")
Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 5
Von Hippel-Lindau status influences phenotype of liver cancers arising from PTEN loss
Authors Sendor A, Hacker K, Chen S, Corona AL, Sen O, Chiang D, Snavely A, Rogers A, Montgomery S, Rathmell WK, McRee A
Received 5 August 2014
Accepted for publication 15 December 2014
Published 24 February 2015 Volume 2015:5 Pages 61—71
DOI https://doi.org/10.2147/GICTT.S72274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Adam B Sendor,1 Kathryn E Hacker,1 Shufen Chen,1 Armando L Corona,1 Oishee Sen,1 Derek Y Chiang,1 Anna Snavely,1 Arlin B Rogers,2 Stephanie A Montgomery,1 W Kimryn Rathmell,1 Autumn J McRee1
1Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, USA; 2Section of Pathology, Department of Biomedical Sciences, Cummings School of Veterinary Medicine, Tufts University, Boston, MA, USA
Background: PTEN loss contributes to the development of liver diseases including hepatic steatosis and both hepatocellular carcinoma (HCC) and cholangiocarcinoma (CC). The factors that influence the penetrance of these conditions are unclear. We explored the influence of sustained hypoxia signaling through co-deletion of Pten and Vhl in a murine model.
Methods: We used a CreER-linked Keratin 18 mouse model to conditionally delete Pten, Vhl or both in somatic cells of adult mice, evaluating the resultant tumors by histology and gene expression microarray. Existing sets of gene expression data for human HCC and CC were examined for pathways related to those observed in the murine tumors, and a cohort of human CC samples was evaluated for relationships between HIF-1α expression and clinical outcomes.
Results: Both Pten deletion genotypes developed liver tumors, but with differing phenotypes. Pten deletion alone led to large hepatic tumors with widespread hepatosteatosis. Co-deletion of Pten and Vhl with the Keratin 18 promoter resulted in reduced steatosis and a reduced tumor burden that was characterized by a trabecular architecture similar to CC. Genes associated with hepatic steatosis were coordinately expressed in the human HCC dataset, while genes involved in hypoxia response were upregulated in tumors from the human CC dataset. HIF-1α expression and overall survival were examined in an independent cohort of human CC tumors with no statistical differences uncovered.
Conclusion: Pten deletion in Keratin 18 expressing cells leads to aggressive tumor formation and widespread steatosis in mouse livers. Co-deletion of Vhl and Pten results in lower tumor burden with gene expression profiling suggesting a switch from a profile of lipid deposition to an expression profile more consistent with upregulation of the hypoxia response pathway. A relationship between tumor hypoxia signaling and altered hepatic steatotic response suggests that competing influences may alter tumor phenotypes.
Keywords: Von Hippel-Lindau (VHL), phosphatase and tension homologue deleted on chromosome 10 (PTEN), cholangiocarcinoma (CC), hepatocellular-cholangiocarcinoma (HCC)
Introduction
Primary liver malignancies, including both hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (CC), are diagnosed in approximately 30,000 people in the United States each year with almost 20,000 deaths.1 These tumors generally demonstrate resistance to chemotherapy and carry particularly dismal prognoses. An aggressive form of primary liver cancer has been described sharing features of both HCC and CC;2 however, the developmental origin of these tumors has remained elusive. While theories have been postulated regarding a potential common cell of origin, little is known regarding the molecular pathogenesis of primary liver tumors and the signal transduction pathways that lead to more aggressive phenotypes.3
The Pten gene, a well-known negative regulator of the phosphatidylinositol 3 kinase (PI3K)/AKT pathway, is key in regulating cell survival, apoptosis and protein translation, and has been described in the tumorigenesis of both HCC and CC.4 Activation of this pathway results in activation of the mammalian target of rapamycin (mTOR) pathway, leading to the transcription of genes involved in angiogenesis and survival.5 Therefore, Pten loss results in constitutive activation of the PI3K/AKT pathway. Varying protein expression patterns of AKT and mTOR have been reported in CC.6,7 Low intra-tumoral PTEN expression has been associated with shorter overall survival (OS) when compared to tumors with high PTEN expression.8 Concomitant deletion of Pten and SMAD4 (a mediator of TGF-β and a frequently altered tumor suppressor in CC) produces murine tumors with intrahepatic CC with evidence of increased mTOR pathway activation.9 Similarly, higher levels of p-AKT implicate this pathway in the development of HCC.10 In a small study of human HCC specimens, PI3K expression was detected in all cases reviewed with reduced or absent Pten expression.11
Data also implicate the hypoxia inducible factor (HIF) family of transcription factors and oxidative damage in liver tumors. HIF is induced by hypoxia, resulting in the transcriptional activation of target genes involved in the cellular adaptation to hypoxia.12 HIFα subunits are normally degraded in the presence of oxygen, but are stabilized under hypoxic conditions or in the setting of pVHL loss. In the liver, HIF-1α has been associated with protection against hepatic steatosis in response to liver injury.13,14 Human CC tumors have been reported to overexpress both reactive oxygen and nitrogen species, which correlate with HIF-1α expression in these tumors.15 The role of HIF stabilization in these cancers as a protective factor or a determinant of more aggressive disease remains unclear.
