")
Back to Journals » International Journal of Nanomedicine » Volume 12
Wheat germ agglutinin-conjugated liposomes incorporated with cardiolipin to improve neuronal survival in Alzheimer’s disease treatment
Authors Kuo YC, Lin CY, Li JS, Lou YI
Received 22 November 2016
Accepted for publication 31 January 2017
Published 2 March 2017 Volume 2017:12 Pages 1757—1774
DOI https://doi.org/10.2147/IJN.S128396
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Yung-Chih Kuo,1 Che-Yu Lin,1 Jay-Shake Li,2 Yung-I Lou3
1Department of Chemical Engineering, 2Department of Psychology, National Chung Cheng University, Chia-Yi, 3Department of Accounting, Providence University, Taichung, Taiwan, Republic of China
Abstract: Curcumin (CRM) and nerve growth factor (NGF) were entrapped in liposomes (LIP) with surface wheat germ agglutinin (WGA) to downregulate the phosphorylation of kinases in Alzheimer’s disease (AD) therapy. Cardiolipin (CL)-conjugated LIP carrying CRM (CRM-CL/LIP) and also carrying NGF (NGF-CL/LIP) were used with AD models of SK-N-MC cells and Wistar rats after an insult with β-amyloid peptide (Aβ). We found that CRM-CL/LIP inhibited the expression of phosphorylated p38 (p-p38), phosphorylated c-Jun N-terminal kinase (p-JNK), and p-tau protein at serine 202 and prevented neurodegeneration of SK-N-MC cells. In addition, NGF-CL/LIP could enhance the quantities of p-neurotrophic tyrosine kinase receptor type 1 and p-extracellular signal-regulated kinase 5 for neuronal rescue. Moreover, WGA-grafted CRM-CL/LIP and WGA-grafted NGF-CL/LIP significantly improved the permeation of CRM and NGF across the blood–brain barrier, reduced Aβ plaque deposition and the malondialdehyde level, and increased the percentage of normal neurons and cholinergic activity in the hippocampus of AD rats. Based on the marker expressions and in vivo evidence, current LIP carriers can be promising drug delivery systems to protect nervous tissue against Aβ-induced apoptosis in the brain during the clinical management of AD.
Keywords: liposome, Alzheimer’s disease, β-amyloid, neurodegeneration, blood–brain barrier, wheat germ agglutinin
Introduction
Alzheimer’s disease (AD) is a commonly encountered neurodegenerative disorder of the brain with progressive memory impairment, loss of cognitive function, and personality change, leading to irreversible dementia.1,2 The AD pathology originates from the temporal lobes and spreads gradually over the hippocampus and amygdala of the partial lobes, revealing its main characteristics of abnormal aggregation of β-amyloid peptide (Aβ), abundant senile plaques around neurons, and neurofibrillary tangles through extensive phosphorylation of tau protein.3–6 In the neurodegeneration pathway, mitogen-activated protein kinase (MAPK) is a typical enzyme family comprising various kinases, which are capable of conjugating covalently with serine, threonine, and tyrosine by phosphate linkage.7 In fact, MAPK-catalyzed phosphorylation plays a central role in regulating the function of protein kinase, phospholipase, transcription factor, and cytoskeleton via the capture of phosphoryl groups.8 This phosphorylation is also regarded as one of the most crucial stages during the alteration of cellular activity, including gene expression, mitosis, metabolism, and apoptosis.9 Thus, an understanding of the kinase pathway is critical to disclosing AD progression and can be of great help in developing an innovative medicinal strategy to avoid AD deterioration.
Liposomes (LIP) have excellent biocompatibility and a high degree of similarity to cell membrane in composition and structure.10 LIP can entrap hydrophobic and hydrophilic drugs for sustained release and facilitate pharmaceutical bioavailability. For example, the liposomal bilayer can encapsulate hydrophobic drugs such as curcumin (CRM), while hydrophilic nerve growth factor (NGF) can be confined to the aqueous core of LIP. CRM, a yellow polyphenol with antioxidative and anti-inflammatory benefits, can affect the expression of phosphorylated p38 (p-p38) and phosphorylated c-Jun N-terminal kinase (p-JNK), 2 essential MAPK members.11,12 Since hydrophobic CRM has low solubility in water and high degradability in basic medium, encapsulation of CRM in drug carriers can enhance its bioavailability.13,14 In addition, NGF can promote the ability of neurotrophic tyrosine kinase receptor type 1 (TrkA) to retard neuronal apoptosis.15,16 However, maintaining an effective level of NGF using a dose of free drug is not easy. A delivery system for sustained release of NGF can stabilize NGF and prolong its function.17 Moreover, to dock Aβ for improved neuroprotection, CRM and NGF may be loaded in LIP-containing cardiolipin (CL).18 This is due to a strong affinity of CL to Aβ.19 Thus, inclusion of CL may enhance the ability of LIP to target Aβ around apoptotic neurons. It was also reported that incorporation of wheat germ agglutinin (WGA) could support the targeted delivery of nanoparticles across the blood–brain barrier (BBB).20 WGA-modified nanoparticles could also be delivered to the brain after intranasal administration via the nose–brain transport pathway.21 Since the pathogenesis of AD is in the central nervous system (CNS), the use of CL-conjugated LIP (CL/LIP) modified with WGA (WGA-CL/LIP) may benefit AD management.
The aim of this study was to unveil the phosphorylation during AD treatment with CL/LIP loaded with CRM (CRM-CL/LIP) and CL/LIP loaded with NGF (NGF-CL/LIP). Although the neurodegeneration actuated by AD pathology has been widely investigated, the influence of CL/LIP on related kinase expressions remains undetermined. Hence, an understanding of the effect of CL/LIP carrying antiapoptotic agents on the pathological pathway can be advantageous to AD therapy. In this study, the effect of WGA-CL/LIP with a BBB-targeting capacity on AD pathology in vivo was also investigated. An AD model of Wistar rats was established by an injection of Aβ into the cornu ammonis area 1 (CA1) and was used to assess plaque formation, degeneration of Nissl bodies, acetylcholinesterase (AChE) activity, and lipid peroxidation (LPO) in the brain.
Materials and methods
Materials
Cholesterol (CHOL), CRM, soybean phosphatidylcholine (SPC), fluorescein isothiocyanate-conjugated dextran 70000 (FITC-D[70000]), Dulbecco’s phosphate-buffered saline (DPBS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), sodium azide, propidium iodide (PI), gelatin, formalin, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), sodium dodecyl sulfate (SDS), polyacrylamide gel (PAG), hydrogen peroxide (30%), 4′,6-diamidino-2-phenylindole (DAPI), thionin, and acetic acid were purchased from Sigma-Aldrich (St Louis, MO, USA). 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1′,3′-bis[1,2-dimyristoyl-sn-glycero-3-phospho]-sn-glycerol (CL), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG[2000]), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG[2000]-CA), and polycarbonate membrane were obtained from Avanti Polar Lipids (Alabaster, AL, USA). The Coomassie (Bradford) protein assay kit, Halt protease inhibitor cocktail, and tissue protein extraction reagent were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Chloroform, acetone, tetrahydrofuran, and dimethyl sulfoxide (DMSO) were obtained from JT Baker (Phillipsburg, NJ, USA). Human Aβ1–42 powders and SK-N-MC cell culture medium were purchased from Life Technologies (Carlsbad, CA, USA). The human NGF kit, lysis buffer, anti-JNK (phospho-T183&Y185) (anti-p-JNK), goat anti-rabbit immunoglobulin G (IgG) (heavy and light, horseradish peroxidase [H&L, HRP]) with rhodamine conjugate, anti-Aβ monoclonal antibody, malondialdehyde (MDA) assay kit (lysis buffer, butylated hydroxytoluene [BHT], thiobarbituric acid [TBA], standard), and AChE assay kit (lysis buffer, acetylthiocholine [AThCh], and bovine serum albumin [BSA], standard) were obtained from Abcam (Cambridge, MA, USA). Sample buffer, anti-TrkA (phospho) (anti-p-TrkA), 3,3′-diaminobenzidine (DAB) substrate, and Entellan® new mounting medium were purchased from Millipore (Billerica, MA, USA). Tris-buffered saline with Tween 20 (TBST), anti-GAPDH, anti-p38(phospho-Thr180&Tyr185) (anti-p-p38), anti-tau(phospho-serine 202) (anti-p-Ser202), and antiextracellular signal-regulated kinase 5 (phospho-Thr218&Tyr220) (anti-p-ERK5) were obtained from Cell Signaling (Danvers, MA, USA). Sephadex G-100, polyvinylidene fluoride (PVDF), enhanced chemiluminescence (ECL), and select ECL were purchased from GE Healthcare (Buckinghamshire, England). Tris-hydroxymethyl aminomethane (tris) was obtained from Riedel-de Haen (Seelze, Germany), Triton-X-100 from Acros (Morris, NJ, USA), methanol from Mallinckrodt Baker (Phillipsburg, NJ, USA), ultrapure water from Barnstead (Dubuque, IA, USA), N-hydroxysuccinimide (NHS) from Alfa Aesar (Ward Hill, MA, USA), WGA from Medicago AB (Uppsala, Sweden), human β-nerve growth factor (hβ-NGF) from Alomone Labs (Jerusalem, Israel), membrane tube (regenerated cellulose for dialysis, 50 kDa) from Spectrum Laboratories (Rancho Dominguez, CA, USA), citric acid from Showa (Tokyo, Japan), human brain-microvascular endothelial cells (HBMECs) from Biocompare (South San Francisco, CA, USA), human astrocytes (HAs) from Sciencell (Carlsbad, CA, USA), 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) from Biological Industries (Beit Haemek, Israel), aqueous mounting medium from Bio SB (Santa Barbara, CA, USA), o-xylene from Fluka (Buchs, Switzerland), ethanol (99.8%) from Tedia (Fairfield, OH, USA), SK-N-MC cells from American Type Tissue Collection (Rockville, MD, USA), transfer buffer from Bio-Rad (Hercules, CA, USA), casein-based blocking reagent (CAS block) from Invitrogen (Waltham, MA, USA), male Wistar rats from BioLasco (Taipei, Taiwan), atropin from Hebei Depond Animal Health Technology (Hebei, China), sodium pentobarbital from Siegfried (Taoyuan, Taiwan), and tissue-tek optimal cutting temperature (OCT) compound from Sakura Finetek (Torrance, CA, USA).
