")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Virtual Screening and Optimization of Novel mTOR Inhibitors for Radiosensitization of Hepatocellular Carcinoma
Authors Feng YQ, Gu SX , Chen YS, Gao XD, Ren YX, Chen JC , Lu YY, Zhang H, Cao S
Received 13 February 2020
Accepted for publication 9 April 2020
Published 8 May 2020 Volume 2020:14 Pages 1779—1798
DOI https://doi.org/10.2147/DDDT.S249156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Ying-Qi Feng,1,* Shuang-Xi Gu,1,* Yong-Shou Chen,1,* Xu-Dong Gao,2 Yi-Xin Ren,1 Jian-Chao Chen,3 Yin-Ying Lu,2 Heng Zhang,1 Shuang Cao1,4
1Key Laboratory for Green Chemical Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan 430072, People’s Republic of China; 2Comprehensive Liver Cancer Department, The Fifth Medical Center, Chinese PLA General Hospital, Beijing 100039, People’s Republic of China; 3Key Laboratory of Structure-Based Drug Design and Discovery, Shenyang Pharmaceutical University, Ministry of Education, Shenyang 110016, People’s Republic of China; 4National Engineering Research Center for the Emergency Drug, Beijing Institute of Pharmacology and Toxicology, Beijing 100850, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shuang Cao; Heng Zhang
Key Laboratory for Green Chemical Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan 430072, People’s Republic of China
Tel/ Fax +86-18701418117; +86-15994288097
Email [email protected]; [email protected]
Background: Radiotherapy has an ameliorative effect on a wide variety of tumors, but hepatocellular carcinoma (HCC) is insensitive to this treatment. Overactivated mammalian target of rapamycin (mTOR) plays an important part in the resistance of HCC to radiotherapy; thus, mTOR inhibitors have potential as novel radiosensitizers to enhance the efficacy of radiotherapy for HCC.
Methods: A lead compound was found based on pharmacophore modeling and molecular docking, and optimized according to the differences between the ATP-binding pockets of mTOR and PI3K. The radiosensitizing effect of the optimized compound ( 2a) was confirmed by colony formation assays and DNA double-strand break assays in vitro. The discovery and preclinical characteristics of this compound are described.
Results: The key amino acid residues in mTOR were identified, and a precise virtual screening model was constructed. Compound 2a, with a 4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine scaffold, exhibited promising potency against mTOR (mTOR IC50=7.1 nmol/L (nM)) with 126-fold selectivity over PI3Kα. Moreover, 2a significantly enhanced the sensitivity of HCC to radiotherapy in vitro in a dose-dependent manner.
Conclusion: A new class of selective mTOR inhibitors was developed and their radiosensitization effects were confirmed. This study also provides a basis for developing mTOR-specific inhibitors for use as radiosensitizers for HCC radiotherapy.
Keywords: virtual docking, HCC, kinase inhibitor, mTOR, radiosensitizer
Introduction
Hepatocellular carcinoma (HCC), a primary liver malignancy, is the third leading cause of cancer-related deaths worldwide.1,2 Many HCCs are diagnosed at an advanced stage and so can only be treated conservatively. Radiotherapy, a non-invasive therapy that can selectively destroy various tumor tissues, is a promising strategy for HCC treatment.3 However, it has been reported that advanced HCC is usually insensitive to ionizing radiation (IR); moreover, the liver can only safely endure IR doses of 90 Gy or less, which is not sufficient for cancer control.4,5 Therefore, it is necessary to develop new strategies to enhance radiotherapeutic efficacy in treating HCC.
mTOR is a serine/threonine protein kinase with a molecular weight of approximately 280 kDa. mTORC1 and mTORC2 are two multiprotein complexes of mTOR, composing of discrete protein and partners. They play an important part in multiple signaling pathways and affects a wide range of physiological functions, including cell growth, proliferation, protein synthesis, autophagy, metabolism, and survival.6 Overactive mTOR is one of the causes of cell over-proliferation and malignant transformation, and inhibition of mTOR has anti-tumor, immunosuppressive, and anti-aging effects.7,8
Reported mTOR inhibitors include selective mTOR inhibitors and mTOR/PI3K dual inhibitors. Wye-125132, Torin1, and way-600 are classic selective mTOR inhibitors. However, nearly all researches using selective mTOR inhibitors alone for anti-tumor treatment are only in clinical stage I or II until now.9–11 Some studies have shown that use of selective mTOR inhibitors alone has a limited inhibitory effect on tumors because of the compensatory effect of certain pathways, especially in the treatment of HCC.12–17 The mTOR/PI3K dual inhibitors include BEZ235 and GDC-0980.18,19 Although the anti-tumor effects of these inhibitors are better than those of selective mTOR inhibitors, the inhibition of two important pathways, mTOR and PI3K, usually leads to significant side effects. Therefore, development of mTOR/PI3K dual inhibitors has also been relatively slow in recent years.18,20
Selective mTOR inhibitors have been shown to enhance the radiosensitivity of cancer cells.21–23 There have been some reports of the use of mTOR inhibitors in radiotherapy.24,25 For example, Liu et al and Wang et al studied the sensitization effects of PI3K/mTOR dual inhibitors in pancreatic cancer and glioma cancer;26,27 and Kahn et al and Hayman et al reported that selective mTOR inhibitors could enhance the sensitivity of pancreatic carcinoma and glioblastoma cells to radiotherapy.21,28 However, there have been few studies of small-molecule selective mTOR inhibitors in radiosensitization of HCC. This may be because selective mTOR inhibitors were previously thought to be ineffective in the treatment of HCC, based on the few reports published in this field. Thus, we decided to focus on this question in the present work.
In addition to converting the application fields, discovering compounds with diverse structures is another approach to developing mTOR inhibitors.29 In this study, based on 2a (with a 4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine scaffold), we describe the identification and characterization of a new type of mTOR inhibitor and the process of optimizing the lead compound.30 Virtual screening and pharmacophore modeling were used to identify the new scaffold of the lead compound. A highly aggressive HCC cell line, MHCC97-H, was used as an in vitro tumor model, and 60Co-γ IR was used to treat MHCC97-H cells to mimic radiotherapy for HCC.31,32
Materials and Methods
Chemistry
General Procedure for the Preparation of 1a–1d, 2a–2j, and 3a–3h
Step 1: Preparation of methyl 3-((5-amino-1H-1,2,4-triazol-3-yl)thio)propanoate (Intermediate 1)
Methyl 3-bromopropanoate (14.36 g, 0.086 mol) was dissolved in absolute ethyl alcohol (40 mL) and set aside. NaOH (3.44 g, 0.086 mol) was dissolved in 200 mL purified water in a 500-mL reactor and stirred until completely dissolved. 3-amino-5-mercapto-1,2,4-triazole (10.00 g, 0.086 mol) was added and stirred. After 30 min, the solution of methyl 3-bromopropanoate and ethyl alcohol was added. The mixture was reacted overnight at room temperature. Upon completion, the product was extracted with ethyl acetate, washed with saturated salt water, and dried over Na2SO4. Methyl 3-((5-amino-1H-1,2,4-triazol-3-yl)thio)propanoate (11.65 g, yield: 67%) was obtained by reduced pressure distillation.
Step 2: Preparation of N-(5-methylpyridin-3-yl)-3-oxobutanamide (Intermediate 2)
5-methylpyridin-3-amine (10.00 g, 0.092 mol) was mixed with 200 mL methylbenzene in a 500-mL reactor and stirred until completely dissolved. Then, ethyl acetoacetate (11.97 g, 0.092 mol) was added to the reactor.33,34 The reaction mixture was heated to 100°C for 10 h, left to stand overnight at room temperature, then precipitated. The slurry was filtered, and the filter cake was rinsed with benzinum and collected. The product was vacuum dried at 40°C for 30 min to obtain N-(5-methylpyridin-3-yl)-3-oxobutanamide (13.26 g, yield: 75%).
Step 3: Preparation of compound methyl 3-((7-(3-methoxyphenyl)-5-methyl-6-((5-methylpyridin-3-yl)carbamoyl)-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)thio)propanoate (2a)
N-(5-methylpyridin-3-yl)-3-oxobutanamide (5.00 g, 0.026 mol) was mixed with absolute ethyl alcohol (80 mL) in a 500-mL reactor and stirred until completely dissolved. 3-methoxybenzaldehyde (3.54 g, 0.026 mol) and methyl 3-((5-amino-1H-1,2, 4-triazol-3-yl)thio) propanoate (5.26 g, 0.026 mol) were subsequently added into the reactor. The mixture was heated to 80°C for 3 h, and white precipitate appeared. The slurry was filtered, and the filter cake was rinsed with absolute ethyl alcohol (3×50 mL) and collected. The reaction product was vacuum evaporated at 40°C for 20 min to obtain compound 2a (5.40 g, yield: 42%). m.p.: 221.7–222.4°C. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1 H), 9.87 (s, 1H), 8.47 (d, J = 2.3 Hz, 1H), 8.11–8.05 (m, 1H), 7.81 (d, J = 2.3 Hz, 1H), 7.25 (t, J = 7.9 Hz, 1H), 6.85 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 6.83–6.71 (m, 2H), 6.50–6.44 (m, 1H), 3.68 (s, 3H), 3.60 (s, 3H), 3.17 (td, J = 7.0, 2.2 Hz, 2H), 2.73 (t, J = 6.9 Hz, 2H), 2.21 (d, J = 24.9 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 172.12, 165.61, 159.72, 157.85, 149.07, 145.08, 142.41, 138.81, 137.53, 135.70, 133.26, 130.26, 127.22, 119.55, 113.55, 113.50, 103.87, 60.44, 55.47, 51.99, 34.45, 26.57, 18.34, 18.31, 17.82. MS (ESI) m/z: 495.57 [M+H]+. Anal. C24H26N6O4S: C 58.29, H 5.30, N 16.99. Found: C 58.32, H 5.30, N 17.02. (NMR data for all newly synthesized compounds can be found in the Supplementary Material).