Mouse models of primary liver tumors are rare and specific molecular contributors to the phenotypic determinants of liver tumors are largely unknown. In order to better elucidate the pathways integral to the development of intrahepatic malignancies, our group generated a mouse model of liver tumors via conditional deletion of Pten and Vhl alone or in combination in adult animals using a Keratin 18 creER recombinase promoter, which is expressed in the bile duct epithelium and activates recombination on treatment with tamoxifen for both genes. Pten mutation has previously been implicated in both cancers. We used deletion of Vhl as a strategy to constitutively stabilize HIF factors in the absence of hypoxia. Liver tumors demonstrated a wide range of tumor phenotypes and penetrance. Expression of genes in the HIF pathway and genes related to fatty liver were examined in the mouse tumors as well as in an established dataset of human HCC and CC to identify commonalities between mouse genotypes and human tumor phenotypes. We also explored associations between clinicopathologic characteristics and clinical outcomes in an independent cohort of human CC samples that were evaluated for protein expression of HIF-1α. The findings of this study suggest that different genetic or molecular changes in susceptible cell populations in the liver influence the development of hepatosteatosis as well as the pathogenesis of a spectrum of malignant histologies.
Materials and methods
Mouse model
Mice were bred and maintained in the Laboratory Animal Facility of the University of North Carolina (UNC) using protocols approved by the UNC Institutional Animal Care and Use Committee. Transgenic mice with Keratin18 (K18)-linked CreERTM 16 were bred with a loxP-flanked Pten (PtenloxP/loxP)17 and loxP-flanked Vhl alleles (VhlloxP/loxP).18 Multiple rounds of cross-breeding resulted in the generation of mice with the following genotypes: K18 CreER;VhlloxP/loxP, K18 CreER; PtenloxP/loxP, and K18 CreER;PtenloxP/loxP;VhlloxP/loxP on a mixed genetic background (BALB/c, C57BL/6). Mice were genotyped following methods described previously.18,19
To induce Pten and/or Vhl loss by CreER recombination in somatic cells of adult animals, mice aged 6 weeks were injected with 1 mg tamoxifen (T5648; Sigma-Aldrich Co, St Louis, MO, USA) in sunflower seed oil (S5007; Sigma-Aldrich Co) daily for 5 days. Mice were sacrificed at 1 year, or when palpable masses were identified or animals showed signs of distress. Following recombination, mice were referred to as Vhl-/-, Pten-/-, and Pten-/-;Vhl-/-.
Histology and immunohistochemistry (IHC)
Tissues harvested from Vhl-/-, Pten-/-, and Pten-/-;Vhl-/- mice were fixed in 10% formalin and embedded in paraffin. Sections (4 μm) were stained with hematoxylin and eosin for histologic evaluation. Antigen retrieval was performed by boiling in citrate buffer (pH 6.0; BioGenex, Fremont, CA, USA) for 30 minutes. Endogenous peroxidase activity was quenched in 3% H2O2 for 15 minutes. Human samples were stained with anti-phosphoAKT (1:50, Cell Signaling Technology 211S, Danvers, MA, USA), and anti-HIF-1α (1;200, Novus Biologicals LLC, Littleton, CO, USA). Detection for all antibodies was performed using the Vector ABC Elite Kit and a Vector DAB kit for substrate detection (Vector Labs, Burlingame, CA, USA).
Gene expression analysis
RNeasy (74104; Qiagen NV, Venlo, the Netherlands) was used to extract ribonucleic acid (RNA) from flash frozen macrodissected mouse tumors and submitted to the UNC Genomic Core for quality check, amplification, labeling, and hybridization against reference RNA (Stratagene Universal Mouse Reference RNA 740100-41) on Agilent Whole Mouse Gene Expression (4×44 K) Microarray (G2519F; Agilent Technologies, Santa Clara, CA, USA). Expression data were refined to include only known members of the HIF pathway or only a set of genes related to fatty liver compiled from previously published data.20–23 Using either the list of HIF pathway genes or the set of fatty liver genes, clustering was performed using Cluster 3.0 (http://www.bonsai.ims.u-tokyo.ac.jp/~mdehoon/software/cluster/). Heat maps were constructed in Java Treeview (http://www.jtreeviewsourceforge.net/). These data have been deposited in GEO (Gene expression omnibus), accession number GSE55874.
Human gene expression data for HCC, CC, and mixed HCC/CC were described previously.24 Mixed HCC/CC expression data were excluded from the analysis to simplify the analysis. As described above, expression data were refined to include only known components of the hypoxia response pathway or only a set of genes related to fatty liver disease. Boxplots used to compare expression of specific genes were constructed in R v2.4.1 (http://www.rproject.org).