Preparation of CRM-CL/LIP
The mole percentages of CHOL, DPPC, and SPC in the lipids were, respectively, 50%, 25%, and 25% for LIP loaded with CRM (CRM-LIP). The mole percentages of CL, CHOL, DPPC, and SPC in the lipids were, respectively, 20%, 40%, 20%, and 20% for CRM-CL/LIP. The lipids were mixed with an additional 3% (mol) DSPE-PEG(2000) and 2% (mol) CRM. To prepare fluorescent LIP, 0.1% (w/v) FITC-D(70000) was added to the mixture. The mixture was dissolved in chloroform and condensed using a rotary evaporator (Panchum, Kaohsiung, Taiwan). The deposited solid thin film was hydrated with DPBS and compressed through a polycarbonate membrane containing pores 100 nm in size using an extruder set (Avanti Polar Lipids). The effluents of CRM-CL/LIP were added to methanol at 37°C for 10 min, analyzed using a reverse-phase BDS Hypersil C-18 column warmed by a column heater (Alltech, Deerfield, IL, USA) at 37°C in a high-performance liquid chromatograph (HPLC; Jasco, Tokyo, Japan) and detected using an ultraviolet (UV) detector (Jasco) at 430 nm. The mobile phase utilized ultrapure water containing citric acid at pH 3 associated with a tetrahydrofuran gradient from 5% to 40% (v/v) for 20 min and was driven using 2 high pressure pumps (Jasco) in a series at a flow rate of 1 mL/min. The encapsulation efficiency of CRM was defined as ([weight of encapsulated CRM]/[weight of total CRM]) ×100%.
Preparation of WGA-CRM-CL/LIP
The components of the lipid bilayer of WGA-grafted CRM-LIP (WGA-CRM-LIP) and WGA-grafted CRM-CL/LIP (WGA-CRM-CL/LIP) were similar to those of CRM-LIP and CRM-CL/LIP except that DSPE-PEG(2000) was replaced by DSPE-PEG(2000)-CA. CRM-LIP and CRM-CL/LIP containing functional carboxyl groups were grafted with WGA in EDC/NHS at the molar ratio of DSPE-PEG(2000)-CA to WGA of 10:1 at 4°C for 12 h. Unloaded WGA in the suspension was separated with a Sephadex G-100 column. In addition, 2 mL of effluent containing WGA-CRM-LIP and WGA-CRM-CL/LIP were collected at 25°C.
Preparation of NGF-CL/LIP and WGA-NGF-CL/LIP
The preparation of LIP loaded with NGF (NGF-LIP) and NGF-CL/LIP was analogous to that of CRM-LIP and CRM-CL/LIP, except that no CRM was incorporated and hβ-NGF of 50 ng/mL was used in the hydration. The steps for grafting WGA on NGF-LIP and NGF-CL/LIP were analogous to those for CRM-LIP and CRM-CL/LIP (described in the “Preparation of WGA-CRM-CL/LIP” section). The effluents of NGF-CL/LIP were treated with 1% (v/v) Triton-X-100 at 4°C for 1 h and analyzed with a human NGF kit using an enzyme-linked immunosorbent assay (ELISA) spectrofluorometer (BioTek, Winooski, VT, USA) at 450 nm. The encapsulation efficiency of NGF was defined as ([weight of encapsulated NGF]/[weight of total NGF]) ×100%.
Particle size and zeta potential
The cumulant Z-average diameter and zeta potential of LIP carriers were examined using a zetasizer 3000 HSA with a photon correlation spectroscope and a laser Doppler velocimeter (Malvern Instruments Ltd, Malvern, UK) at 25°C. To a buffer containing 0.1 M of Tris at pH 7.4, 1 mg/mL of LIP suspension was added, and 3 mL of the preparation was used in the detection.
Morphology
The surface of the LIP carriers was investigated using a field-emission scanning electron microscope (Jeol, Tokyo, Japan). The concentration of the suspension was 1 mg/mL, and 10 μL of the suspension was vibrated ultrasonically for 1 min, uniformly loaded on a coverslip, dried at 25°C for 24 h, vacuum-dried, glued with carbon paint, and covered with platinum using sputter coating at an accelerating voltage of 3 kV for 90 s. The structure of the LIP carriers was analyzed using a transmission electron microscope (Jeol). Then, 10 μL of the suspension was distributed on a 200-mesh copper grid with carbon coating and treated with 10 μL of 2% (w/v) phosphotungstic acid (PTA) solution for 2 min.
Dissolution profile of CRM and NGF from LIP carriers
To a 3.3 30 cm2 membrane tube in a flask with 20 mL of DPBS at pH 7.4, 1 mg/mL of LIP carriers in 2 mL of DPBS containing 0.05% sodium azide at pH 7.4 was added and shaken in a bath-reciprocal shaker at 50 rpm and 37°C for 2 days. Then, 100 μL of the sample containing CRM or NGF was treated with 1% (v/v) Triton-X-100 at 4°C for 1 h. The volume of the medium was compensated with 100 μL of fresh DPBS when sampling. To evaluate the quantity of released CRM, the sample was treated with methanol at 37°C for 30 min, analyzed using a HPLC (Jasco) with a reverse-phase BDS Hypersil C-18 column (Thermo Hypersil-Keystone, Bellefonte, PA, USA) at 37°C, and detected using an UV detector (Jasco) at 430 nm. The mobile phase used ultrapure water (Barnstead) containing citric acid at pH 3 with a tetrahydrofuran gradient from 5% to 40% (v/v), driven using 2 high pressure pumps (Jasco) at a flow rate of 1 mL/min for 20 min. To assess the release of NGF, the sample was analyzed with a human NGF kit using an ELISA spectrofluorometer at 450 nm.
Stability of LIP carriers
The LIP carriers (1 mg/mL) in Tris buffer at 0.1 M and pH 7.4 were stored at 4°C for 30 days. The variation in the cumulant Z-average diameter in the suspension was evaluated using a zetasizer 3000 HSA with a photon correlation spectroscope and a laser Doppler velocimeter at 25°C.
TEER and BBB permeability for PI after treating with LIP carriers
The method for propagating HBMECs, HAs, and SK-N-MC cells and the method for establishing an in vitro BBB model comprising a monolayer of HBMECs regulated by HAs in a transwell (BD Falcon, Franklin Lakes, NJ, USA) has been described previously.22,23 Free drugs or 0.025% (w/v) LIP carriers in culture medium were added to the upper donor chamber and placed in a humidified CO2 incubator (NuAire, Plymouth, MN, USA) at 37°C for 4 h. A Millicell system (Millipore) was used to evaluate the electrical resistances of cell-free and monolayer-containing membrane. The transendothelial electrical resistance (TEER) of the monolayer was defined as (electrical resistance of monolayer-containing membrane − electrical resistance of cell-free membrane) × surface area of the membrane. After incubating with LIP carriers, the upper donor chamber was filled with fresh medium containing 0.025% (w/v) PI and cultured in the CO2 incubator for 5 h. The quantity of PI in the lower receiver chamber was determined using an ELISA spectrofluorometer at an excitation of 485 nm and emission of 590 nm. The permeability for PI was defined as ([flux of PI]/[concentration of PI in the lower receiver chamber]). The reciprocal of BBB permeability for PI was evaluated by subtracting the reciprocal of the permeability for PI across cell-free membrane from that across monolayer-containing membrane.
BBB permeability for CRM and NGF using LIP carriers
Then, 0.025% (w/v) LIP carriers were added to the donor chamber in the humidified CO2 incubator for 5 h. To monitor the CRM and NGF levels, 20 μL of liquid in the receiver chamber was sampled every 2.5 h, treated with 1% (v/v) Triton-X-100 at 4°C for 1 h, and analyzed using the method for determining the quantity of released CRM and NGF (described in the “Dissolution profile of CRM and NGF from LIP carriers” section). The volume in the receiver chamber was compensated with 20 μL of fresh medium at the sampling point. The BBB permeability of CRM and NGF was defined analogously to that of PI (described in the “TEER and BBB permeability for PI after treating with LIP carriers” section).