The title compound was synthesized according to the general procedure to give a white power. (total yield: 16.2%). m.p.: 232.2–233.4°C. 1H NMR (600 MHz, DMSO-d6) δ 10.34 (s, 1H), 9.90 (s, 1H), 8.68 (s, 1H), 8.24 (d, J = 4.6 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.30 (dd, J = 8.4, 4.7 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.79 (d, J = 7.7 Hz, 1H), 6.74 (s, 1H), 6.48 (s, 1H), 3.69 (s, 3H), 3.37 (t, J = 6.3 Hz, 2H), 3.01 (h, J = 7.0, 6.5 Hz, 2H), 2.19 (s, 3H), 1.84 (p, J = 6.8 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.65, 159.72, 158.30, 149.01, 144.73, 142.52, 141.60, 137.70, 136.08, 130.24, 126.95, 123.98, 119.51, 113.49, 103.79, 70.65, 60.40, 58.28, 55.47, 29.74, 28.30, 17.83. MS (ESI) m/z: 467.69 [M+H]+. Anal. C23H26N6O3S: C 59.21, H 5.62, N 18.01. Found: C 59.25, H 5.64, N 18.05.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 18.8%). m.p.: 227.4–229.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 9.93 (s, 1H), 8.68 (d, J = 2.5 Hz, 1H), 8.23 (dd, J = 4.7, 1.4 Hz, 1H), 7.94 (ddd, J = 8.4, 2.6, 1.5 Hz, 1H), 7.34–7.20 (m, 2H), 6.85 (dd, J = 8.2, 2.5 Hz, 1H), 6.82–6.71 (m, 2H), 6.49 (s, 1H), 4.87 (t, J = 4.6 Hz, 1H), 3.93–3.70 (m, 4H), 3.33 (s, 2H), 3.02 (td, J = 7.2, 2.1 Hz, 2H), 2.54–2.47 (m, 1H), 2.19 (s, 3H), 1.92 (td, J = 7.6, 4.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.64, 159.73, 158.17, 149.04, 144.73, 142.49, 141.60, 137.71, 136.08, 130.25, 126.95, 123.98, 119.50, 113.54, 113.46, 103.79, 102.68, 64.76, 60.40, 55.47, 34.13, 26.09, 17.83. MS (ESI) m/z: 495.35 [M+H]+. Anal. C24H26N6O4S: C 58.29, H 5.30, N 16.99. Found: C 58.26, H 5.31, N 17.01.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 20.3%). m.p.: 229.5–229.2°C. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 9.94 (s, 1H), 8.68 (d, J = 2.5 Hz, 1H), 8.23 (dd, J = 4.7, 1.5 Hz, 1H), 7.94 (ddd, J = 8.4, 2.6, 1.5 Hz, 1H), 7.34–7.20 (m, 2H), 6.85 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 6.83–6.71 (m, 2H), 6.49 (s, 1H), 3.68 (s, 3H), 3.60 (s, 3H), 3.18 (td, J = 7.0, 2.1 Hz, 2H), 2.73 (t, J = 6.9 Hz, 2H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.11, 165.63, 159.74, 157.87, 149.08, 144.75, 142.42, 141.61, 137.65, 136.07, 130.25, 126.96, 123.98, 119.52, 113.58, 113.47, 103.82, 60.43, 55.47, 51.97, 34.46, 26.58, 17.83. MS (ESI) m/z: 481.52 [M+H]+. Anal. C23H24N6O4S: C 57.49, H 5.03, N 17.49. Found: C 57.45, H 5.02, N 17.47.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 7.6%). m.p.: 222.3–223.6°C. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.95 (s, 1H), 8.68 (dd, J = 2.6, 0.7 Hz, 1H), 8.43–8.36 (m, 2H), 8.23 (dd, J = 4.7, 1.5 Hz, 1H), 7.94 (ddd, J = 8.4, 2.6, 1.5 Hz, 1H), 7.34–7.22 (m, 4H), 6.88 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 6.83–6.72 (m, 2H), 6.48 (d, J = 1.1 Hz, 1H), 4.26 (d, J = 14.1 Hz, 1H), 4.18 (d, J = 14.1 Hz, 1H), 3.69 (s, 3H), 2.19 (d, J = 0.9 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.61, 159.75, 157.33, 149.91, 149.09, 147.79, 144.75, 142.37, 141.60, 137.58, 136.05, 130.26, 126.95, 124.21, 123.99, 119.64, 113.61, 113.54, 103.82, 60.51, 55.50, 33.86, 17.84. MS (ESI) m/z: 486.22 [M+H]+. Anal. C25H23N7O2S: C 61.84, H 4.77, N 20.19. Found: C 61.88, H 4.76, N 20.16.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 11.4%). m.p.: 240.3–242.5°C. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 9.94 (s, 1H), 7.51 (ddd, J = 11.4, 3.4, 1.9 Hz, 1H), 7.36–7.20 (m, 3H), 6.91–6.80 (m, 2H), 6.84–6.70 (m, 2H), 6.46 (d, J = 1.2 Hz, 1H), 3.68 (s, 3H), 3.60 (s, 3H), 3.17 (td, J = 7.1, 2.5 Hz, 2H), 2.73 (t, J = 6.8 Hz, 2H), 2.17 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.11, 165.45, 163.69, 161.29, 159.72, 157.83, 149.09, 142.38, 141.21, 141.10, 137.34, 130.76, 130.66, 130.24, 119.54, 115.63, 113.55, 113.49, 110.32, 110.11, 106.78, 106.52, 104.07, 60.47, 55.45, 51.97, 34.46, 26.57, 17.79. MS (ESI) m/z: 498.42 [M+H]+. Anal. C24H24FN5O4S: C 57.94, H 4.86, N 14.08. Found: C 57.59, H 4.87, N 14.11.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 6.9%). m.p.: 254.3–256.1°C. 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 9.61 (s, 1H), 7.47–7.38 (m, 2H), 7.24 (t, J = 7.9 Hz, 1H), 6.89–6.80 (m, 3H), 6.81–6.70 (m, 2H), 6.43 (s, 1H), 3.69 (d, J = 5.9 Hz, 6H), 3.60 (s, 3H), 3.17 (td, J = 7.0, 2.6 Hz, 2H), 2.77–2.66 (m, 2H), 2.18–2.13 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.12, 164.74, 159.70, 157.71, 155.83, 149.22, 142.45, 136.14, 132.49, 130.15, 121.63, 119.58, 114.21, 113.51, 113.48, 104.50, 60.59, 55.64, 55.47, 51.97, 34.47, 26.57, 17.70. MS (ESI) m/z: 510.67 [M+H]+. Anal. C25H27N5O5S: C 59.93, H 5.34, N 13.74. Found: C 59.89, H 5.33, N 13.72.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 9.4%). m.p.: 212.7–213.0°C. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.95 (s, 1H), 8.13 (t, J = 2.0 Hz, 1H), 7.81 (ddd, J = 8.2, 2.3, 1.1 Hz, 1H), 7.64 (dt, J = 7.8, 1.3 Hz, 1H), 7.42 (t, J = 7.9 Hz, 1H), 7.25 (t, J = 7.9 Hz, 1H), 6.88–6.76 (m, 2H), 6.76 (dd, J = 2.5, 1.6 Hz, 1H), 6.49 (d, J = 1.1 Hz, 1H), 3.68 (s, 3H), 3.60 (s, 3H), 3.17 (td, J = 7.0, 2.4 Hz, 2H), 2.77–2.66 (m, 2H), 2.54 (s, 3H), 2.21–2.16 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 198.07, 172.12, 165.43, 159.71, 157.81, 149.07, 142.41, 139.82, 137.69, 137.31, 130.23, 129.53, 124.41, 123.88, 119.58, 119.08, 113.55, 113.52, 104.03, 60.45, 55.45, 51.98, 34.45, 27.17, 26.56, 17.82. MS (ESI) m/z: 522.24 [M+H]+. Anal. C25H27N5O5S: C 59.87, H 5.22, N 13.43. Found: C 59.89, H 5.33, N 13.72.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 15.2%). m.p.: 229.8–231.6°C. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 10.07 (s, 1H), 7.89 (d, J = 8.3 Hz, 2H), 7.68 (d, J = 8.4 Hz, 2H), 7.24 (t, J = 7.9 Hz, 1H), 6.84 (dd, J = 8.2, 2.5 Hz, 1H), 6.81–6.71 (m, 2H), 6.49 (s, 1H), 3.67 (s, 3H), 3.60 (s, 3H), 3.22–3.11 (m, 2H), 2.73 (t, J = 7.0 Hz, 2H), 2.18 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 196.94, 172.12, 165.58, 159.71, 157.86, 149.03, 143.82, 142.41, 137.80, 132.19, 130.26, 129.81, 119.52, 119.09, 113.55, 113.46, 103.98, 60.45, 55.45, 51.99, 34.44, 26.88, 26.56, 17.83. MS (ESI) m/z: 522.27 [M+H]+. Anal. C26H27N5O5S: C 59.87, H 5.22, N 13.43. Found: C 59.75, H 5.21, N 13.42.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 16.7%). m.p.: 224.5–226.7°C. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.87 (s, 1H), 8.47 (d, J = 2.3 Hz, 1H), 8.08 (dd, J = 1.9, 0.9 Hz, 1H), 7.84–7.78 (m, 1H), 7.25 (t, J = 7.9 Hz, 1H), 6.89–6.75 (m, 2H), 6.74 (dd, J = 2.5, 1.6 Hz, 1H), 6.47 (d, J = 1.1 Hz, 1H), 4.86 (t, J = 4.6 Hz, 1H), 3.93–3.80 (m, 2H), 3.83–3.70 (m, 2H), 3.68 (s, 3H), 3.02 (td, J = 7.2, 2.2 Hz, 2H), 2.25 (s, 3H), 2.21–2.15 (m, 3H), 1.92 (ddd, J = 8.3, 7.1, 4.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.62, 159.72, 158.15, 149.03, 145.07, 142.48, 138.83, 137.59, 135.71, 133.24, 130.25, 127.23, 119.52, 113.53, 113.50, 103.85, 102.68, 64.76, 60.42, 55.47, 34.13, 26.08, 18.34, 17.81. MS (ESI) m/z: 509.45 [M+H]+. Anal. C25H28N6O4S: C 59.04, H 5.55, N 16.52. Found: C 59.37, H 5.56, N 16.49.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 23.1%). m.p.: 227.3–238.9°C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.93 (s, 1H), 7.55–7.46 (m, 1H), 7.36–7.20 (m, 3H), 6.91–6.80 (m, 2H), 6.84–6.70 (m, 2H), 6.46 (d, J = 1.1 Hz, 1H), 4.86 (t, J = 4.6 Hz, 1H), 3.93–3.78 (m, 2H), 3.80–3.70 (m, 2H), 3.68 (s, 3H), 3.01 (tt, J = 8.4, 4.3 Hz, 2H), 2.19–2.14 (m, 3H), 1.92 (td, J = 7.6, 4.