Human samples, survival data, and statistical analysis
Twenty-seven paraffin-embedded CC samples collected between June 2001 and November 2009 were identified using the UNC Tumor Registry. Clinicopathologic data, including patients’ demographics, tumor stage and tumor characteristics as well as treatment course (surgical resection, chemotherapy, and/or radiotherapy) and survival outcome were obtained from the medical record after obtaining approval for waiver of consent from the UNC Biomedical IRB Committee of the Office of Human Research Ethics.
IHC was evaluated using a proportion score for neoplastic cells on a scale of 0 to 5, and an intensity score for neoplastic cells expressed on a scale of 0 to 3. When intensity varied within a given neoplasm, the average intensity was judged. A combined score (proportion score + intensity score) of at least 5 was used to determine positive protein expression.
OS was defined as the time from the date of diagnosis to death from any cause. OS was described using the method of Kaplan–Meier. OS was compared between the positive and negative HIF-1α groups using the log-rank test. Additionally, Fisher’s exact tests were used to compare clinicopathologic characteristics between the positive and negative HIF-1α groups.
Results
Somatic loss of Pten alone or in combination with Vhl deletion produces distinct tumor phenotypes in the liver
No tumor formation or evidence of hepatic injury was observed in Vhl-/- mice aged 12 months, although some expansion of vascular sinusoids was appreciated (Figure 1A and B). However, both Pten-/- and Pten-/-;Vhl-/- mouse populations formed tumors located exclusively in the liver with overt tumor phenotype onset (palpable mass, weight loss, ruffed coat) at approximately 9–12 months. No abnormalities were observed in other tissues. Tumors were detectable only on histologic evaluation in the majority of Pten-/-;Vhl-/- animals, but were grossly visible as numerous massive tumors in Pten-/- mice. Pten-/-;Vhl-/- mouse tumors were small and widely dispersed (Figure 1C–E), and displayed a trabecular morphology (Figure 1D and E, white arrows) with large, unencapsulated, expansile and partially infiltrative masses of neoplastic cells replacing and compressing normal remaining hepatocytes, consistent with histologic findings of a CC. Tumors from Pten-/- mice, however, histologically showed highly variable neoplastic cells that typically arose within foci of hepatic alteration (Figure 1F–H). The phenotype was indicative of a poorly-differentiated carcinoma with mixed glandular and solid morphologies (Figure 1F, black arrows), and may represent a hepatocellular or mixed cell neoplasm.
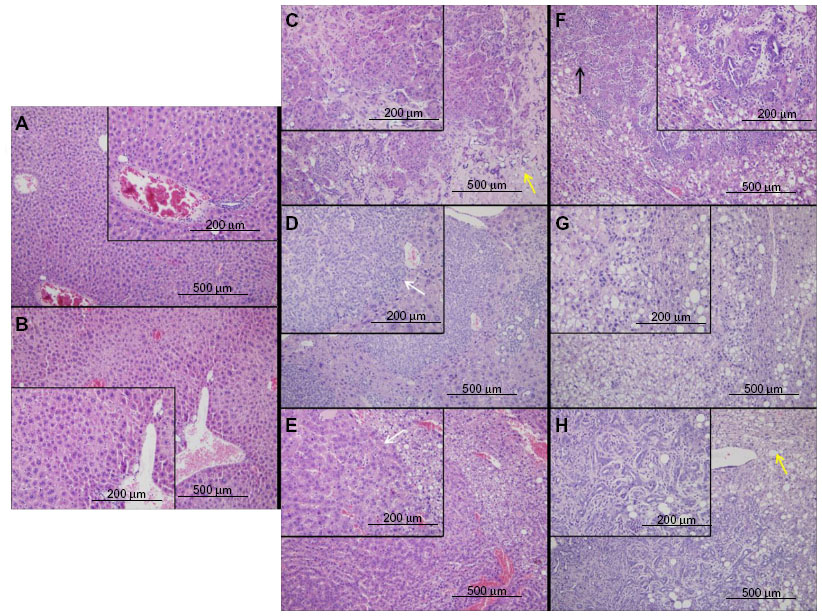 | Figure 1 Liver and tumor tissue from mice with different genotypes show differing phenotypes on histological examination. |
Mouse Vhl-/-, Pten-/-;Vhl-/- and Pten-/- tumors show different levels of apparent fatty liver development
Vhl-/- livers showed no areas of fatty liver disease. However, both Pten-/- and Pten-/-;Vhl-/- livers displayed some lipid accumulation. In the Pten-/-;Vhl-/- livers, this was manifested as small foci of steatosis, with lipid filled vesicles in hepatocytes generally in the vicinity of areas of tumor formation (Figure 1C, yellow arrows). Pten-/- livers, on the other hand, were nearly completely replaced by hepatocytes showing macrovesicular steatosis, without evidence of inflammation, in all areas regardless of the presence of tumor cells. Areas of lipid accumulation and fatty change are indicated by yellow arrows (Figure 1F–H).