Toxicity of LIP carriers
HBMECs or SK-N-MC cells were seeded at a density of 7.5×103 cells/well on gelatin-pretreated 96-well MicroWell plates (Nalge Nunc, Rochester, NY, USA) in a humidified CO2 incubator for 8 h, cultured with 0.025% (w/v) LIP carriers in the humidified CO2 incubator for 12 h, and reacted with 50 μL of XTT per well containing 2% (v/v) activation solution in the humidified CO2 incubator for 4 h. After the assay with XTT, the viability of HBMECs and SK-N-MC cells was determined using an ELISA spectrofluorometer at 450 nm and defined as ([optical density of HBMECs or SK-N-MC cells after treating with LIP carriers − optical density of XTT)/(optical density of HBMECs or SK-N-MC cells − optical density of XTT]).
Total protein in lysed SK-N-MC cells
The expanded SK-N-MC cells, at a density of 1×105 cells/well in 12-well plates, were washed with DPBS, incubated with 200 μL of lysis buffer per well for 30 min, and preserved in a freezer (Panasonic Healthcare, Gunma, Japan) at −80°C. The 2 μL sample was mixed with 198 μL of ultrapure water. The 150 μL of diluted solution was reacted with 150 μL of Coomassie protein assay kit in 96-well plates, calibrated with an albumin standard, and evaluated using an ELISA spectrofluorometer at 595 nm. The sample was heated to 95°C and distributed into vials with total protein of 25 μg in 25% (v/v) sample buffer per vial.
LIP carrier treatment for SK-N-MC cells
For the expression of p-p38, p-JNK, and p-Ser202, the expanded SK-N-MC cells were treated with free CRM, CRM-LIP, and CRM-CL/LIP at the final volume of 2.25 mL/well and fibrillar Aβ1–42 at final concentration of 10 μM. Briefly, Aβ1–42 was dissolved in HFIP at a concentration of 1 mg/mL, incubated at room temperature for 1 h, sonicated in a water bath for 10 min, and dried under a gentle stream of nitrogen. DMSO was used to resuspend the peptide. The solution was incubated at room temperature for 12 min, aliquoted into smaller volumes, and stored at −80°C. For a working solution, 900 μL of DPBS were added to the peptide stock solution and incubated at room temperature for 2 h to allow peptide aggregation. The final quantity of CRM was controlled at 0.8 μg CRM/well. In addition, SK-N-MC cells were treated with free NGF, NGF-LIP, and NGF-CL/LIP at analogous conditions for analyzing the expression of p-TrkA and p-ERK5, except that the final quantity of NGF was 4.7 ng NGF/well.
Western blot of p-p38, p-JNK, p-Ser202, p-TrkA, and p-ERK5
For SDS-PAG electrophoresis, the voltage was maintained at 60 V for 15–20 min and then at 120 V for 30 min, when proteins in the sample migrated to the separating gel. The proteins in the gel were transferred to a methanol-treated PVDF membrane in transfer buffer at 400 mA and 4°C for 90 min. The PVDF membrane with transferred proteins was washed with TBST for 15 min, blocked with 5% (w/v) skim milk for 1 h, and washed with TBST for 15 min. The transferred proteins were reacted, respectively, with antiphospho-protein antibody (anti-p-p38, anti-p-JNK, anti-p-Ser202, anti-p-TrkA, and anti-p-ERK5) and anti-GAPDH in a dilution ratio of 1:1,000 at 4°C for 12 h. The PVDF membrane was washed with TBST for 15 min, incubated with goat anti-rabbit IgG (H&L, HRP) in a dilution with a ratio of 1:2,000 at 25°C for 1.5 h, washed with TBST for 15 min, treated with ECL for anti-GAPDH and with select ECL for other antibodies for 1 min, and analyzed using a Luminescence Image Analysis System (Fujifilm, Tokyo, Japan). The quantity of the specific protein band in the lanes was analyzed using the ImageJ (version 1.50; National Institutes of Health, Bethesda, MD, USA) program.
Immunochemical staining of p-p38, p-JNK, and p-ERK5
SK-N-MC cells were seeded at a density of 1×104 cells/well in a 24-well flat-bottom tissue culture microtiter plate pretreated with gelatin for 12 h; then, 10 μM of fibrillary Aβ1–42 and fluorescent LIP carriers were added, and the SK-N-MC cells were cultured in a humidified CO2 incubator for 24 h. The culture of SK-N-MC cells was fixed with 0.5 mL of 10% (v/v) formalin at 25°C for 5 min, immersed in 0.5% (v/v) Triton-X-100 at 25°C for 10 min, treated with 200 μL of CAS block at 25°C for 10 min, reacted with antiphospho-protein antibody (anti-p-p38, 1:800; anti-p-JNK, 1:200; and anti-p-ERK5, 1:200) at 4°C for 12 h, incubated with goat anti-rabbit IgG (H&L, HRP) with rhodamine conjugate (1:200) at 25°C for 1 h in darkness, counterstained with 0.5% (w/v) DAPI in 0.1% (v/v) Triton-X-100 at 25°C for 3 min in darkness, and incubated with aqueous mounting medium. The fluorescent staining was visualized using a confocal laser scanning microscope (LSM 510; Zeiss, Oberkochen, Germany) at excitation of 350 nm (blue), 490 nm (green), and 540 nm (red) and emission of 475 nm (blue), 520 nm (green), and 565 nm (red).
AD rat model
Male Wistar rats (250–280 g, 8 weeks old) were housed in the Animal Laboratory of National Chung Cheng University in accordance with institutional guidelines and the guidelines of the Animal Protection Committee of the Council of Agriculture of the Republic of China. The animal experiments followed the Affidavit of Approval of Animal Use Protocol approved by the Institutional Animal Care and Use Committee of National Chung Cheng University. Before intravenous (iv) injection, the rats were fed sanitized food and water and fasted for 12 h. In every independent experiment to examine brain tissue, 7 rats were used as a group with Aβ1–42 and relevant treatments. The rats were treated with atropin (0.4 mg/kg, 0.4 mg/mL) by intraperitoneal (ip) injection for 3 min and anesthetized with sodium pentobarbital (60 mg/kg, 60 mg/mL) by ip injection. The anteroposterior (AP), mediolateral (ML), and dorsoventral (DV) domains were defined via lambda and bregma. The rat brain was fixed using a stereotaxic apparatus with bragma as the reference (origin) to find the LV-D3V zone. The subfield location of CA1 in the hippocampus was controlled by subtracting 0.35 cm (backward direction), 0.2 cm (right direction), and 0.27 cm (deep direction) from the defined AP, ML, and DV points of stereotaxic coordinates.24 The injection coordinates were based on an atlas of the rat brain.25 Before injection, Aβ1–42 was prepared and dissolved according to the method described in the “LIP carrier treatment for SK-N-MC cells” section. After drilling, 3.3 mg/mL of Aβ1–42 with 15 μL of a total volume were injected into the DV domain at a flow rate of 2 μL/min for 7.5 min; the needle remained in the injection position for 5 min. The cut on the head was stitched up, and the AD rat model was established after housing for 1 week. The control animals were sham rats injected with saline solution only.
LIP carrier treatment for AD rats
After anesthetization and fixation, the suspension containing 1 mL and 0.88 μg/mL of free CRM, 1.25 mL and 0.8 mg/mL of WGA-CRM-LIP, and 1 mL and 0.8 mg/mL of WGA-CRM-CL/LIP was administered by iv injection via a tail vein. The dosage of the 3 kinds of CRM preparation was the same. This treatment was carried out every 2 days, and the rats underwent the complete therapy 3 times before euthanization.
Immunohistochemical staining of Aβ plaque
The brain of the rats was taken out, washed, frozen on dry ice, embedded in tissue-tek OCT compound, and stored at −80°C for 30 min. The hippocampus was sectioned at −20°C using a cryostat microtome (Leica, Wetzlar, Germany) for samples of 10 μm, fixed in acetone for 5 min, immersed in 0.1% (v/v) hydrogen peroxide in methanol for 1 min, and treated with CAS block for 1 h. The pretreated CA1 section was reacted with anti-Aβ monoclonal antibody (1:100) at 4°C for 12 h, incubated with goat anti-rabbit IgG (H&L, HRP) (1:200) at room temperature for 45 min, and developed with DAB substrate at room temperature for 5 min. The sample was dehydrated in ethanol at concentrations increasing from 75% to 99.8%, immersed in o-xylene for 3 min, and sealed with Entellan® new mounting medium. The distribution of Aβ plaque in the hippocampus was visualized using an inverted microscope (Nikon, Tokyo, Japan).
Nissl staining
The procedure for pretreatment of tissue samples for Nissl staining was similar to that for Aβ plaque staining. After fixation with acetone, the sectioned sample was immersed in 75% ethanol at 37°C for 8 h, reacted with 0.1% (w/v) thionin for 20 min, treated with 150 μL of acetic acid in 100 mL of 95% ethanol for 2 min, dehydrated in ethanol at increasing concentrations, immersed in o-xylene for 3 min, and sealed with Entellan® new mounting medium. The optical image of the sample was obtained using the inverted microscope.