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.47, 163.69, 161.30, 159.72, 158.14, 149.05, 142.45, 141.23, 141.12, 137.40, 130.76, 130.66, 130.24, 119.52, 115.64, 113.53, 113.49, 110.31, 110.10, 106.78, 106.52, 104.05, 102.68, 64.76, 60.44, 55.46, 34.13, 26.08, 17.78. MS (ESI) m/z: 512.84 [M+H]+. Anal. C25H26FN5O4S: C 58.70, H 5.12, N 13.69. Found: C 58.73, H 5.13, N 13.73.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 17.8%). m.p.: 251.9–253.1°C. 1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 9.61 (s, 1H), 7.47–7.38 (m, 2H), 7.24 (t, J = 7.9 Hz, 1H), 6.88–6.79 (m, 3H), 6.81–6.70 (m, 2H), 6.46–6.41 (m, 1H), 4.86 (t, J = 4.6 Hz, 1H), 3.93–3.80 (m, 2H), 3.83–3.72 (m, 2H), 3.69 (d, J = 6.3 Hz, 6H), 3.01 (tt, J = 8.3, 4.3 Hz, 2H), 2.18–2.13 (m, 3H), 1.92 (ddd, J = 8.1, 7.1, 4.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.76, 159.69, 158.01, 155.82, 149.19, 142.52, 136.19, 132.51, 130.15, 121.63, 119.56, 114.21, 113.48, 104.48, 102.69, 64.76, 60.56, 55.64, 55.46, 34.14, 26.09, 17.69. MS (ESI) m/z: 524.55 [M+H]+. Anal. C26H29N5O5S: C 59.64, H 5.58, N 13.38. Found: C 59.52, H 5.57, N 13.40.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 13.6%). m.p.: 212.5–213.8°C. 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 9.92 (s, 1H), 8.10 (t, J = 1.9 Hz, 1H), 7.78 (ddd, J = 8.1, 2.2, 1.0 Hz, 1H), 7.62 (dt, J = 7.8, 1.3 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.22 (t, J = 7.9 Hz, 1H), 6.82 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 6.81–6.70 (m, 2H), 6.49–6.44 (m, 1H), 4.84 (t, J = 4.6 Hz, 1H), 3.91–3.68 (m, 4H), 3.30 (d, J = 1.8 Hz, 3H), 2.99 (tt, J = 8.3, 4.2 Hz, 2H), 2.51 (s, 3H), 2.16 (s, 3H), 2.00–1.85 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 198.07, 165.45, 159.70, 158.11, 149.02, 142.49, 139.83, 137.69, 137.37, 130.23, 129.52, 124.40, 123.86, 119.56, 119.08, 113.52, 104.01, 102.67, 64.76, 60.43, 55.44, 34.12, 27.17, 26.07, 17.82. MS (ESI) m/z: 536.71 [M+H]+. Anal. C27H29N5O5S: C 60.55, H 5.46, N 13.08. Found: C 60.49, H 5.44, N 13.04.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 19.2%). m.p.: 224.7–226.3°C. 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 10.07 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.0 Hz, 1H), 6.88–6.80 (m, 1H), 6.81–6.71 (m, 2H), 6.50 (s, 1H), 4.86 (t, J = 4.7 Hz, 1H), 3.86 (d, J = 6.5 Hz, 2H), 3.77 (d, J = 6.2 Hz, 2H), 3.67 (s, 3H), 3.01 (tt, J = 9.0, 4.6 Hz, 2H), 2.18 (s, 3H), 1.92 (tt, J = 7.7, 4.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 196.92, 165.60, 159.71, 158.16, 148.99, 143.84, 142.48, 137.87, 132.18, 130.26, 129.81, 119.49, 119.09, 113.52, 113.46, 103.96, 102.66, 64.76, 60.42, 55.45, 34.11, 26.88, 26.07, 17.84. MS (ESI) m/z: 536.32 [M+H]+. Anal. C27H29N5O5S: C 60.55, H 5.46, N 13.08. Found: C 60.69, H 5.47, N 13.07.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 11.7%). m.p.: 236.7–238.1°C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.86 (s, 1H), 9.44 (s, 1H), 8.48 (d, J = 2.3 Hz, 1H), 8.08 (dd, J = 1.9, 0.8 Hz, 1H), 7.85–7.79 (m, 1H), 7.10 (t, J = 7.8 Hz, 1H), 6.69–6.55 (m, 3H), 6.43 (d, J = 1.1 Hz, 1H), 3.61 (s, 3H), 3.24–3.12 (m, 2H), 2.73 (t, J = 7.0 Hz, 2H), 2.25 (s, 3H), 2.20–2.14 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.12, 165.60, 157.95, 157.78, 149.13, 145.04, 142.41, 138.88, 137.38, 135.74, 133.23, 130.03, 127.27, 117.80, 115.67, 114.11, 104.15, 60.48, 51.99, 34.48, 26.58, 18.35, 17.77. MS (ESI) m/z: 481.53 [M+H]+. Anal. C23H24N6O4S: C 57.49, H 5.03, N 17.49. Found: C 57.58, H 5.02, N 17.45.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 18.9%). m.p.: 259.6–261.1°C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 9.81 (s, 1H), 9.46 (s, 1H), 8.46 (d, J = 2.3 Hz, 1H), 8.07 (d, J = 1.9 Hz, 1H), 7.80 (d, J = 2.3 Hz, 1H), 7.10–7.01 (m, 2H), 6.73–6.64 (m, 2H), 6.40 (s, 1H), 3.60 (s, 3H), 3.15 (d, J = 5.0 Hz, 1H), 2.77–2.66 (m, 2H), 2.24 (s, 3H), 2.17 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.13, 165.74, 157.79, 157.55, 148.81, 145.01, 138.81, 137.15, 135.75, 133.22, 131.33, 128.85, 127.20, 115.66, 104.25, 60.16, 51.98, 34.48, 26.57, 18.34, 17.76. MS (ESI) m/z: 481.21 [M+H]+. Anal. C23H24N6O4S: C 57.49, H 5.03, N 17.49. Found: C 57.62, H 5.05, N 17.53.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 20.4%). m.p.: 231.7–233.5°C. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 9.88 (s, 1H), 8.46 (d, J = 2.3 Hz, 1H), 8.07 (dd, J = 1.9, 0.9 Hz, 1H), 7.82–7.76 (m, 1H), 7.37–7.19 (m, 4H), 6.50 (d, J = 1.1 Hz, 1H), 3.60 (s, 3H), 3.16 (dd, J = 7.0, 2.1 Hz, 1H), 2.72 (t, J = 6.9 Hz, 2H), 2.24 (s, 3H), 2.21–2.16 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.11, 165.61, 157.85, 149.05, 145.07, 140.89, 138.84, 137.45, 135.68, 133.23, 129.06, 128.68, 127.43, 127.23, 103.99, 60.63, 51.98, 34.46, 26.57, 18.33, 17.80. MS (ESI) m/z: 465.10 [M+H]+. Anal. C23H24N6O3S: C 59.47, H 5.21, N 18.09. Found: C 59.31, H 5.19, N 18.07.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 7.8%). m.p.: 235.8–236.9°C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.86 (s, 1H), 8.47 (d, J = 2.4 Hz, 1H), 8.07 (dd, J = 2.0, 0.8 Hz, 1H), 7.84–7.77 (m, 1H), 7.12 (s, 4H), 6.46 (d, J = 1.1 Hz, 1H), 3.60 (s, 3H), 3.17 (dd, J = 7.1, 1.3 Hz, 1H), 2.72 (t, J = 7.0 Hz, 2H), 2.24 (s, 6H), 2.18 (d, J = 0.9 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.12, 165.64, 157.74, 148.95, 145.03, 138.78, 138.01, 137.38, 135.73, 133.23, 129.58, 127.40, 127.16, 104.04, 60.37, 51.98, 34.47, 26.56, 21.13, 18.34, 17.80. MS (ESI) m/z: 479.69 [M+H]+. Anal. C24H26N6O3S: C 60.23, H 5.48, N 17.56. Found: C 60.37, H 5.49, N 17.59.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 23.7%). m.p.: 236.4–238.1°C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.91 (s, 1H), 8.41 (d, J = 2.3 Hz, 1H), 8.06 (dd, J = 2.0, 0.8 Hz, 1H), 7.79–7.73 (m, 1H), 7.28–7.18 (m, 1H), 7.21–7.08 (m, 3H), 6.70 (d, J = 1.2 Hz, 1H), 3.60 (s, 3H), 3.15 (td, J = 7.1, 1.2 Hz, 2H), 2.71 (t, J = 7.0 Hz, 2H), 2.37 (s, 3H), 2.25–2.16 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 172.10, 165.70, 157.65, 148.81, 145.05, 138.87, 138.65, 137.14, 136.33, 135.64, 133.27, 131.02, 128.59, 128.55, 127.03, 126.81, 103.94, 57.84, 51.97, 34.44, 26.60, 19.14, 18.32, 17.69. MS (ESI) m/z: 479.83 [M+H]+. Anal. C24H26N6O3S: C 60.23, H 5.48, N 17.56. Found: C 60.35, H 5.47, N 17.53.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 15.1%). m.p.: 237.4–238.9°C. 1H NMR (400 MHz, DMSO-d6) δ 6.83 (d, J = 1.6 Hz, 2H), 3.60 (s, 3H), 3.25–3.08 (m, 2H), 2.72 (td, J = 7.1, 1.5 Hz, 2H), 2.27–2.15 (m, 12H). 13C NMR (101 MHz, DMSO-d6) δ 172.11, 165.68, 157.65, 148.97, 145.08, 140.81, 138.90, 138.01, 137.13, 135.68, 133.23, 130.20, 127.31, 125.24, 104.09, 60.68, 51.97, 34.44, 26.60, 21.34, 18.34, 17.82. MS (ESI) m/z: 493.70 [M+H]+. Anal. C25H28N6O3S: C 60.96, H 5.73, N 17.06. Found: C 61.25, H 5.72, N 17.09.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 12.8%). m.p.: 241.5–242.7°C. 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 9.87 (s, 1H), 8.07 (d, J = 1.9 Hz, 1H), 7.78 (s, 1H), 7.39–7.06 (m, 3H), 6.50 (s, 1H), 3.60 (s, 3H), 3.16 (d, J = 2.6 Hz, 1H), 2.71 (s, 1H), 2.21 (d, J = 24.4 Hz, 6H).13C NMR (101 MHz, DMSO-d6) δ 172.11, 165.55, 161.07, 157.96, 148.91, 145.12, 138.85, 137.57, 137.11, 135.62, 133.26, 129.70, 129.62, 127.26, 116.00, 115.78, 103.76, 59.93, 51.98, 34.43, 26.55, 18.33, 17.82. MS (ESI) m/z: 483.65 [M+H]+. Anal. C23H23FN6O3S: C 57.25, H 4.80, N 17.42. Found: C 57.38, H 4.80, N 17.45.