Mouse Vhl-/-, Pten-/-;Vhl-/- and Pten-/- tumors differ in expression of HIF target genes and in expression of fatty liver-related genes
To verify the expected relative expression changes corresponding with Vhl deletion, we examined the tumors of Pten-/- and Pten-/-;Vhl-/- mice by microarray expression profiling. Direct assessment of HIFα protein was not possible due to high levels of nonspecific interactions in multiple antibodies tested. We observed an expected decrease in Vhl gene expression in the Pten-/-;Vhl-/- samples, compared to the single deletion mutant, Pten-/- (Figure 2A). A corresponding increase in expression of a panel of canonical HIF target genes was also observed in those tumors harboring a Vhl deletion (Figure 2A).
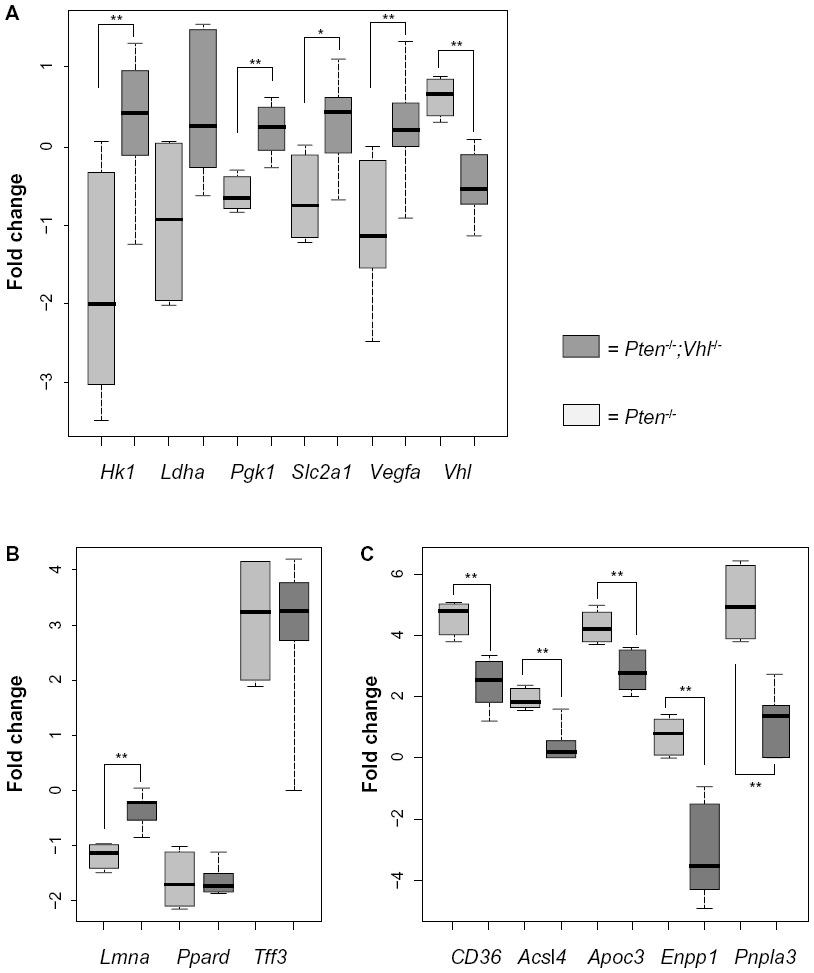 | Figure 2 Genes related to hypoxia and fatty liver are differentially expressed between mouse genotypes. |
Because the two liver genotypes displayed markedly varying amounts of hepatic steatosis, we also examined the expression of murine orthologs of genes previously identified as downregulated (Lmna, Ppard, Tff3), or upregulated (CD36, csl4, Apoc3, Enpp1, Pnpla3) in human fatty liver disease.20–23 Genes reported to be downregulated in humans were either reduced or unchanged in the Pten-/- mouse tumors relative to Pten-/-;Vhl-/- tumors (Figure 2B). Those genes reported to be upregulated in human fatty liver disease were more highly expressed in Pten-/- relative to Pten-/-; Vhl-/- (Figure 2C), consistent with the differences observed histologically.
Fatty liver gene expression differs significantly between human HCC and CC
Given that the livers from the Pten-/- and Pten-/-;Vhl-/- mice showed varying degrees of lipid disposition, but shared histologic features within the spectrum of tumors ranging from CC to HCC, we investigated the differences in expression of genes involved in fatty liver disease between human HCC or CC in an established and publicly available dataset.24 This investigation considers the possibility that the genes associated with hepatic steatosis might be more prevalent in human HCC as compared with CC. Indeed, when we examined a larger cohort of fatty liver genes,20–23 we observed a significantly reduced signal in the CC samples relative to HCC, shown in the heat map (Figure 3A). Specifically exploring the expression of the same panel of genes found to be coordinately up- or downregulated in the mouse tumors, we observed changes in gene expression consistent with fatty liver disease primarily in the HCC sample dataset (Figure 3B and C). These findings are suggestive of liver damage represented by steatosis as being linked with HCC in this animal model. The relationship of these levels of expressed genes to the non-malignant hepatocyte compartment cannot be determined from these studies.