AChE activity
Approximately 5 mg of homogenized brain tissue was treated with 100 μL of lysis buffer, 100 μL of tissue protein extraction reagent, and 1 μL of Halt protease inhibitor cocktail and centrifuged at 2,000× g at 4°C for 10 min. Then, 50 μL of the supernatant was reacted with 50 μL of AThCh and 50 μL of 0.1% BSA in 96-well plates at 25°C for 30 min and detected using an ELISA spectrofluorometer at 410 nm. The AChE activity in the rat brain (mU/mg tissue) was calibrated with the AChE standard and 0.1% BSA.
LPO assay
Approximately 10 mg of homogenized brain tissue was treated with 300 μL of lysis buffer and 3 μL of BHT, and centrifuged at 12,000× g at 4°C for 5 min. Then, 200 μL of the supernatant was reacted with 600 μL of TBA at 95°C for 1 h. The resultant 200 μL of solution at 25°C in 96-well plates was detected using an ELISA spectrofluorometer at 532 nm. The MDA level in the rat brain (MDA nmol/mg tissue) was calibrated with the MDA standard.
Statistics
Data presented here were mean ± standard deviation. The statistical significance between groups was determined with a one-way analysis of variance followed by Tukey’s honest significant difference test.
Results and discussion
Property of LIP carriers
As shown in Table 1, an inclusion of CL increased the particle size, the absolute value of the zeta potential, and the encapsulation efficiency of CRM and of NGF. The reasons for these results are described as follows. First, CL was incorporated in lipid bilayers and could extend the curvature of the membrane. Second, CL, bearing a negative charge, could confer its charge to LIP. Third, CL, carrying 4 hydrophobic tails, could enhance the affinity of the lipid bilayer to CRM via hydrophobic interaction. Fourth, anionic CL could benefit the confinement of positively charged NGF in an internal aqueous core via electrical attraction. Fifth, CRM-LIP and NGF-LIP without CL contained 50% CHOL in a lipid mixture. An inclusion of CL decreased the molar percentage of CHOL from 50% to 40% and led to a decrease in membrane rigidity. In addition, the diameter of particles encapsulating CRM was larger than that of particles encapsulating NGF. This was because CRM could increase the particle curvature; however, NGF in an aqueous core did not have such an effect. The encapsulation efficiency of CRM was higher than that of NGF. This was because hydrophobic CRM favored the vacant space among lipids in bilayers; however, hydrophilic NGF stayed more easily in the external aqueous phase (Table 1).
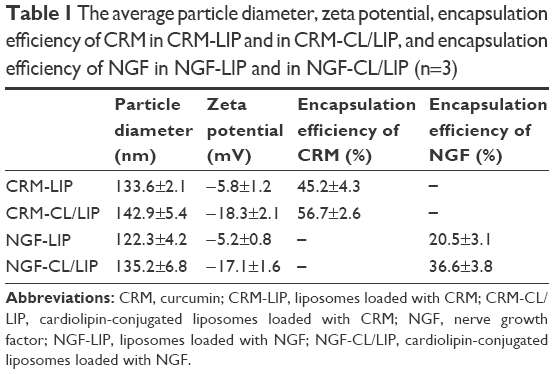 | Table 1 The average particle diameter, zeta potential, encapsulation efficiency of CRM in CRM-LIP and in CRM-CL/LIP, and encapsulation efficiency of NGF in NGF-LIP and in NGF-CL/LIP (n=3) |
Figure 1 shows the typical biomedical and pharmaceutical properties of LIP carriers. In Figure 1A, the spheroidal particles have a slightly uneven periphery. The sizes of these particles are about 130 nm as shown in Figure 1A (a) and (c) and 145 nm in Figure 1A (b) and (d), consistent with those listed in Table 1. The minor difference in the diameter between Figure 1A and Table 1 was due to the drying of the sample before imaging. The counterstaining of PTA also yielded the dark morphology as shown in Figure 1A (c) and (d). No initial bursts of CRM and NGF were found in the release profiles (Figure 1B). Collisions among LIP carriers might favor the dissolution of CRM and NGF in the early stage. The dissolution of CRM from CRM-LIP was faster than that from CRM-CL/LIP. This was because the affinity of CRM to CL was stronger than that to other lipids. Also, the absolute value of the zeta potential of CRM-CL/LIP was higher than that of CRM-LIP (Table 1), rendering a reduced collision and less damage to the bilayer of the former. In addition, the release of CRM was faster than that of NGF. This was because CRM could escape from the bilayer only by overcoming the hydrophobic attraction of lipids; however, NGF had to shun the hydrophobic repulsion of lipids to penetrate the bilayer and then migrate through the membrane. As shown in Figure 1C, the particle size enlarged moderately after storage at 4°C. This was because DPPC and SPC could bind to CHOL. This linkage potently braced the bilayer structure for a high degree of mechanical strength and eschewed vesicular fusion.26 Moreover, the increase in the average diameter of WGA-CRM-CL/LIP was insignificant, in general, suggesting a diminished collision frequency and a stabilized colloidal system. This was because surface WGA induced steric repulsion and hindered WGA-CRM-CL/LIP from aggregation.27 As shown in Figure 1D, the order in TEER was CRM ≅ WGA-CRM-CL/LIP > CRM-CL/LIP and NGF > NGF-CL/LIP ≅ WGA-NGF-CL/LIP. The order in BBB permeability for PI was CRM ≅ WGA-CRM-CL/LIP < CRM-CL/LIP and NGF < NGF-CL/LIP ≅ WGA-NGF-CL/LIP. These orders suggested that CL/LIP might slightly imperil the BBB; nonetheless, WGA could protect the structural integrity of the BBB, although WAG evoked a targeting effect on HBMECs and intensified the interaction between WGA-CL/LIP and the cells. A high TEER and a low BBB permeability for PI revealed that the impact of LIP carriers after permeating the BBB did not elicit severe leakage from the tight junction.28 In Figure 1E, CL/LIP mildly impeded the delivery of CRM across the BBB. Nevertheless, the targeting capacity of WGA-CRM-CL/LIP considerably promoted BBB permeability for CRM, compared with free CRM. Considering the permeation of NGF, NGF-CL/LIP slightly reduced BBB permeability for NGF, and the modification of WGA on NGF-CL/LIP also substantially ameliorated the permeability. It is well-known that the drug efficacy of CRM is hampered by its high degradability and poor plasma solubility, and it is liable to be decomposed in the gastrointestinal tract and liver.29 Furthermore, exogenous NGF is regarded as an important bioactive element with inadequate bioavailability in the brain because it is hydrophilic and bulky with a molecular weight of 27 kDa.30 The current LIP carriers with WGA and CL could be practical formulations to enhance the usability of CRM and NGF in the brain. As shown in Figure 1F, the order in the cytotoxicity was CRM ≅ WGA-CRM-CL/LIP > CRM-CL/LIP and NGF ≅ WGA-NGF-CL/LIP > NGF-CL/LIP. Viability was higher than 89%, although CRM-CL/LIP and NGF-CL/LIP were relatively toxic to HBMECs and SK-N-MC cells in these formulations. Based on the 2 orders, surface WGA could constrain adverse responses of lipids, and the cytotoxicity of CL/LIP and WGA-CL/LIP loaded with CRM and NGF was in a tolerable range. The data in Table 1 and Figure 1 reveal traits of CRM- and NGF-encapsulated LIP appropriate to rescuing injured neuron-like cells such as SK-N-MC cells. Hence, we further analyzed the effect of LIP carriers on relevant kinases expressed by SK-N-MC cells after an insult with Aβ.