The title compound was synthesized according to the general procedure to give a white power. (total yield: 10.9%). m.p.: 235.4–237.2°C. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 9.89 (s, 1H), 8.45 (d, J = 2.3 Hz, 1H), 8.08 (dd, J = 1.9, 0.8 Hz, 1H), 7.78 (d, J = 2.3 Hz, 1H), 7.47–7.25 (m, 1H), 7.23–6.96 (m, 3H), 6.50 (s, 1H), 3.60 (s, 3H), 3.17 (td, J = 7.0, 3.3 Hz, 2H), 2.71 (t, J = 7.0 Hz, 2H), 2.32–2.11 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 172.10, 165.52, 163.81, 161.38, 158.09, 148.98, 145.16, 143.54, 143.48, 138.84, 137.75, 135.59, 133.28, 131.16, 131.08, 127.27, 123.75, 123.72, 115.77, 115.56, 114.37, 114.15, 103.43, 60.11, 51.97, 34.42, 26.56, 18.33, 17.86. MS (ESI) m/z: 483.78 [M+H]+. Anal. C23H23FN6O3S: C 57.25, H 4.80, N 17.42. Found: C 57.42, H 4.81, N 17.46.
Molecular Modeling
Pharmacophore Model
The “GALAHAD” module available in SYBYL X2.1 (Tripos International, St. Louis, MO, USA) was used with default settings to develop the pharmacophore.35 Seven molecules (AZD2014, WYE-354, GDC-0349, WYE-687, OSI-027, WAY-600, and INK128) were chosen for pharmacophore modeling on the basis of their high activities and diverse structures. Strain energy, steric overlap, and pharmacophoric similarity were used to evaluate the rationality of pharmacophores; the model with the optimal values was chosen for analysis.
Molecular Docking
Molecular docking was implemented using the surflex-docking package of Sybyl-X 2.1. A cocrystal structure of mTOR with ADP (4JSV) was obtained from the Protein Data Bank. Before docking, 4JSV was prepared by removing water and magnesium ions and extracting the ligand. Addition of hydrogen and charges and treatment of the terminal residues were also performed on 4JSV. Then, the “protomol” was generated using the ligand-based mode, and an appropriate binding pocket was formed. The reliability of the surflex-docking was validated by redocking the original ligand (ADP) into the binding pocket. Next, all of the candidate compounds were docked into the binding pocket, and 20 possible docked conformations were obtained with different scores.
All molecular modeling figures were drawn using PyMOL (http://www.pymol.org).36
Biological Evaluations
mTOR Enzyme Assay
LANCE® ultra time-resolved fluorescence resonance energy transfer (TR-FRET) assay (Invitrogen, Carlsbad, CA, USA) was used to determine the mTOR kinase activities of all the compounds following the manufacturer’s instructions, with compound GSK2126458 (Selleck, China) as positive control.37 Briefly, mTOR enzyme (0.1 μg/mL, Invitrogen, Carlsbad, CA, USA), ATP (3 μmol/L (μM)), GFP-4EBP1 peptide (0.4 μM), and test compounds were diluted in kinase buffer (50 mM HEPES pH 7.5, 1 mM EGTA, 3 mmol/L (mM) MnCl2, 10 mM MgCl2, 2 mM DTT, and 0.01% Tween-20). The reaction was performed in black 384-well proxiplates (Corning, New York, NY, USA) at room temperature for 1 h, then stopped by adding EDTA to 10 mM. Tb-antiphospho-4EBP1 (Thr37/46) antibody (PerkinElmer, Fremont, CA, USA) was added to each well, and the mixture was incubated at room temperature for 30 min. Test compound concentrations were 10,000, 2500, 625, 156.25, 39.06, 9.77, 2.44, 0.61, 0.15, 0.04 and 0.01 nM. The final DMSO concentration was 1%. A Spectramax 190 reader (Molecular Devices, Valley, CA, USA) was used to measure the intensity of the light in TR-FRET mode (excitation 320 nm, emission 665 nm). All compounds were tested twice, and the results were expressed as the average IC50 (inhibitory concentration 50%) of the two experiments. The experiments were performed according to the methods described by Cao et al.6 The inhibition/dose-response plots of B170422, 1c, and 2a are provided in the Supplementary Materials (Figure S1).
PI3Kα Enzyme Assay
PI3Kα (p110α/p85α), PI3Kβ (p110β/p85α), PI3Kγ (p120γ) and PI3Kδ (p110δ/p85α) were purchased from Promega Corporation (Madison, Wisconsin, USA, #V1691). The inhibition activities of 2a against PI3Kα, PI3Kβ, PI3Kγ, PI3Kδ were determined using ADP-Glo kinase assays (Madison, Wisconsin, USA, #V1690). Per the manufacturer’s protocol, reagents were prepared as follows. 10 µL of PI3K enzyme were diluted in 310 μL 2.5× kinase reaction buffer (Promega, #V1691) to give 2.5× kinase solutions. 50 μL PIP2:3PS substrate and ATP were added to 100 µL 10× lipid dilution buffer and 250 µL water to give a 2.5× PIP2:3PS lipid kinase substrate working solution. 25 μL of ultra-pure ATP (10 mM) were diluted in 975 µL water to give a 250-µM ATP water solution.
Tested compounds were serially diluted to the desired concentrations at the time of the experiments, and 1 μL of each dilution was added to a 384-well plate (Corning, New York, NY, USA), together with 4 mL of the PIP2:3PS lipid kinase substrate working solution. Then, 4 μL of kinase solution was added to each well of the assay plate except the control well, to which 4 μL of 1× kinase reaction buffer was added instead (enzyme control group). When the kinase reaction started, 1 μL of 250 μM ATP was added to the assay plate, then covered, mixed for 30–60 s, and incubated for 1 h at 23°C (room temperature). Ten microliters of ADP-Glo™ reagent (Promega, Madison, WI, USA, #V1691) was added to the reaction mixture to stop the enzyme reaction and deplete unconsumed ATP. After 40 min, 20 μL kinase detection reagent (Promega, Madison, WI, USA, #V1691) was added to the reaction mixture to convert ADP to ATP. Test compound concentrations were 10,000, 2500, 625, 156.25, 39.06, 9.77, 2.44, 0.61, 0.15, 0.04 and 0.01 nM. The final DMSO concentration was 1%. The mixture was shaken for 1 min and equilibrated for 40 min before being examined using a luminescence plate reader. Finally, conversion data were collected using FlexStation (Molecular Devices, USA) and RLU (relative light unit) values were converted to inhibition values using the following formula: (max − sample RLU)/(max − min) × 100%, where “max” is the RLU of the dimethyl sulfoxide (DMSO) control and “min” is the RLU of the no-enzyme control. All compounds were tested twice, and the results were expressed as the average IC50 of the two experiments. Compound GSK2126458 (Selleck, China) was used as the positive control. The experiments were performed according to the methods described by Cao et al.6 The inhibition/dose-response plots of B170422, 1c, and 2a are listed in the Supplementary Materials (Figure S1 and S2).
Cell-Survival Inhibition Assay
The present work did not include any materials obtained directly from human participants and only used MHCC97-H cells purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The usage of the cell lines was permitted by the ethics committee of the Fifth Medical Center, General Hospital of the Chinese PLA (previously named the Beijing 302 Hospital). All experiments were performed according to the Declaration of Helsinki (World Health Organization).38
For proliferation analysis, cells were seeded in 96-well plates (5 × 103 cells per well) (Corning, NY, USA). Cells were cultured in DMEM (complete Dulbecco’s modified Eagle’s medium, Invitrogen, USA) with 10% FBS (fetal bovine serum, Invitrogen, USA) at 37°C with 5% CO2 for 24 h. 2a was dissolved in DMSO and diluted with DMEM to a final DMSO concentration of 1%. Next, cells were treated with 2a at a series of concentrations (10, 3, 1, 0.3, 0.1, 0.03, 0.01 and 0.003 μM) for the indicated durations. Following incubation for 12 h, cells were harvested for MTT assays (Amresco, Washington, USA) according to the manufacturer’s instructions. Absorbance was measured using a multi-functional micro-plate reader at 490 nm. Inhibition rates were calculated as follows: (optical density (O.D.) 490 of the control group - O.D. 490 of the administration group)/(O.D. of the 490 control group - O.D. 490 of the blank group) × 100%. Relative cell survival rates were calculated as: 100% - inhibition rate (%). Assays were performed three times with similar results.39–41
Colony Formation
For the colony formation experiments, MHCC97-H cells (2 × 103 cells per well) were seeded in six-well plates (Corning, USA) and cultured for 3–4 weeks. A DMSO solution of 2a (30 nM concentration of 2a; final concentration of DMSO: 1%) was added to the cells and incubated for 12 h, then the cells were treated with the appropriate dose (0, 2, 4 or 8 Gy) of 60Co-γ IR for 5 min.42 Results are shown as images or histograms for the two groups: solvent control + fractionated radiation and 2a + fractionated radiation. Inhibition rates were calculated as: [(control group′s O.D.546nm)–(administration group′s O.D. 546nm)]/(control group’s O.D. 546 nm) × 100%.
Immunocytochemistry (Cellular Immunofluorescence)
For the γ-H2aX staining (green fluorescence), MHCC97-H cells (5 × 103 cells per well) were seeded into 96-well plates (Corning, USA) and cultured for 3–4 weeks. The DMSO solution of 2a (30 nM concentration of 2a; final concentration of DMSO: 1%) was added to the cells and incubated for 12 h, then the cells were treated with a 4-Gy dose of 60Co-γ IR for 5 min. Plates were fixed with 3% paraformaldehyde for 30 min and then permeabilized by Triton X-100 (0.5%) treatment at 4°C for 10 min. Next, plates were blocked with 10% bovine serum albumin diluted in PBS (phosphate-buffered saline). After blocking, plates were incubated with FITC conjugated antibody (anti-γ-H2AX [1:500]) diluted in PBS at 37°C for 1 h in the dark. Fluorescent signals were visualized using a fluorescence microscope. The nuclei of MHCC97-H cells were stained with Hoechst33342 (blue fluorescence). Results are shown as images or histograms for the four groups: 0 Gy IR + solvent control (DMSO diluted with DMEM to 1% concentration) treatment group, 2a treatment group, 4-Gy dose IR treatment group, and 4-Gy dose IR + 2a treatment group. The FITC intensity was quantitatively analyzed using the ImageJ software.