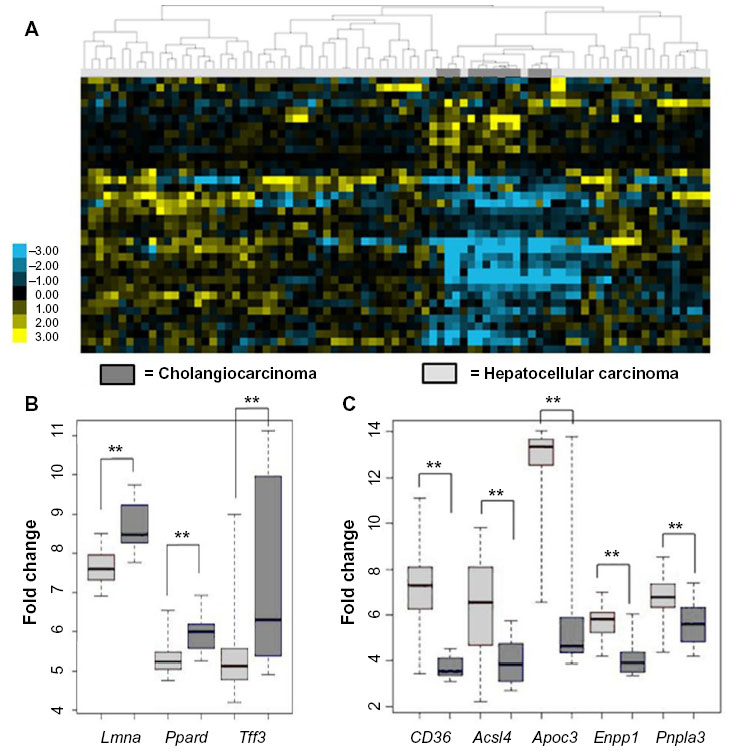 | Figure 3 Human HCC and CC gene expression datasets show different regulation of genes related to fatty liver. |
Human HCC and CC differentially express HIF target genes
In a similar analysis, we examined the expression of common HIF target genes in human HCC and CC tumors. Vhl has not been implicated as a disease gene in either human HCC or CC, however, multiple mechanisms can alter hypoxia signaling. Using the same panel of canonical HIF target genes examined in the Pten-/- and Pten-/-;Vhl-/- mice, we also observed a significant increase in the expression of hypoxia target genes hexokinase and SLC2A1 in the human CC dataset as compared with HCC (Figure 4A). The expression level of VHL is unchanged, as expected, suggesting that any activation of hypoxia response signaling likely occurs via physiologic hypoxia signaling.
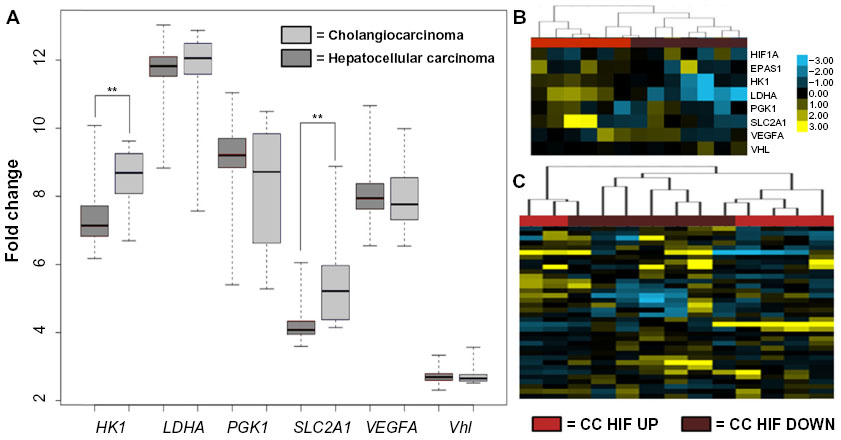 | Figure 4 Genes specific to the hypoxia response show variable expression between human HCC and CC. |
The histologic features of Pten-/-;Vhl-/- tumors were most consistent with CC, however, both genotypes of mouse tumors had elements of the trabecular or glandular architecture common to CC. We therefore sought to determine if hypoxia signaling might influence the severity of intrahepatic CC disease. Using the publicly available dataset, we indeed observed that a subset of CC tumors displayed upregulation of HIF target genes (Figure 4B). Furthermore, when we classified CC tumors according to their hypoxia gene signature (CC HIF UP or CC HIF DOWN), we observed correspondence with fatty liver genes such that those tumors with upregulation of HIF targets displayed downregulation of fatty liver genes and vice versa (Figure 4C). This finding suggests that tumor hypoxia, or the expression of genes associated with tumor hypoxia, may influence the development of hepatic steatosis in the development of liver tumors.