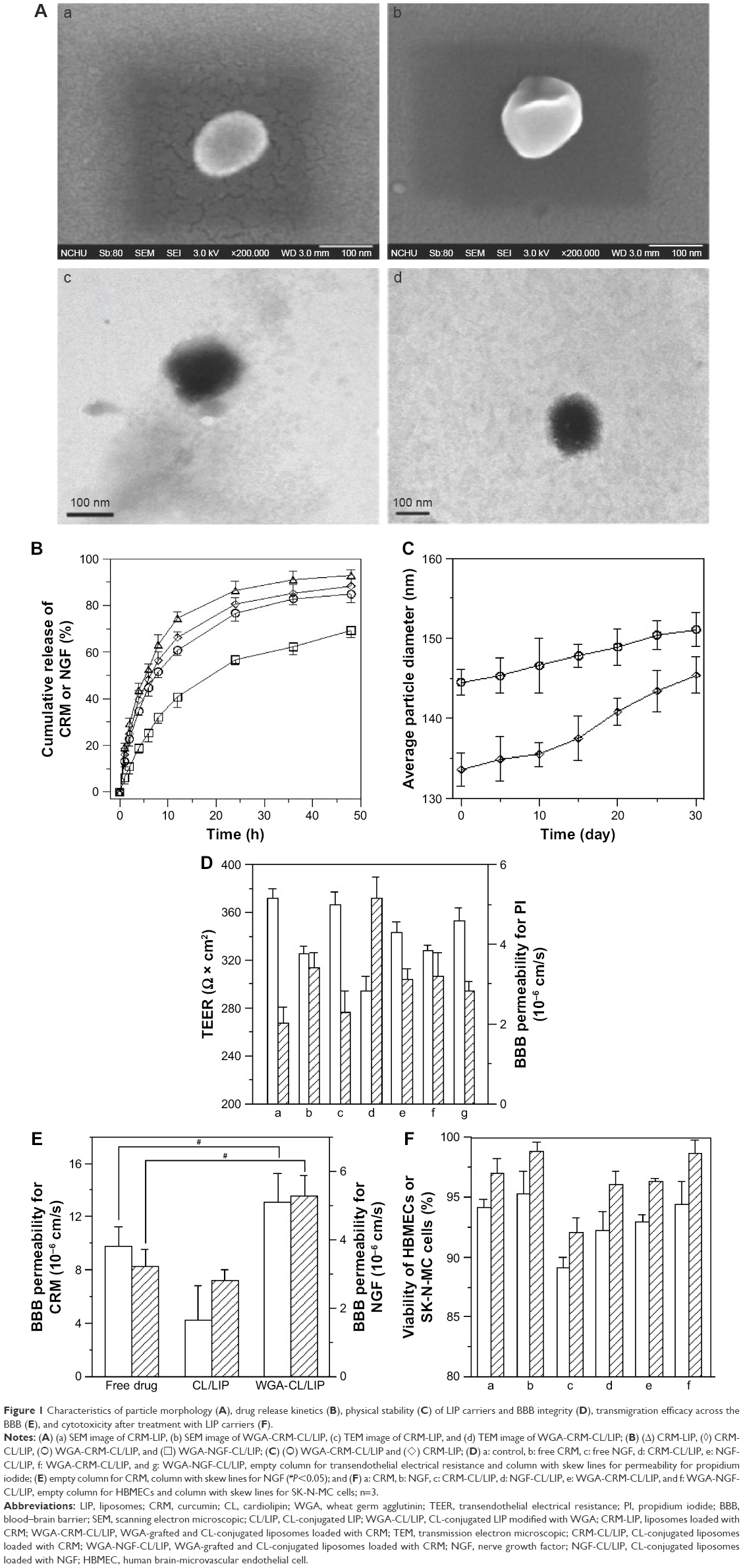 | Figure 1 Characteristics of particle morphology (A), drug release kinetics (B), physical stability (C) of LIP carriers and BBB integrity (D), transmigration efficacy across the BBB (E), and cytotoxicity after treatment with LIP carriers (F). |
Phosphorylation of p38, JNK, and Tau protein at Ser202, TrkA, and ERK5
Figure 2A and B shows, respectively, the Western blot and relative protein ratio of p-p38 and p-JNK in the neuronal system. As indicated in the 2 figures, induction with Aβ substantially provoked the expression of p-p38 and p-JNK by SK-N-MC cells, compared with the control group. The Western blots shown in Figure 2A were consistent with those reported in the literature.31,32 After the insult with Aβ, the 3 formulations (CRM, CRM-LIP, and CRM-CL/LIP) reduced the average quantity of p-p38 and p-JNK, in general. Free CRM yielded a relatively minor decrease in the p-p38 and p-JNK levels, compared with the Aβ group. The reason for this insignificant difference was that free CRM might actuate nuclear translocation of p-p38 and p-JNK during apoptotic degeneration. Thus, after treating Aβ-insulted SK-N-MC cells with free CRM, p-p38 and p-JNK might not be straightforwardly expressed in the cytoplasm. Figure 2C and D, respectively, shows the Western blot and relative protein ratio of tau protein at p-Ser202. As indicated in the 2 figures, the difference in the expression of p-Ser202 between the Aβ group and the control group was obvious, even if the duration of the insult with Aβ was not very long. Compared with the Aβ group, the Aβ + CRM group could not significantly diminish the level of p-Ser202. This demonstrated an unsatisfactory hindrance of free CRM from the synthesis of tau protein, although it has been found that free CRM of 500 ppm might reduce the phosphorylation of tau protein.33 However, CRM-LIP and CRM-CL/LIP significantly reduced the average quantity of p-Ser202, suggesting that the 2 vehicles could accelerate the delivery of CRM to SK-N-MC cells for downregulating the expression of p-Ser202. In addition, the effect of CRM-LIP and CRM-CL/LIP on the quantity of p-Ser202 was nearly equivalent. This similarity in expression resulted from the following: first, CRM-LIP and CRM-CL/LIP were biocompatible with SK-N-MC cells (data shown in Figure 1E) and could not strongly incite the production of p-Ser202 without the induction with Aβ. Second, the release rate of CRM from CRM-LIP was faster than that from CRM-CL/LIP due to the affinity of CRM to CL (data shown in Figure 1B). Thus, the hydrophobic attraction between CRM and CL increased the difficulty in releasing CRM from CRM-CL/LIP to rescue SK-N-MC cells. Third, CRM-CL/LIP had the capacity to bind to Aβ for topical release of CRM around SK-N-MC cells. Thus, binding of CL to Aβ increased a local concentration of CRM released from CRM-CL/LIP to rescue SK-N-MC cells. This reason, however, was contradictory to the second one. Figure 2E and F shows, respectively, the Western blot and relative protein ratio of p-TrkA and p-ERK5. As indicated in the 2 figures, the insult with Aβ apparently decreased the expression of p-TrkA, compared with the control group. This expression of p-TrkA indicated that the self-protective capability of SK-N-MC cells was inadequate after an insult with Aβ. It has been observed that an insult with Aβ significantly enhanced the TrkA level for 8 h; nevertheless, the TrkA level was reduced at 16 h after the insult.34 In addition, the average quantity of p-TrkA was significantly enhanced in the Aβ + NGF group, Aβ + NGF-LIP group, and Aβ + NGF-CL/LIP group, compared with the Aβ group. This manifested the ability of NGF, NGF-LIP, and NGF-CL/LIP to salvage SK-N-MC cells from neurodegeneration. As seen in Figure 2E and F, the addition of Aβ reduced the expression of p-ERK5, compared with the control group. Treatment with NGF yielded a level of p-ERK5 comparable to the Aβ group, suggesting that the effect of free NGF on the expression of p-ERK5 was inconsequential. This weak influence resulted primarily from the fact that the degradation of NGF could be faster than the enhancement of the expression of p-ERK5. It has been found that in rat pheochromocytoma cells, the maximal impact of free NGF on the expression of p-ERK5 occurred at 5 min; however, this stimulation deteriorated with time.35 In Figure 2E and F, the expression of p-ERK5 in the Aβ + NGF-LIP group was also comparable to that in the Aβ group. Nonetheless, the expression of p-ERK5 in the Aβ + NGF-CL/LIP group was significantly greater than that in the Aβ group. This was because the controlled release of NGF from NGF-CL/LIP could protect NGF against degradation in the microenvironment and provide a sufficient probability for interacting with SK-N-MC cells to adequately express p-ERK5. Moreover, NGF-CL/LIP might trigger membrane fusion in the ERK5 pathway. In addition to the continuous release and membrane fusion, the Aβ-targeting capacity of NGF-CL/LIP enhanced the possibility of focally releasing NGF to promote the p-ERK5 level and recover SK-N-MC cells.