Results and Discussion
Identification of Lead Compound
Structure-based virtual screening can be used for efficient and rapid screening of huge compound libraries, whereas virtual docking is more appropriate for accurate screening of smaller samples. In order to find compounds with new scaffolds, pharmacophore modeling and molecular docking were used to screen our library of 2063 small compounds in this study.43,44 The compounds were obtained from several research groups and most of them have not been previously reported; among them, 1276 compounds were serine/threonine kinase inhibitors with structural diversity.
Seven typical selective mTOR inhibitors (AZD2014, WYE-125132, GDC-0349, WYE-687, OSI-027, WAY-600, and INK128) were used to establish the pharmacophore model, which was validated using active and inactive molecules, respectively.12,14,21,28,45,46 The characteristics of molecules were further modified to generate a new pharmacophore with three main features: one projection of a hydrogen bond donor (magenta), two projections of hydrogen acceptors (green), and four hydrophobic features (cyan) (Figure 1A).47 The pharmacophore model was used to search our database of 2063 compounds, and 237 compounds were isolated.
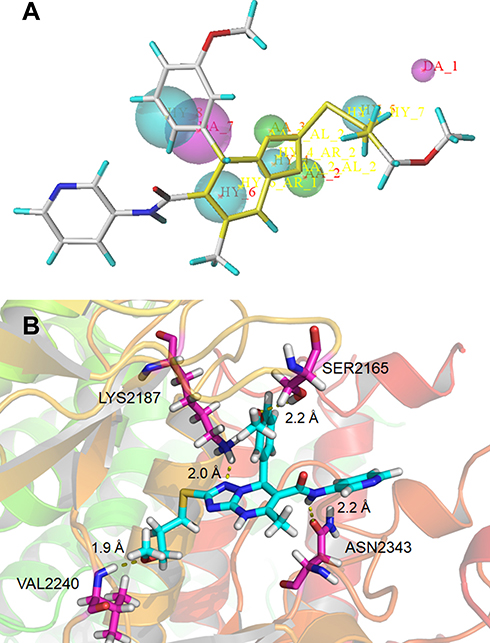 |
Figure 1 (A) Pharmacophore model based on the reported mTOR inhibitors and the lead compound B170422 screened out by the pharmacophore model; (B) schematic of the binding mode of B170422 with mTOR. |
Before molecular docking, a redocking study was carried out to validate the reliability of the docking model. ADP (the original ligand) was redocked into the binding site using surflex-docking, and the redocked conformation was compared with the original crystallographic conformation of the ADP. As shown in Figure S3 in the Supplementary Materials, the redocked ADP and the crystal ADP in the complex were almost completely superimposable, indicating that the surflex-docking method and the parameters used were reasonable and reliable. Then, 237 compounds were virtually docked to mTOR and ranked according to various molecular characteristics, including hydrophobicity, polarity, and entropy (Figure 1B). The 50 top-ranked compounds were picked out, of which, 35 compounds were retained after manual selection based on visual inspection. The inhibitory activities of these compounds on mTOR at 10 μM were examined using enzyme inhibitory activity tests. Five hits were selected out with mTOR-inhibition rates greater than 50% at 10 μM; their structures are shown in Figure 2.
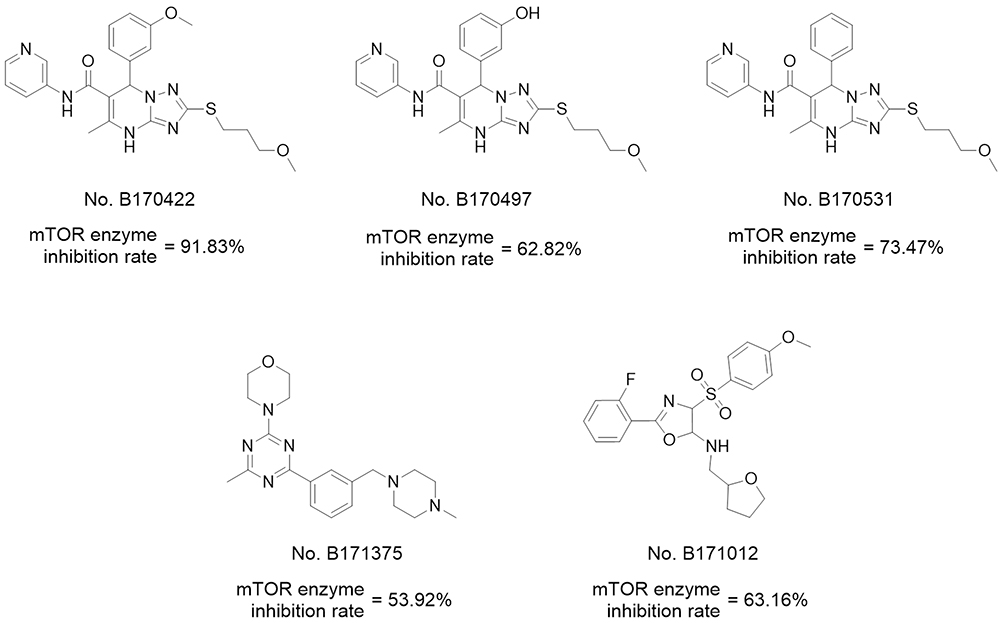 |
Figure 2 Hits obtained by virtual screening. |
Of the five hits (Figure 2), three with 4.7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine structures showed the greatest activity, and B170422 (1a) showed optimal activity with an mTOR inhibition rate of 91.83% (mTOR IC50=167 nM, PI3K IC50=1578 nM). Thus, B170422 was structurally optimized as a lead compound to improve its inhibitory activity and selectivity towards mTOR.
Structural Analysis of Lead Compound
Before analyzing the structure–function relationship of lead compound (B170422), the targeted protein was researched. Both mTOR and PI3K belong to the phosphoinositide 3-kinase-related kinase (PIKK) family and share a highly conserved ATP-binding pocket, with 68% residue similarity in the kinase catalytic domain, enabling many mTOR inhibitors (including BEZ235 and GDC-0980) to also have inhibitory effects on PI3K.18,19,48 The ATP-binding sites of the α, β, δ, and γ subunits of PI3K have similar sequences and three-dimensional protein structures. PI3Kα, the most common subtype of the PI3K family, was selected for further study to investigate the selectivity of the compounds.
The three-dimensional crystal structures of the ATP binding pocket of mTOR and PI3Kα were compared to determine how selectivity of B170422 could be improved. There was high similarity at the bottom of the pocket, but structural differences were observed at the pocket opening. PI3Kα has a hairpin structure (marked in yellow in Figure 3), composed of LEU766, GLU767, and GLU768, which causes the β-sheet unit of PI3Kα to be inclined further forward than the equivalent unit in mTOR. The ATP-binding pocket of PI3Kα is partially concealed, which makes the binding pocket opening of PI3Kα smaller than that of mTOR. According to our measurements, the opening width of the ATP-binding pocket of PI3Kα was 6.88 Å, whereas that of mTOR was 8.73 Å, a 27% difference in opening width. This suggests that it would be more difficult for a larger compound to enter the binding pocket of PI3Kα compared with that of mTOR. In fact, according to previous reports, PI3K-selective inhibitors do have more flattened molecular structures in most cases, whereas mTOR-specific inhibitors tend to have larger molecular spatial structures. Some molecules can enter the binding pocket of mTOR but cannot enter that of PI3Kα owing to these structural differences. The structural differences between mTOR and PI3Kα may account for some compounds’ selective inhibitory effect on mTOR, which has been confirmed by some inhibitors (LY294002, PI-103, TGX-221, Torin1, Wye-125132, and Way-600), whose structures are shown in Figure 4.20,49,50
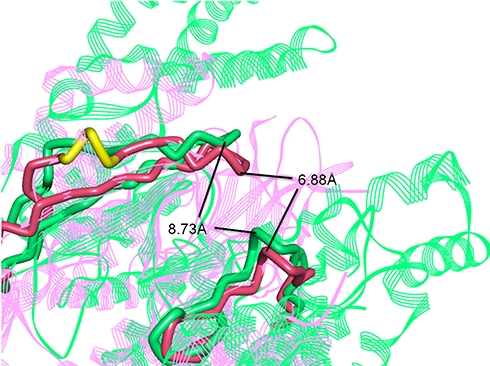 |
Figure 3 Superposition of three-dimensional protein crystal structures of mTOR and PI3Kα. Red: mTOR (4JSV); green: PI3Kα (6OAC). The ATP binding pocket of mTOR and PI3Kα is represented by solid tube; the other parts are represented by ribbon. |
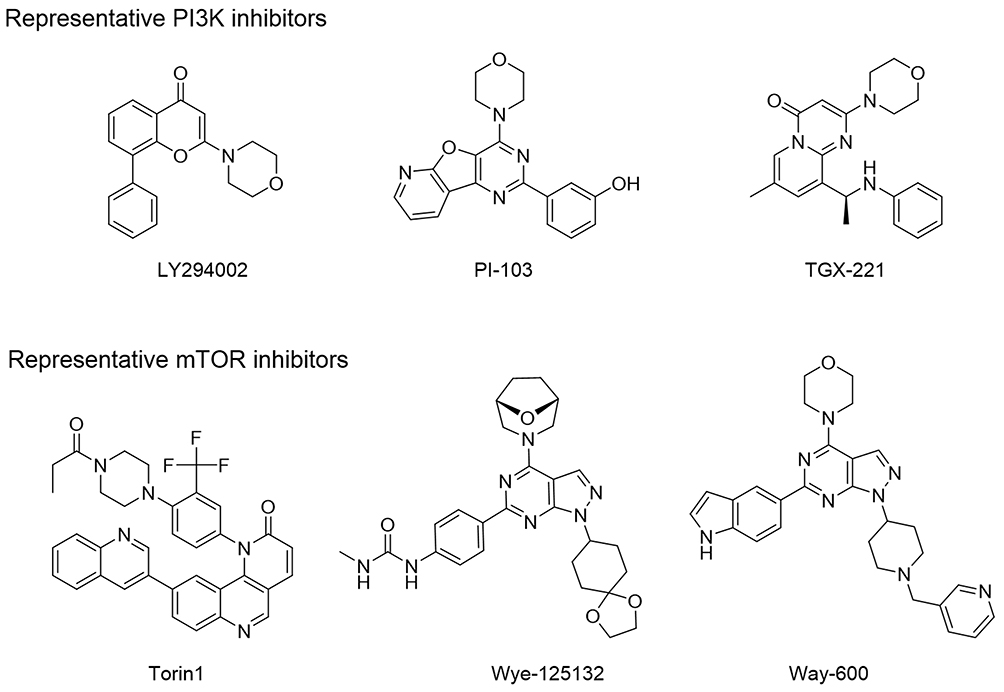 |
Figure 4 Representative mTOR inhibitors and PI3K inhibitors. |
These differences explain the selectivity of B170422 towards mTOR. There is a 90° angle between the benzene ring and the scaffold (4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine) in B170422, making it larger in size and thus less able to enter the relatively narrow cavity of PI3Kα.