An independent cohort of human CC samples express variable levels of HIF-1α
To more directly examine the role of HIF expression in influencing tumor phenotype, we identified a cohort of CC cases at UNC for protein analysis. The clinicopathologic characteristics of 27 CC patients and their tumors are summarized in Table 1. Via IHC, 56% of these tumors expressed HIF-1α. Univariate analysis of OS in these patients based on HIF-1α expression was not statistically significant (60 versus 30.9 months, P=0.402) (Figure 5). In comparing the clinicopathologic variables of these two groups, patients whose tumors displayed positive HIF-1α expression were more likely to have resectable tumors with only 13% being un-resectable compared to 50% of the HIF-1α negative tumors (P=0.053; Table 2). The small sample size and exploratory nature of this study begs for future studies aimed at determining whether targeting the HIF pathway has any bearing on clinical outcomes.
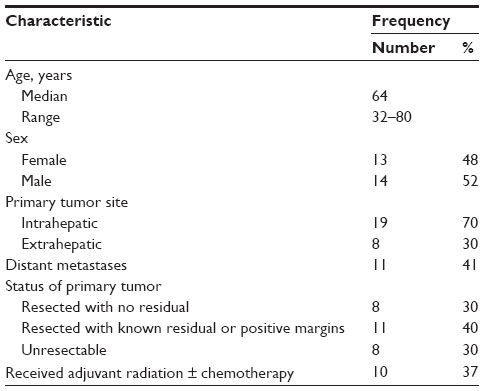 | Table 1 Clinicopathologic characteristics of an independent cohort of human cholangiocarcinoma samples |
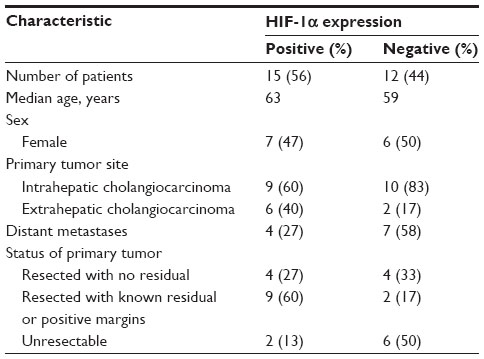 | Table 2 Clinicopathologic characteristics of human cholangio-carcinoma samples based on HIF-1α expression |
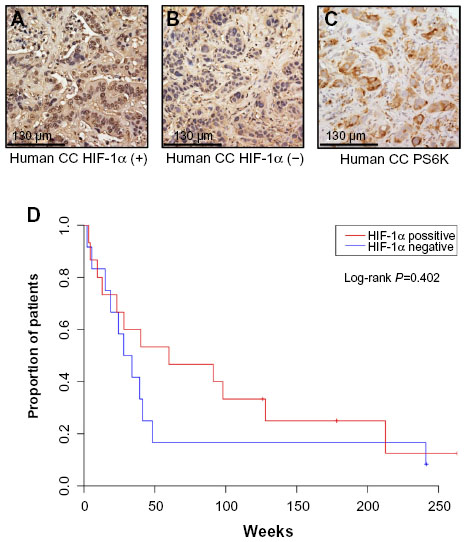 | Figure 5 HIF-1α is variably expressed in a UNC cohort of human CC and may influence disease severity. |
Discussion
This study describes three unique phenotypes of liver histology based on the conditional deletion of Pten and/or Vhl in a mouse model. The somatic deletion of these genes using a promoter targeting the biliary tree in adult animals suggests that Keratin 18 expressing cells contribute substantially in adulthood to the liver as a whole. While mice with Vhl loss alone displayed no visible pathology in the liver, those with Pten loss (either as a single insult or in combination with Vhl deletion) developed a spectrum of liver tumors with various degrees of steatosis. These findings suggest that Vhl loss alone is not sufficient or necessary for the pathogenesis of liver cancer but that Pten inactivation is a key contributor to the development of malignancy. This is congruent with other studies that have described increased PI3K and AKT expression, both downstream effectors of Pten loss, in human HCC and CC.25,26 The novel contribution of this study is revealing that this effect is modulated by deregulation of hypoxia response signaling.