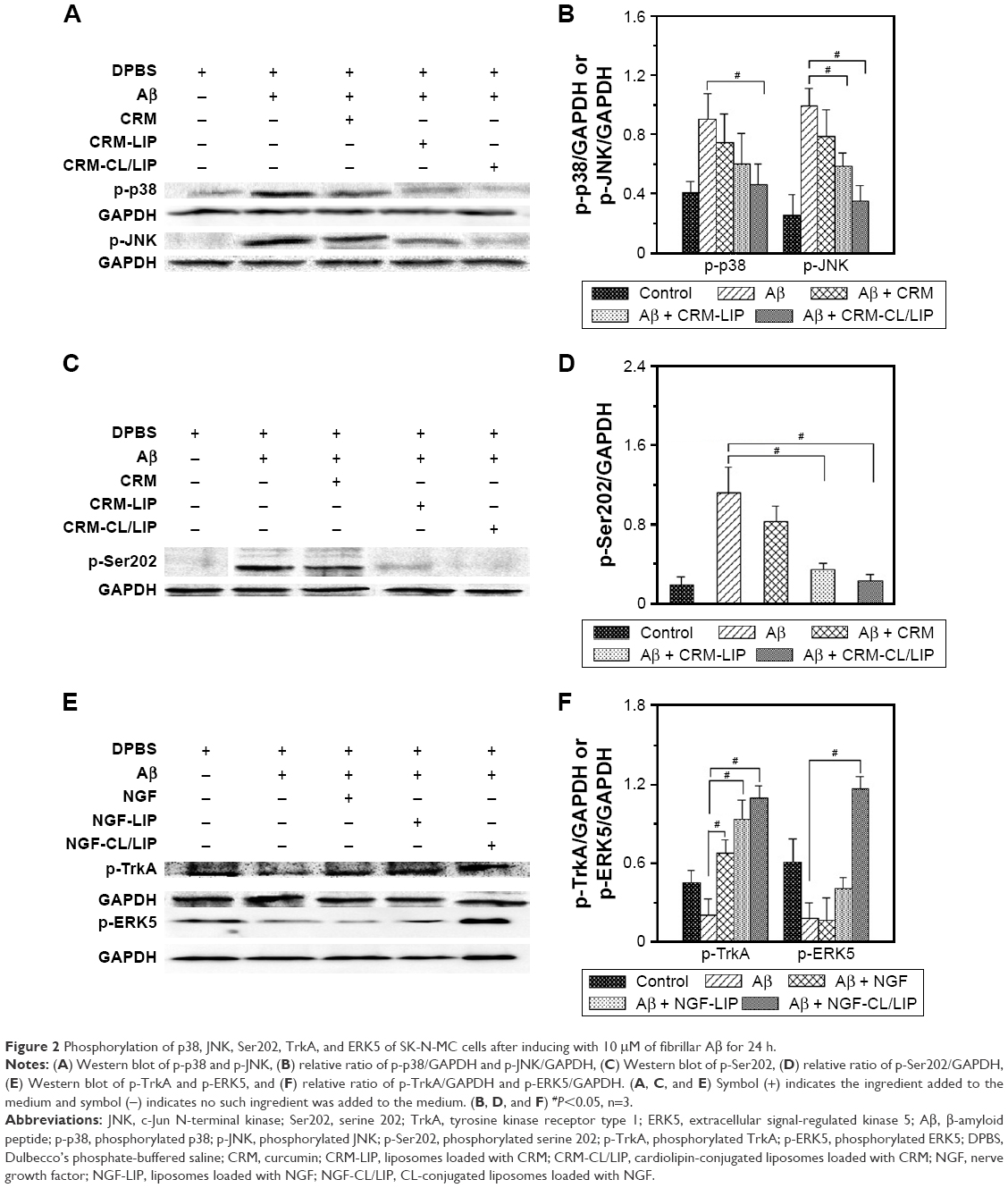 | Figure 2 Phosphorylation of p38, JNK, Ser202, TrkA, and ERK5 of SK-N-MC cells after inducing with 10 μM of fibrillar Aβ for 24 h. |
Fluorescent images of the degenerated neuronal system
Figure 3 shows the immunochemical staining of p-p38, p-JNK, and p-ERK5 during apoptosis of SK-N-MC cells after the insult with Aβ. As indicated in Figure 3A(b) and B(b), the red dots of p-p38 and p-JNK were amalgamated with the nuclei. In addition, treatment with free CRM could not substantially reduce the expression of p-p38 and p-JNK (Figure 3A[c] and B[c]). As shown in Figure 3A(d,e) and B(d,e), CRM-LIP and CRM-CL/LIP reduced the red intensity. Moreover, the green carriers interconnected with SK-N-MC cells and migrated across the cell membrane to the cells. LIP carriers could approach the nuclei of aggregated cells, causing the green dots and blue dots to almost completely coincide (Figure 3A and B[d,e]). A study on the apoptotic regulators of cortical neurons concluded that neurodegenerative pathways were prone to translocate intracellular p-p38 and p-JNK to nuclei.36 As seen in Figure 3C(b), the insult with Aβ decreased the quantity of p-ERK5. After rescue with free NGF, the increase in the expression of p-ERK5 was unclear (Figure 3C[c]). As exhibited in Figure 3C(d), NGF-LIP significantly enhanced the synthesis of p-ERK5, compared with treatment with NGF. Furthermore, NGF-CL/LIP was more efficient than NGF-LIP in the production of p-ERK5 in SK-N-MC cells (Figure 3C[d,e]). These images and the data shown in Figure 2 are consistent. In addition, the upregulation of p-ERK5 in SK-N-MC cells was also consistent with the viability data of our previous study.37 Figure 4 summarizes the role of LIP carriers in the withdrawal and enhancement of the neural survival/apoptosis factor investigated in this study.
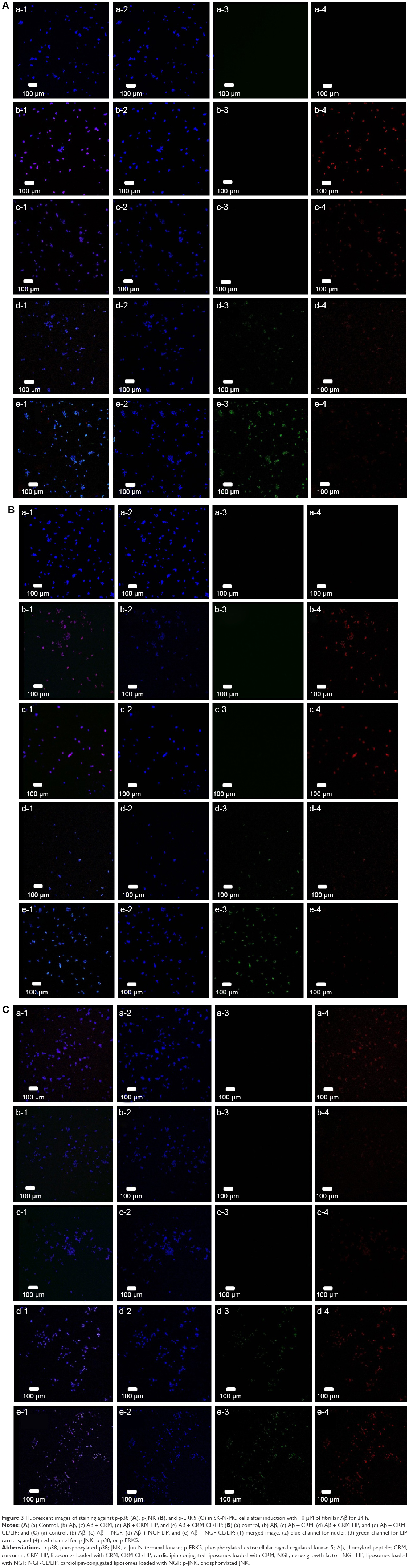 | Figure 3 Fluorescent images of staining against p-p38 (A), p-JNK (B), and p-ERK5 (C) in SK-N-MC cells after induction with 10 μM of fibrillar Aβ for 24 h. |
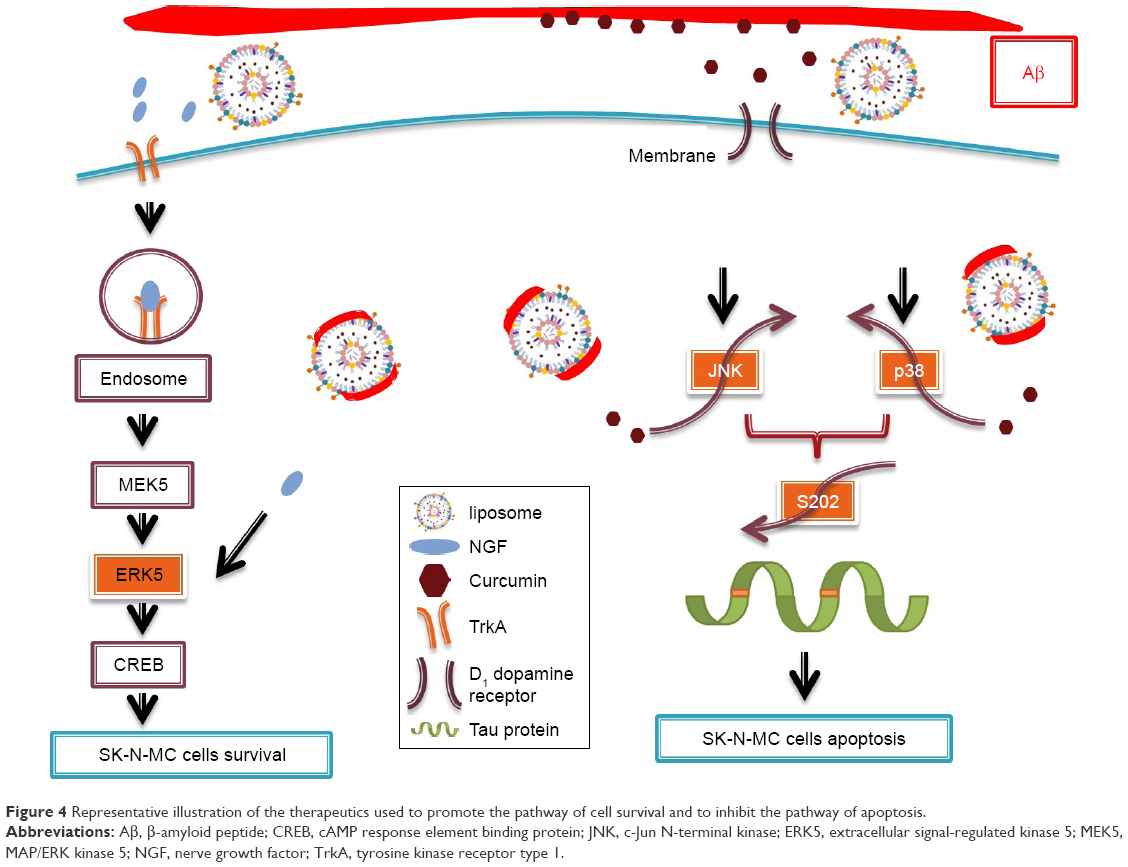 | Figure 4 Representative illustration of the therapeutics used to promote the pathway of cell survival and to inhibit the pathway of apoptosis. |
Formation of Aβ plaque and salvage of neurons in the hippocampus of AD rats treated with LIP carriers
Figure 5A shows the stained images of deposited Aβ in the rat brains. The stains of Aβ plaque shown in Figure 5A (b-1) and (b-2) were more evident than those in Figure 5A (a-1) and (a-2), demonstrating the satisfactory establishment of the AD model in vivo.38 In this study, the deposition of Aβ plaque in the hippocampus was consistent with that reported in the literature using transgenic mice.39 In addition, the quantity of Aβ plaque in Figure 5A (c-1) and (c-2) was less than that in Figure 5A (b-1) and (b-2). This suggested that free CRM was capable of partially eliminating the deposited Aβ. Compared with Figure 5A (c-1) and (c-2), (d-1) and (d-2) exhibited a lower intensity of Aβ plaque. This result could be attributed to 2 factors. First, the encapsulated CRM was protected by LIP against rapid degradation in the blood.40 Second, WGA on CRM-LIP could target N-acetylglucosamine on endothelia and increase the quantity of CRM transported across the BBB.41 When comparing Figure 5A (d-1) and (d-2) with Figure 5A (e-1) and (e-2), one could conclude that the capability of WGA-CRM-CL/LIP to remove Aβ plaque was greater than that of WGA-CRM-LIP. This could be explained by the fact that the incorporation of CL in LIP favored Aβ docking and benefited the local delivery of CRM around Aβ-insulted tissue. Figure 5B shows the images of Nissl staining in rat brains. As indicated