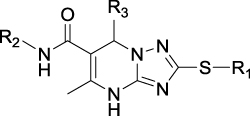
Hydrogen bonding is a key factor in ensuring the activity of B170422. Molecular docking showed that B170422 formed hydrogen bonds with SER2165, LYS2187, VAL2240, and ASN2343 in mTOR. Molecular dynamics (MD) simulations were used to further confirm the four amino acids making a great contribution to the stable binding between B170422 and the protein (the MD simulations are described in detail in the Supplementary Materials). Next, we modified the structure of B170422 without compromising the four hydrogen bonds between compounds and residues. Based on the interactions with SER2165, VAL2240, and ASN2343, we modified B170422 at three sites: R1, R2, and R3 (Table 1).
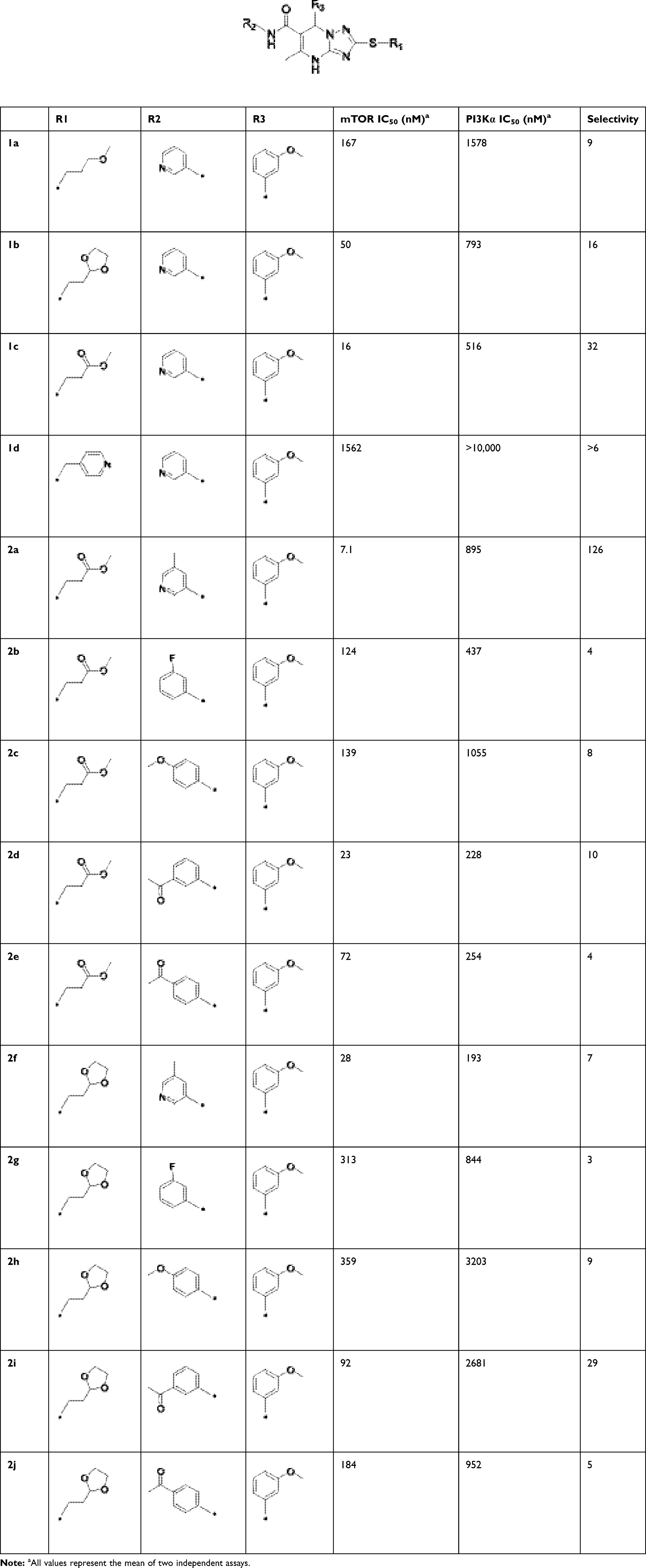 |
Table 1 Structures, Enzyme Inhibitory Activities, and Selectivities of 1a–1d and 2a–2j |
Structural Modification and Structure–Activity Relationship
To improve the activity of B170422, R1 was first modified and four different groups were transferred onto it (Table 1). These selected side chains (groups) were all flexible and had at least one hydrogen bond acceptor to form a hydrogen bond with VAL2240 in mTOR. An enzyme inhibitory activity test was immediately performed after these compounds were obtained. The greatest effect on mTOR was observed with 1c, which had a methyl acetate structure (mTOR IC50=16 nM). The molecular docking schematic (Table 1) shows that the side chain needed appropriate hydrophobicity and length to extend into the narrow hydrophobic cavity in mTOR.47
Previous studies have shown that the amide groups on R2 can interact with ASN2343 in mTOR to form a hydrogen bond. The R2 group occupied a wide hydrophobic cavity at the binding pocket, so a large group needed to be added to R2. Five different R2 groups were introduced to clarify the influence of R2 groups on the compound’s activity, and a series of new compounds (2a–2j) were obtained.
As shown in Table 1, 2a had greater inhibitory activity than 2b, 2c, 2d, or 2e. This phenomenon was also observed between 2f and 2g–2j. Given the high activity of 1c, we concluded that the activity of the pyridine derivative was better than that of the phenyl derivative in R2. Detailed structure–activity relationship information for 2a is provided in Figure S8 in the Supplementary Materials. Among the compounds studied, 2a had the greatest inhibitory effect on mTOR, with an IC50 value of 7.1 nM (126 times higher than the effect on PI3Kα).
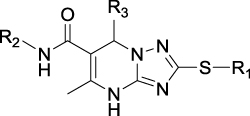
After R1 and R2 were determined, different substituent groups were introduced into R3 (Table 2). The docking of B170422 with mTOR and PI3Kα showed that R3 possessed a structure vertical to the scaffold, which was the main reason for the specific binding of B170422 with the cavity in mTOR. The substituent group on the benzene ring (R3) further increased the size, making it more difficult for B170422 to enter the PI3Kα cavity and increasing its selectivity for mTOR. On the other hand, the amino acid residue SER2165 in mTOR stabilized the spatial configuration of R3 through hydrogen interactions.
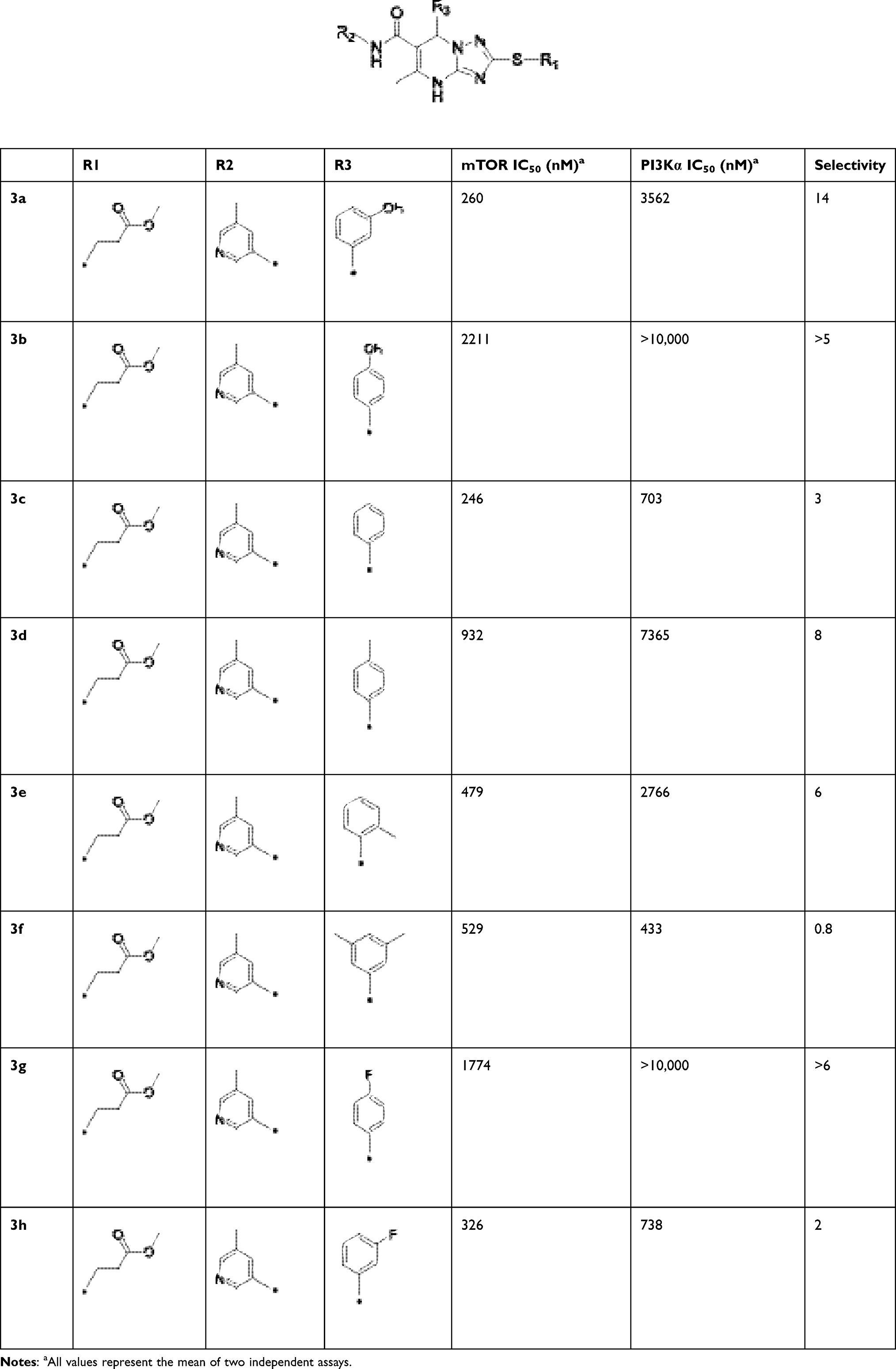 |
Table 2 Structures, Enzyme Inhibitory Activities, and Selectivities of 3a–3h |
Based on the above research on R3, a series of benzene ring derivatives with different substituent groups were introduced into R3. Enzymatic activity tests showed that 2a with a hydrogen bond receptor more potently inhibited mTOR. However, replacing the hydrogen bond acceptor with hydrogen bond donors (as in 3a and 3b) on the benzene ring decreased the activity of 2a, because the conformational change in R3 weakened the affinity between the derivatives and mTOR. We also observed that the derivatives with a para-substituent group on R3 (3b, 3d, and 3g) had low inhibitory activity against mTOR, probably owing to the steric hindrance of R3 making it difficult for derivatives to enter into the cavity of mTOR. In general, 2a showed the best inhibitory activity against mTOR among these compounds.