The deletions in these mice took place in cells expressing cre-ER from the Keratin 18 promoter. Although Keratin 18 is widely expressed in a variety of epithelial compartments, in the liver we only observed expression in bile duct epithelial cells (Figure S1), and we observed tumor development in no other organs of these animals. The deletion of Pten and/or Vhl in the Keratin 18 expressing cells in the liver suggests that a range of liver tumor features can arise from possible vulnerable cells in the bile duct. From this small compartment of target cells, we observed widespread effects on both the non-malignant liver parenchyma in addition to supporting the development of neoplasms in the liver. Both Pten-/- and Pten-/-; Vhl-/- mice showed some degree of hepatic steatosis, with Pten-/- mice demonstrating widespread fatty replacement of normal liver and Pten-/-; Vhl-/- demonstrating largely normal hepatic tissue with occasional foci of steatosis. In a previously described Pten liver-specific knockout model, apolipoprotein B, a known regulator of lipogenesis that serves as a lipid transporter, was reduced, leading to increased intrahepatic triglyceride formation and pronounced hepatosteatosis.27 Our model suggests that concomitant loss of Vhl in the setting of Pten deletion results in an attenuated fatty liver phenotype; however, the metabolic features supporting the preservation of normal architecture remain unclear.
Our analysis also revealed that genes associated with hepatic steatosis were coordinately expressed in both the Pten-/- samples and gene expression data available for human HCC. Pten-/-;Vhl-/- tumors did not display this association, and neither was it observed in the expression data derived from human CC. Conversely, genes canonical to hypoxia signaling were upregulated in Pten-/-;Vhl-/- tumors as would be predicted, as well as in a significant subset of CC. Taken together, these distinct gene sets separating tumors originating from a common susceptible tissue and driven by Pten deletion, but modified by Vhl loss, implicate integrated pathophysiologic signals ultimately contributing to the tumor phenotype.
In our cohort of human CC samples, we found that intra-tumoral expression of HIF-1α correlated with increased resectability and showed a trend toward improved OS, suggesting that even within this histologic disease entity, hypoxia signaling may contribute to the final disease phenotype. This finding differs from other published reports in which intra-tumoral expression of HIF-1α has traditionally correlated with localized hypoxia and tissue necrosis, as well as higher stage, larger tumors and worsened prognosis.28–30 However, HIF-1α can display tumor suppressive activity in other tumor settings, suggesting that the influence of HIF-1α on tumor phenotype may be highly context-dependent.31–33 Alternatively, components of hypoxic physiology could override tumor suppressive signals.
Conclusion
This study is the first to examine how Pten and Vhl inactivation contributes to the molecular pathogenesis of primary cancers of the liver. The Vhl gene is not reported to be a common target in any liver cancers,34–36 and our model indicates that Vhl inactivation alone is not sufficient for tumor formation, but that conditional loss of Pten in a subset of cells results in tumor formation that roughly phenocopies HCC in humans. In this setting, Vhl deletion serves as a surrogate for constitutive activation of the hypoxia response pathway, and the loss of both Pten and Vhl predisposes to a CC phenotype of cancer. Interestingly, the histologic appearance of the liver parenchyma, as well as the tumor spectrum, is altered with concomitant deletion of Vhl. Further studies are needed to further understand the contributions of hypoxia signaling to the development of hepatic steatosis and tumorigenesis. Studies revealing the cell of origin for these tumors and the factors that contribute to disease phenotype can provide targets that will potentially lead to more effective therapies to modulate these aggressive malignancies.
Acknowledgments
The authors would like to acknowledge the support for this project from generous donations by the Stegman family, and grant funding from the UNC Lineberger Cancer Center (AJM), the UNC Calabresi K12 Career Development Grant (AJM), PREP Scholar Program R25 GM089569 (ALC), K24 CA172355 (WKR) and R01 CA121781 (WKR, SC). In addition, we thank Dr Bert O’Neil and members of the Rathmell lab for many helpful discussions. Finally, we thank the Gene Expression Core Facility and Animal Histopathology Core Facility, both supported by the UNC Lineberger Cancer Center (P30 CA016086 – Cancer Center Core Support Grant; Cancer Center Support; Developmental Funds; Tissue Procurement Facility; Translational Pathology Lab; Program Leadership; staff investigators).
Disclosure
ABS, KEH, SC, ALC, OS, AS, ABR, SAM, WKR and AJM have no relevant disclosures. DC is now an employee of Novartis Pharmaceuticals.