in this figure, regular neurons displayed an ellipsoid-shaped morphology and the purple dots with angles were pathological cells. The morphology of apoptotic neurons in these images was consistent with that observed in the literature.42 The AD rats (Figure 5B[b]) exhibited more degenerated neurons in the brain than the normal rats (Figure 5B[a]). Compared with Figure 5B(b), Figure 5B(c) evidenced a minor improvement in neural rescue by free CRM after the insult with Aβ. As revealed in Figure 5B(d,e), WGA-CRM-LIP and WGA-CRM-CL/LIP retarded neurodegeneration considerably. In addition, the number of regular neurons shown in Figure 5B (a-3), (b-3), (c-3), (d-3), and (e-3) were, respectively, 166, 42, 92, 113, and 138. The number of regular neurons shown in Figure 5B (a-4), (b-4), (c-4), (d-4), and (e-4) were, respectively, 60, 15, 38, 45, and 54. Thus, the typical neurons shown in Figure 5B(e) had recovered almost to the level of the control group (Figure 5B[a]). These images were consistent with the staining of Aβ plaque (Figure 5A). Moreover, the effect of the 3 CRM preparations on healthy animals would be minor, because the healthy brain had sufficient neurons and was not in an inflammatory state, so the anti-inflammatory CRM, WGA-CRM-LIP, and WGA-CRM-CL/LIP would have little effect. Also, the lipid ingredients in LIP carriers were mainly found in cell membrane and had a low toxicity to brain neurons. Thus, the control animals in this study were sham rats with iv injection of saline solution.
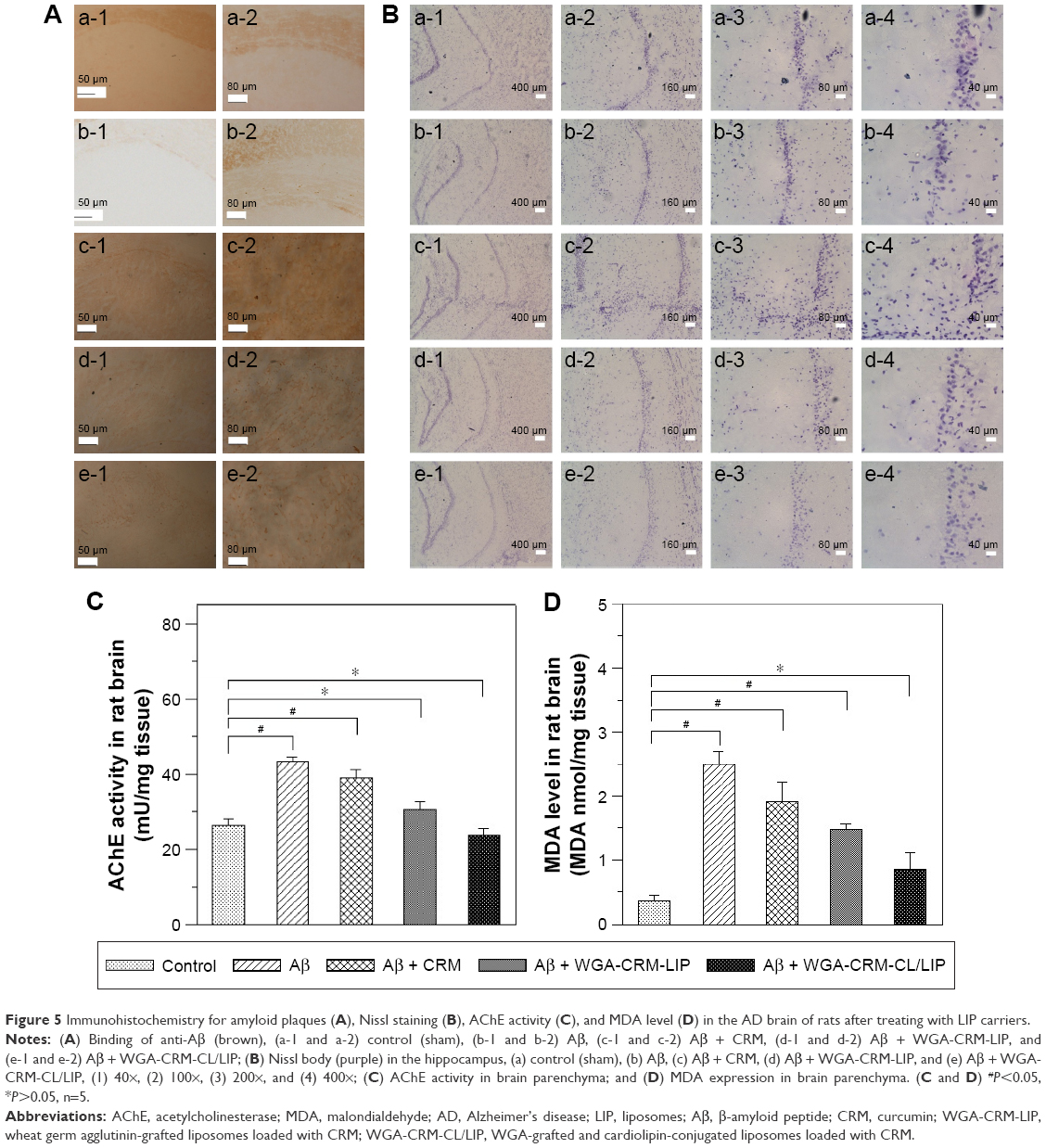 | Figure 5 Immunohistochemistry for amyloid plaques (A), Nissl staining (B), AChE activity (C), and MDA level (D) in the AD brain of rats after treating with LIP carriers. |
Figure 5C shows the AChE activity in the brain of rats with an Aβ-induced memory deficit. As indicated in this figure, the insult with Aβ significantly enhanced AChE activity, testifying to the deterioration in cholinergic function of the neurons. In addition, free CRM was unable to reduce AChE activity. The order of the 3 formulations in AChE activity was CRM > WGA-CRM-LIP > WGA-CRM-CL/LIP. It was concluded that the formation of Aβ plaque was closely correlated with AChE activity in the CNS.6 Thus, the injected Aβ tended to elicit the peroxidation of nervous tissue and enhance the activity of AChE.43 It was also observed that CRM played a role in the inhibition of AChE production.44 As revealed in Figure 5C, the AChE activity of the Aβ + WGA-CRM-CL/LIP group was almost equivalent to that of the control group. This showed that after the insult with Aβ, WGA-CRM-CL/LIP could be efficacious in suppressing the synthesis of AChE and reduce the decomposition of acetylcholine for better neurotransmission. Figure 5D shows the MDA level after treatment with LIP carriers. As indicated in this figure, and compared with the control group, the insult with Aβ significantly increased the quantity of MDA, verifying the characteristics of AD brains. The rationale behind this was that free radicals activated chain reactions for lipid oxidation and generated the by-product MDA, which injured neurons via the toxic MDA-protein adducts.45 The quantity of MDA was in the following order: CRM > WGA-CRM-LIP > WGA-CRM-CL/LIP. In addition, the difference in the quantity of MDA between the Aβ group and the Aβ + CRM group was insignificant, although CRM could reduce the amount of free radicals and suppress MDA production. LPO in the brains of AD patients was found to be higher than that of normal people of the same age.46 It was also intriguing that LPO was found to precede the accumulation of Aβ plaque in Tg2576 mice.47 Thus, the oxidative damage to the brain could generally lead to an AD pathogenesis.