Furthermore, the inhibition activities of 2a against PI3Kβ, PI3Kγ, PI3Kδ were determined by enzymatic assays (PI3Kβ IC50=3103 nM, PI3Kγ IC50=1178 nM, PI3Kδ IC50=660 nM). The analysis results of Western blot assays showed that 2a had a definite inhibitory effect on both mTORC1 and mTORC2, and the inhibitory effect on mTORC1 was stronger than that on mTORC2 (the Western blot assays are described in detail in the Supplementary Materials).
Molecular docking models were used to explain the notable inhibitory activity of 2a. 2a was trapped in the kinase domain of mTOR and interacted with the amino acid residues LYS2187, SER2165, VAL2240, and ASN2343 (Figure 5A). Two hydrophobic features in the core structure of 2a formed strong hydrophobic interactions with residue ILE2356. Another hydrophobic contact was observed in the binding of the alkyl chain in 2a (R1) to residue LEU2185 (Figure 5A). The triazolopyrimidine structure was flattened on one side of the cavity (Figure 5B), while the benzene ring structure was oriented perpendicular to the scaffold to support the other side of the cavity. The ethyl propionate structure was deeply embedded in the hydrophobic cavities of mTOR.
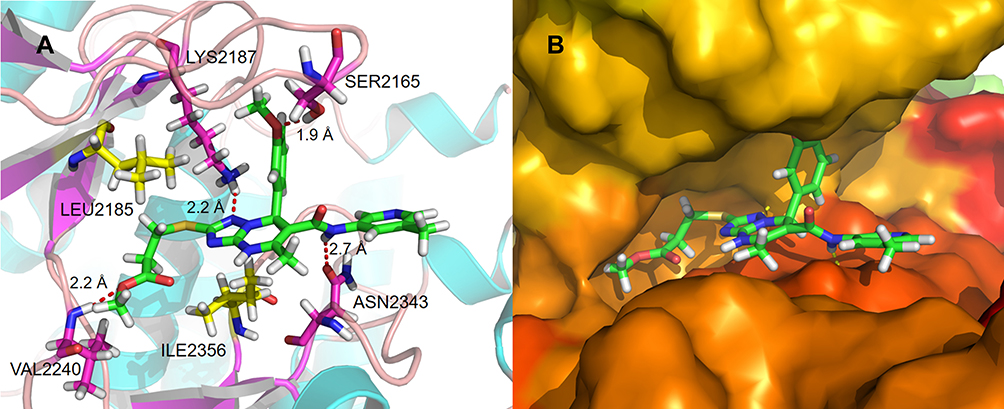 |
Figure 5 (A) Schematic of the binding mode of 2a with mTOR; (B) image of the mTOR surface around 2a. |
Chemistry
The structures of these newly synthesized compounds were all based on a 4.7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine scaffold. Intermediate 1, Intermediate 2, and benzaldehyde derivative were mixed together to synthesize the target compound by a one-step procedure (Scheme 1). The synthesis approach was applied flexibly in accordance with the different groups (R1, R2, and R3), and the overall yield of the process reached 23%. To the best of our knowledge, none of the compounds synthesized have been reported before.
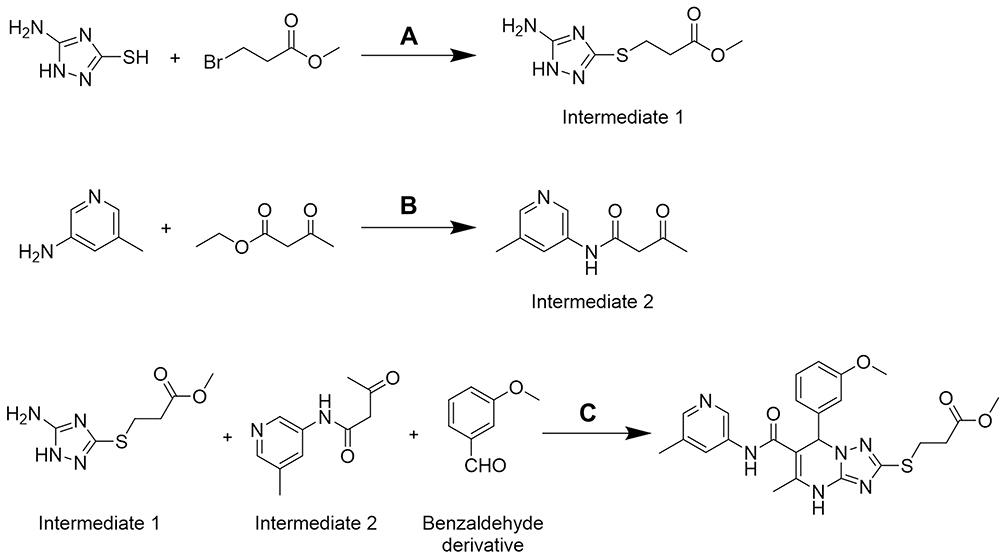 |
Scheme 1 The synthetic route of 2a. Reagents and conditions: (A) NaOH, ethyl alcohol, rt; (B) triethylamine, methylbenzene, 100°C; (C) ethyl alcohol, 80°C. |
In vitro Radiosensitization Experiment
Compounds showing better performance in the enzymatic assays were selected for further research. In order to determine the role of 2a in radiosensitization, the concentration at which 2a could significantly inhibit mTOR kinase while not directly affecting MHCC97-H cells was determined by treating MHCC97-H cells with 2a at different concentrations (Table 3). The results showed that at a concentration of 0.03 μM, 2a had a significant inhibitory effect on mTOR, with almost no effect on the survival of MHCC97-H cells.
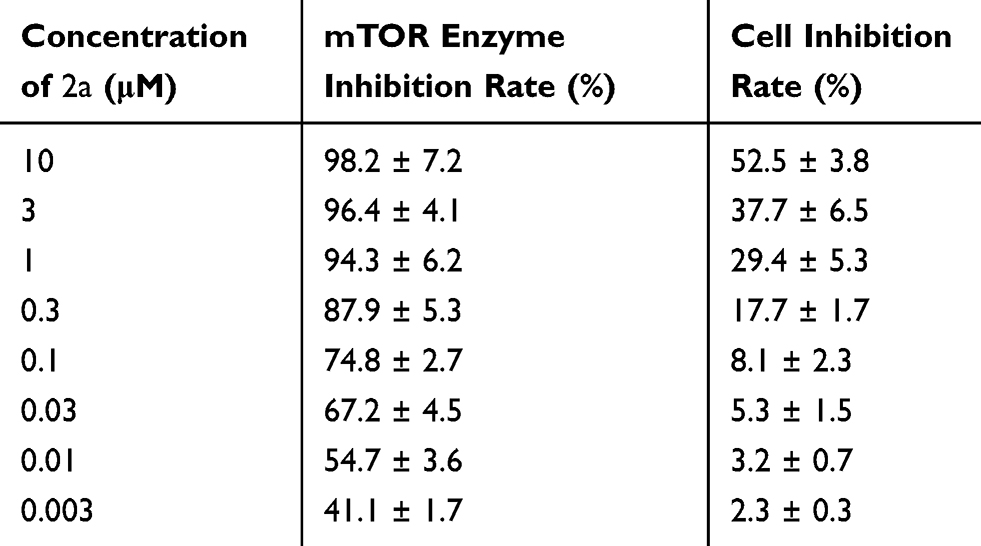 |
Table 3 Comparison of the Inhibitory Activity of 2a on mTOR Enzyme and MHCC97-H Cells at Different Concentrations (Considering the Experimental Error, the Number of Decimal Places Has Been Optimized) |
Compound 2a Enhances in vitro Antitumor Efficacy of IR Therapy
To determine whether 2a could enhance the radiosensitization of MHCC97-H cells, a colony formation assay was performed. As shown in Figure 6, IR treatment slightly attenuated the colony formation of MHCC97-H cells in a dose-dependent manner. After 2a was added, the apoptosis rate of MHCC97-H cells increased significantly at different IR doses. These results showed that treatment with 2a enhanced the sensitivity of MHCC97-H cells to IR therapy.
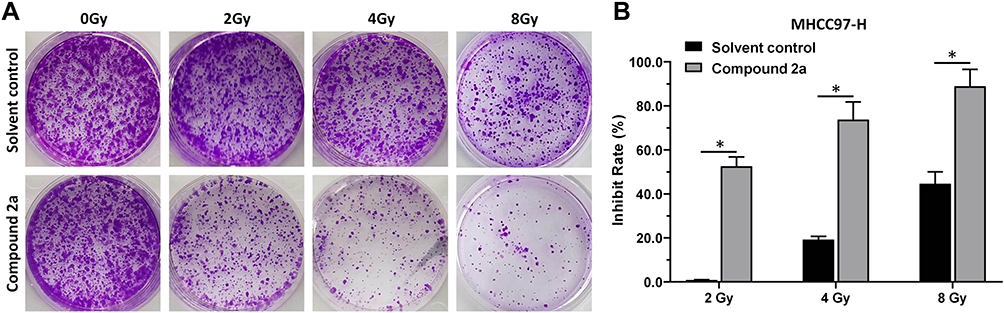 |
Figure 6 Compound 2a enhances the sensitivity of MHCC97-H cells to IR. MHCC97-H cells pretreated with 2a were treated with the indicated dose (0, 2, 4, or 8 Gy) of 60Co-γ IR and were examined by colony formation experiments. (A) Experimental results are shown as images of colonies; (B) experimental results are shown as inhibition rates (mean ± SD). *p<0.05 versus solvent control or 2a. |
Compound 2a Enhances DSBs After IR Exposure
Moreover, the DNA DSBs in MHCC97-H cells were evaluated by γ-H2aX staining, which is a representative indicator of DNA DSBs. As shown in Figure 7A, treatment with 2a alone did not induce the formation of γ-H2aX foci in MHCC97-H cell nuclei. Treatment with a 4-Gy IR dose induced γ-H2AX foci formation (Figure 7B). As expected, treatment with 2a and IR together significantly enhanced the formation of IR-induced γ-H2aX foci.
