – making sense out of sequence. Nat Genet. 1999;21(Suppl 1):56–60. | |
Youn A, Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011;27(2):175–181. | |
BCM.edu [homepage on the Internet]. Cancer Exome Sequencing. Houston: Baylor College of Medicine Medical Genetics Laboratories; 2014. Available from: https://www.bcm.edu/geneticlabs/test_detail.cfm?testcode=9610. Accessed March 19, 2014. | |
Personalgenome.com [homepage on the Internet]. Personal Genome Diagnostics; CancerComplete. Baltimore: Personal Genome Diagnostics; 2014. Available from: http://personalgenome.com/cancercomplete. Accessed March 19, 2014. | |
Teutsch SM, Bradley LA, Palomaki GE, et al; EGAPP Working Group. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genet Med. 2009;11(1):3–14. | |
Veenstra DL, Piper M, Haddow JE, et al. Improving the efficiency and relevance of evidence-based recommendations in the era of whole-genome sequencing: an EGAPP methods update. Genet Med. 2013;15(1):14–24. | |
Gudgeon JM, McClain MR, Palomaki GE, Williams MS. Rapid ACCE: experience with a rapid and structured approach for evaluating gene-based testing. Genet Med. 2007;9(7):473–478. | |
Asan, Xu Y, Jiang H, et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011;12(9):R95. | |
Guo Y, Long J, He J, et al. Exome sequencing generates high quality data in non-target regions. BMC Genomics. 2012;13:194. | |
Clark MJ, Chen R, Lam HY, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29(10):908–914. | |
Hedges DJ, Guettouche T, Yang S, et al. Comparison of three targeted enrichment strategies on the SOLiD sequencing platform. PLoS One. 2011;6(4):e18595. | |
Banerji S, Cibulskis K, Rangel-Escareno C, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–409. | |
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Ellis M, Ding L, Shen D, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486(7403):353–360. | |
Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486(7403):395–399. | |
Stephens PJ, Tarpey PS, Davies H, et al; Oslo Breast Cancer Consortium (OSBREAC). The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. | |
Gracia-Aznarez FJ, Fernandez V, Pita G, et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS One. 2013;8(2):e55681. | |
Thompson ER, Doyle MA, Ryland GL, et al; kConFab. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8(9):e1002894. | |
Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47–52. | |
Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. | |
Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. | |
Lohr JG, Stojanov P, Lawrence MS, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879–3884. | |
Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1398–1403. | |
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | |
Liang H, Cheung LW, Li J, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22(11):2120–2129. | |
Kinde I, Bettegowda C, Wang Y, et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci Transl Med. 2013;5(167):167ra4. | |
Zhao S, Choi M, Overton JD, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110(8):2916–2921. | |
Liu P, Morrison C, Wang L, et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis. 2012;33(7):1270–1276. | |
Xiong D, Li G, Li K, et al. Exome sequencing identifies MXRA5 as a novel cancer gene frequently mutated in non-small cell lung carcinoma from Chinese patients. Carcinogenesis. 2012;33(9):1797–1805. | |
Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. | |
Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–664. | |
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. | |
Lindberg J, Mills IG, Klevebring D, et al. The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur Urol. 2013;63(4):702–708. | |
Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. | |
Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. | |
Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–1157. | |
Sohn I, Jung WY, Sung CO. Somatic hypermutation and outcomes of platinum based chemotherapy in patients with high grade serous ovarian cancer. Gynecol Oncol. 2012;126(1):103–108. | |
Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19(3):368–371. | |
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. Available from: http://www.acmg.net/docs/ACMG_Releases_Highly-Anticipated_Recommendations_on_Incidental_Findings_in_Clinical_Exome_and_Genome_Sequencing.pdf. Accessed March 19, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.