References
Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220–241. | |
Cazals-Hatem D, Rebouissou S, Bioulac-Sage P, et al. Clinical and molecular analysis of combined hepatocellular-cholangiocarcinomas. J Hepatol. 2004;41(2):292–298. | |
Walther Z, Jain D. Molecular pathology of hepatic neoplasms: classification and clinical significance. Patholog Res Int. 2011;2011: 403929. | |
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | |
Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol. 2003;23(17):6139–6149. | |
Herberger B, Puhalla H, Lehnert M, et al. Activated mammalian target of rapamycin is an adverse prognostic factor in patients with biliary tract adenocarcinoma. Clin Cancer Res. 2007;13(16):4795–4799. | |
Javle MM, Yu J, Khoury T, et al. Akt expression may predict favorable prognosis in cholangiocarcinoma. J Gastroenterol Hepatol. 2006; 21(11):1744–1751. | |
Chung JY, Hong SM, Choi BY, Cho H, Yu E, Hewitt SM. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin Cancer Res. 2009;15(2):660–667. | |
Xu X, Kobayashi S, Qiao W, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116(7):1843–1852. | |
Zhou B, Li Y, Deng Q, et al. SRPK1 contributes to malignancy of hepatocellular carcinoma through a possible mechanism involving PI3K/Akt. Mol Cell Biochem. 2013;379(1–2):191–199. | |
Bassullu N, Turkmen I, Dayangac M, et al. The predictive and prognostic significance of c-erb-B2, EGFR, PTEN, mTOR, PI3K, p27, and ERCC1 expression in hepatocellular carcinoma. Hepat Mon. 2012; 12(10 HCC):e7492. | |
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. | |
Yoo W, Noh KH, Ahn JH, et al. HIF-1alpha expression as a protective strategy of HepG2 cells against fatty acid-induced toxicity. J Cell Biochem. 2014;115(6):1147–1158. | |
Nishiyama Y, Goda N, Kanai M, et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J Hepatol. 2012;56(2):441–447. | |
Pinlaor S, Sripa B, Ma N, et al. Nitrative and oxidative DNA damage in intrahepatic cholangiocarcinoma patients in relation to tumor invasion. World J Gastroenterol. 2005;11(30):4644–4649. | |
Wen F, Cecena G, Munoz-Ritchie V, Fuchs E, Chambon P, Oshima RG. Expression of conditional cre recombinase in epithelial tissues of transgenic mice. Genesis. 2003;35(2):100–106. | |
Trotman LC, Niki M, Dotan ZA, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1(3):E59. | |
Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98(4):1583–1588. | |
Chen S, Sanford CA, Sun J, et al. VHL and PTEN loss coordinate to promote mouse liver vascular lesions. Angiogenesis. 2010;13(1):59–69. | |
Guillen N, Navarro MA, Arnal C, et al. Microarray analysis of hepatic gene expression identifies new genes involved in steatotic liver. Physiol Genomics. 2009;37(3):187–198. | |
Westerbacka J, Kolak M, Kiviluoto T, et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes. 2007;56(11):2759–2765. | |
Chiappini F, Barrier A, Saffroy R, et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Lab Invest. 2006;86(2):154–165. | |
Hooper AJ, Adams LA, Burnett JR. Genetic determinants of hepatic steatosis in man. J Lipid Res. 2011;52(4):593–617. | |
Woo HG, Lee JH, Yoon JH, et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 2010;70(8):3034–3041. | |
Yothaisong S, Dokduang H, Techasen A, et al. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumour Biol. 2013;34(6):3637–3648. | |
Zheng L, Gong W, Liang P, et al. Effects of AFP-activated PI3K/Akt signaling pathway on cell proliferation of liver cancer. Tumour Biol. 2014;35(5):4095–4099. | |
Qiu W, Federico L, Naples M, et al. Phosphatase and tensin homolog (PTEN) regulates hepatic lipogenesis, microsomal triglyceride transfer protein, and the secretion of apolipoprotein B-containing lipoproteins. Hepatology. 2008;48(6):1799–1809. | |
Morine Y, Shimada M, Utsunomiya T, et al. Hypoxia inducible factor expression in intrahepatic cholangiocarcinoma. Hepatogastroenterology. 2011;58(110–111):1439–1444. | |
Batmunkh E, Shimada M, Morine Y, et al. Expression of hypoxia-inducible factor-1 alpha (HIF-1alpha) in patients with the gallbladder carcinoma. Int J Clin Oncol. 2010;15(1):59–64. | |
Miyake K, Yoshizumi T, Imura S, et al. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas. 2008;36(3):e1–e9. | |
Mack FA, Rathmell WK, Arsham AM, Gnarra J, Keith B, Simon MC. Loss of pVHL is sufficient to cause HIF dysregulation in primary cells but does not promote tumor growth. Cancer Cell. 2003;3(1):75–88. | |
Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14(6):435–446. | |
Shen C, Beroukhim R, Schumacher SE, et al. Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1(3):222–235. | |
Chan-On W, Nairismagi ML, Ong CK, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45(12):1474–1478. | |
Ahn SM, Jang SJ, Shim JH, et al. A genomic portrait of resectable hepatocellular carcinomas: Implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60(6):1972–1982. | |
Nault JC, Zucman-Rossi J. Genetics of hepatocellular carcinoma: the next generation. J Hepatol. 2014;60(1):224–226. |
Supplementary material
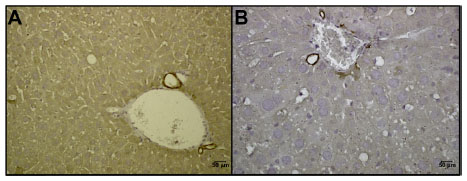 | Figure S1 Keratin 18 staining in normal liver identifies sites of probable tumor origin. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.