Conclusion
CRM-CL/LIP could inhibit the phosphorylation of p38, JNK, and tau protein at Ser202 and avert serious degeneration of Aβ-insulted neurons. In addition, the upregulation of p-TrkA and p-ERK5 by treatment with NGF-CL/LIP promoted neuronal viability after the insult with Aβ. The membrane fusion and Aβ-docking characteristics enabled CRM-CL/LIP and NGF-CL/LIP to interact intensively with neurons. With their brain-targeting capacity, WGA-CRM-CL/LIP and WGA-NGF-CL/LIP could permeate the BBB without severe impairment to the structure of the tight junction. Moreover, WGA-CRM-CL/LIP substantially reduced Aβ plaque deposition and LPO, maintained neural survival, and lowered AChE activity in the hippocampus of AD rats. Hence, LIP carriers can be potential preparations for neuroprotection against apoptosis and enhance the quantity of viable neurons for AD treatment.
Acknowledgment
This work was supported by the Ministry of Science and Technology of the Republic of China under grant number MOST 103-2221-E-194-043-MY3.
Disclosure
The authors report no conflicts of interest in this work.
References
Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2013;314(5800):777–781. | ||
Brambilla D, Le Droumaguet B, Nicolas J, et al. Nanotechnologies for Alzheimer’s disease: diagnosis, therapy, and safety issues. Nanomedicine. 2011;7(5):521–540. | ||
Basso M, Yang J, Warren L, et al. Volumetry of amygdala and hippocampus and memory performance in Alzheimer’s disease. Psychiatry Res. 2006;146(3):251–261. | ||
Anekonda TS, Reddy PH. Can herbs provide a new generation of drugs for treating Alzheimer’s disease? Brain Res Rev. 2005;50(2):361–376. | ||
Hampel H, Frank R, Broich K, et al. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9(7):560–574. | ||
Garcia-Ayllon MS, Silveyra MX, Saez-Valero J. Association between acetylcholinesterase and beta-amyloid peptide in Alzheimer’s cerebrospinal fluid. Chem Biol Interact. 2008;175(1–3):209–215. | ||
Anderson NG, Maller JL, Tonks NK, Sturgill TW. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature. 1990;343(6259):651–653. | ||
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathway mediated by ERK, JNK, and p38 protein kinase. Science. 2002;298(5600):1911–1912. | ||
Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79(1):143–180. | ||
Schnyder A, Huwyler J. Drug transport to brain with targeted liposomes. Neurotherapeutics. 2005;2(1):99–107. | ||
Ono K, Hasegawa H, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J Neurosci Res. 2004;75(6):742–750. | ||
Manogaran E, Ramanathan M, Mohamed AA. Identification of curcumin targets in neuroinflammatory pathways: molecular docking scores with GSK-3β, p38 MAPK, COX, ICE and TACE enzymes. Acta Pol Pharm. 2012;66(2):237–245. | ||
Lee WH, Loo CY, Bebawy M, Luk F, Mason RS, Rohanizadeh R. Curcumin and its derivatives: their application in neuropharmacology and neuroscience in the 21st century. Curr Neuropharmacol. 2013;11(4):338–378. | ||
Priyadarsini KI. The chemistry of curcumin: from extraction to therapeutic agent. Molecules. 2014;19(12):20091–20112. | ||
Bottger D, Ullrich C, Humpel C. Monocytes deliver bioactive nerve growth factor through a brain capillary endothelial cell-monolayer in vitro and counteract degeneration of cholinergic neurons. Brain Res. 2010;1312:108–119. | ||
Kuo YC, Chung CY. TATVHL peptide-grafted alginate/poly(γ-glutamic acid) scaffolds with inverted colloidal crystal topology for neuronal differentiation of iPS cells. Biomaterials. 2012;33(35):8955–8966. | ||
Sakiyama-Elbert SE, Hubbell JA. Controlled release of nerve growth factor from a heparin-containing fibrin-based cell ingrowth matrix. J Control Release. 2000;69(1):149–158. | ||
Gobbi M, Re F, Canovi M, et al. Lipid-based nanoparticles with high binding affinity for amyloid-β1–42 peptide. Biomaterials. 2010;31(25):6519–6529. | ||
Camilleri A, Zarb C, Caruana M, et al. Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols. Biochim Biophys Acta. 2013;1828(11):2532–2543. | ||
Wei X, Chen X, Ying M, Lu W. Brain tumor-targeted drug delivery strategies. Acta Pharm Sin B. 2014;4(3):193–201. | ||
Gao X, Wu B, Zhang Q, et al. Brain delivery of vasoactive intestinal peptide enhanced with the nanoparticles conjugated with wheat germ agglutinin following intranasal administration. J Control Release. 2007;121(3):156–167. | ||
Kuo YC, Lu CH. Effect of human astrocytes on the characteristics of human brain-microvascular endothelial cells in the blood–brain barrier. Colloids Surf B. 2011;86(1):225–231. | ||
Kuo YC, Liang CT. Inhibition of human brain malignant glioblastoma cells using carmustine-loaded catanionic solid lipid nanoparticles with surface anti-epithelial growth factor receptor. Biomaterials. 2011;32(12):3340–3350. | ||
Shi X, Lu X, Zhan L, et al. Rat hippocampal proteomic alterations following intrahippocampal injection of amyloid beta peptide (1–40). Neurosci Lett. 2011;500(2):87–91. | ||
Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 7th ed. Amsterdam, the Netherlands: Elsevier; 2013. | ||
Lopez-Pinto JM, Gonzalez-Rodriguez ML, Rabasco AM. Effect of cholesterol and ethanol on dermal delivery from DPPC liposomes. Int J Pharm. 2005;298(1):1–12. | ||
Michele LD, Zaccone A, Eiser E. Analytical theory of polymer-network-mediated interaction between colloidal particles. Proc Natl Acad Sci U S A. 2012;109(26):10187–10192. | ||
Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood–brain barrier by nanoparticles. J Control Release. 2012;161(2):264–273. | ||
Hefti F. Is Alzheimer disease caused by lack of nerve growth factor? Ann Neurol. 1983;13(1):109–110. | ||
Kuo YC, Lin CC. Rescuing apoptotic neurons from Alzheimer’s disease using wheat germ agglutinin-conjugated and cardiolipin-conjugated liposomes with encapsulated neuron growth factor and curcumin. Int J Nanomed. 2015;10:2653–2672. | ||
Song C, Perides G, Wang D, Liu YF. β-Amyloid peptide induces formation of actin stress fibers through p38 mitogen-activated protein kinase. J Neurochem. 2002;83(4):828–836. | ||
Morishima Y, Gotoh Y, Zieg J, et al. β-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurochem. 2001;21(19):7551–7560. | ||
Ma QL, Zuo X, Yang F, et al. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J Biol Chem. 2013;288(6):4056–4065. | ||
Bulbarelli A, Lonati E, Cazzaniga E, et al. TrkA pathway activation induced by amyloid-beta (Abeta). Mol Cell Neurosci. 2009;40(3):365–373. | ||
Obara Y, Yamauchi A, Takehara S, et al. ERK5 activity is required for nerve growth factor-induced neurite outgrowth and stabilization of tyrosine hydroxylase in PC12 cells. J Biol Chem. 2009;284(35):64–73. | ||
Willaime-Morawek S, Brami-Cherrier K, Mariani J, Caboche J, Brugg B. C-Jun N-terminal kinases/c-Jun and p38 pathways cooperate in ceramide-induced neuronal apoptosis. Neuroscience. 2003;119(2):387–397. | ||
Kuo YC, Wang CT. Protection of SK-N-MC cells against β-amyloid peptide-induced degeneration using neuron growth factor-loaded liposomes with surface lactoferrin. Biomaterials. 2014;35(22):5954–5964. | ||
Oddo S, Caccamo A, Tran L, et al. Temporal profile of amyloid-β (Aβ) oligomerization in an in vivo model of Alzheimer disease – a link between Aβ and tau pathology. J Biol Chem. 2006;281(3):1599–1604. | ||
DeMattos RB, Lu J, Tang Y, et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron. 2012;76(5):908–920. | ||
Tsai YM, Chien CF, Tsai TH. Curcumin and its nano-formulation: the kinetics of tissue distribution and blood–brain barrier penetration. Int J Pharm. 2011;416(1):331–338. | ||
Du J, Lu WL, Ying X, et al. Dual-targeting topotecan liposomes modified with tamoxifen and wheat germ agglutinin significantly improve drug transport across the blood-brain barrier and survival of brain tumor-bearing animals. Mol Pharm. 2009;6(3):905–917. | ||
Nakajima W, Ishida A, Lange MS, et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci. 2000;20(21):7994–8004. | ||
Melo JB, Agostinho P, Oliveira CR. Involvement of oxidative stress in the enhancement of acetylcholinesterase activity induced by amyloid beta-peptide. Neurosci Res. 2003;45(1):117–127. | ||
Ahmed T, Gilanu AH. Inhibitory effect of curcuminoids on acetylcholinesterase activity and attenuation of scopolamine-induced amnesia may explain medicinal use of turmeric in Alzheimer’s disease. Pharmacol Biochem Behav. 2009;91(4):554–559. | ||
Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. 2008;153(1):6–20. | ||
Lovell M, Ehmann W, Butler S, Markesbery W. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology. 1995;45(8):1594–1601. | ||
Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VMY. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. Neuroscience. 2001;21(12):4183–4187. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.