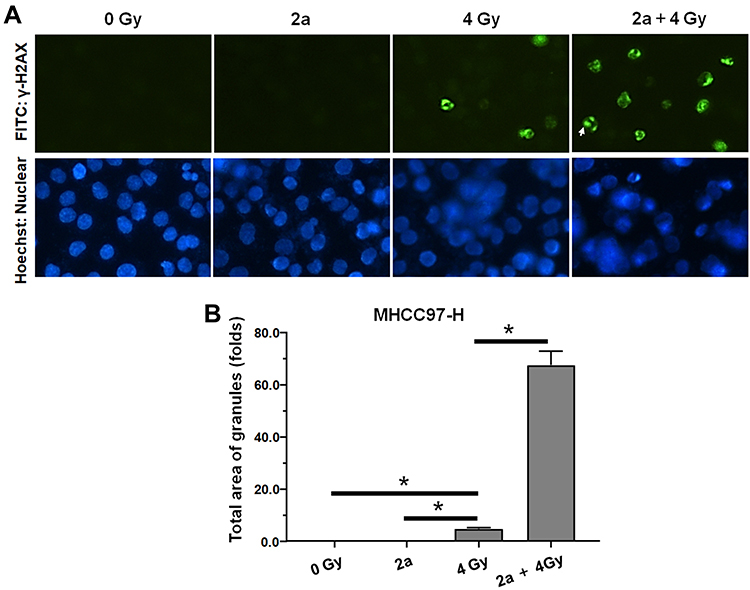 |
Figure 7 Compound 2a enhances DNA DSBs induced by 60Co-γ IR in MHCC97-H cells. (A) 2a enhances the γ-H2aX foci induced by IR (4 Gy) in nuclear of MHCC97-H cells; (B) the total area of granules of γ-H2aX foci (mean ± SD). *p<0.05. |
In these two in vitro experiments, we investigated the effects of 2a on IR therapy applied to HCC cells. The results showed that treatment with 2a at a non-cytotoxic concentration (0.03 μM) enhanced the efficacy of IR against HCC cells in vitro. These results provide a basis for developing mTOR-specific inhibitors as radiosensitizers in radiotherapy for HCC.
Conclusions
Although some progress has been made, HCC still seriously endangers human health and poses a challenge to the public health system. Non-surgical treatment with radiotherapy shows an ameliorative effect on a wide range of tumors, but not on HCC, owing to its insensitivity. Therefore, as novel radiosensitizers, the mTOR inhibitors have development potential in radiotherapy for HCC.
In this study, based on pharmacophore modeling and virtual docking, we obtained a lead compound with a scaffold of 4.7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine. By analyzing the three-dimensional structure of mTOR and comparing it with that of PI3Kα, some key amino acid residues in mTOR were confirmed, including SER2165, LYS2187, VAL2240, and ASN2343, which were used to design highly selective mTOR inhibitors. Then, 20 new compounds were synthesized, among which compound 2a had the greatest effect on mTOR (mTOR IC50=7.1 nM) with 126-fold selectivity over PI3Kα. The radiosensitizing effect of 2a was confirmed by colony formation assays and DNA DSB assays in vitro. This work identified a novel structural radiosensitizer and suggests a novel approach for the treatment of HCC.
Abbreviations
HCC, hepatocellular carcinoma; mTOR, mammalian target of rapamycin; ATP, adenosine triphosphate; PI3K, phosphoinositide 3-kinase; nM, nmol/L; μM, μmol/L; mM, mmol/L; PIKK, phosphoinositide 3-kinase-related kinase; DNA, deoxyribonucleic acid; ADP, adenosine diphosphate; TR-FRET, time-resolved fluorescence resonance energy transfer; IR, ionizing radiation; EDTA, ethylene diamine tetraacetic acid; DMSO, dimethyl sulfoxide; 4EBP1, 4E-binding protein 1; RLU, relative light unit; DMEM, Dulbecco’s modified eagle medium; O.D., optical density; FITC, fluorescein isothiocyanate; PBS, phosphate-buffered saline; MD, molecular dynamics; DSBs, double-strand breaks.
Supplementary Materials
Figure S1–S3; MD simulations (Figure S4–S6); Western blot assays (Figure S7); Figure S8; the 1H-NMR, 13C-NMR and MS spectra of compound 1a–1d, 2a–2j and 3a–3h.
Acknowledgments
We gratefully acknowledge financial support from the program for the National Natural Science Foundation of China-Youth Foundation (No. 81502906), Graduate Innovative Fund of Wuhan Institute of Technology (No. CX2018001) and Natural Science Foundation of Beijing, China (No. 7172205).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zaller N, Brinkley-Rubinstein L. Incarceration, drug use, and infectious diseases: a syndemic still not addressed. Lancet Infect Dis. 2018;18(12):1301–1302. doi:10.1016/S1473-3099(18)30538-3
2. Feng F, Jiang Q, Jia H, et al. Which is the best combination of TACE and Sorafenib for advanced hepatocellular carcinoma treatment A systematic review and network meta-analysis. Pharmacol Res. 2018;135:89–101. doi:10.1016/j.phrs.2018.06.021
3. Liu R, Zhao D, Zhang X, et al. A20 enhances the radiosensitivity of hepatocellular carcinoma cells to (60)Co-gamma ionizing radiation. Oncotarget. 2017;8(54):93103–93116. doi:10.18632/oncotarget.21860
4. Toya R, Murakami R, Baba Y, et al. Conformal radiation therapy for portal vein tumor thrombosis of hepatocellular carcinoma. Radiother Oncol. 2007;84(3):266–271. doi:10.1016/j.radonc.2007.07.005
5. Song LL, Peng Y, Yun J, et al. Notch-1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008;27(44):5833–5844. doi:10.1038/onc.2008.190
6. Cao S, Cao R, Liu X, et al. Design, synthesis and biological evaluation of novel benzothiazole derivatives as selective PI3Kβ inhibitors. Molecules. 2016;21(7):876–890. doi:10.3390/molecules21070876
7. Barra F, Evangelisti G, Ferro Desideri L, et al. Investigational PI3K/AKT/mTOR inhibitors in development for endometrial cancer. Expert Opin Investig Drugs. 2019;28(2):131–142. doi:10.1080/13543784.2018.1558202
8. Corti F, Nichetti F, Raimondi A, et al. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: a review of current evidences and future perspectives. Cancer Treat Rev. 2019;72:45–55. doi:10.1016/j.ctrv.2018.11.001
9. Schmid P, Zaiss M, Harper-Wynne C, et al. Fulvestrant plus vistusertib vs fulvestrant plus everolimus vs fulvestrant alone for women with hormone receptor-positive metastatic breast cancer: the MANTA phase 2 randomized clinical trial. JAMA oncol. 2019;5(11):1556. doi:10.1001/jamaoncol.2019.2526
10. Lapointe S, Mason W, MacNeil M, et al. A phase I study of vistusertib (dual mTORC1/2 inhibitor) in patients with previously treated glioblastoma multiforme: a CCTG study. Invest New Drugs. 2019. doi:10.1007/s10637-019-00875-4
11. Behbakht K, Sill MW, Darcy KM, et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a gynecologic oncology group study. Gynecol Oncol. 2011;123(1):19–26. doi:10.1016/j.ygyno.2011.06.022
12. Yu K, Shi C, Toral-Barza L, et al. Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res. 2010;70(2):621–631. doi:10.1158/0008-5472.CAN-09-2340
13. Atkin J, Halova L, Ferguson J, et al. Torin1-mediated TOR kinase inhibition reduces Wee1 levels and advances mitotic commitment in fission yeast and HeLa cells. J Cell Sci. 2014;127(Pt 6):1346–1356. doi:10.1242/jcs.146373
14. Yu K, Toral-Barza L, Shi C, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69(15):6232–6240. doi:10.1158/0008-5472.CAN-09-0299
15. Li S, Liang Y, Wu M, et al. The novel mTOR inhibitor CCI-779 (temsirolimus) induces antiproliferative effects through inhibition of mTOR in Bel-7402 liver cancer cells. Cancer Cell Int. 2013;13:30. doi:10.1186/1475-2867-13-30
16. Llerena S, Garcia-Diaz N, Curiel-Olmo S, et al. Applied diagnostics in liver cancer. Efficient combinations of sorafenib with targeted inhibitors blocking AKT/mTOR. Oncotarget. 2018;9(56):30869–30882. doi:10.18632/oncotarget.25766
17. de Melo AC, Paulino E, Garces AH. A review of mTOR pathway inhibitors in gynecologic cancer. Oxid Med Cell Longev. 2017;2017:4809751. doi:10.1155/2017/4809751
18. Suzuki Y, Enokido Y, Yamada K, et al. The effect of rapamycin, NVP-BEZ235, aspirin, and metformin on PI3K/AKT/mTOR signaling pathway of PIK3CA-related overgrowth spectrum (PROS). Oncotarget. 2017;8(28):45470–45483. doi:10.18632/oncotarget.17566
19. Sutherlin DP, Bao L, Berry M, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011;54(21):7579–7587. doi:10.1021/jm2009327
20. Djuzenova CS, Fiedler V, Katzer A, et al. Dual PI3K- and mTOR-inhibitor PI-103 can either enhance or reduce the radiosensitizing effect of the Hsp90 inhibitor NVP-AUY922 in tumor cells: the role of drug-irradiation schedule. Oncotarget. 2016;7(25):38191–38209. doi:10.18632/oncotarget.9501
21. Kahn J, Hayman TJ, Jamal M, et al. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol. 2014;16(1):29–37. doi:10.1093/neuonc/not139
22. Alcorn S, Walker AJ, Gandhi N, et al. Molecularly targeted agents as radiosensitizers in cancer therapy-focus on prostate cancer. Int J Mol Sci. 2013;14(7):14800–14832. doi:10.3390/ijms140714800
23. Chang L, Graham PH, Ni J, et al. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit Rev Oncol Hematol. 2015;96:507–517. doi:10.1016/j.critrevonc.2015.07.005
24. Benderli Cihan Y. Role of mTOR signaling pathway proteins and proteins influencing mTOR pathway in resistance to radiotherapy in prostate cancer. J BUON. 2018;23(6):1931–1932.
25. Wen PY, Omuro A, Ahluwalia MS, et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17(9):1275–1283. doi:10.1093/neuonc/nov083
26. Liu T, Sun Q, Li Q, et al. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Mol Cancer Ther. 2015;14(2):429–439. doi:10.1158/1535-7163.MCT-14-0548
27. Wang WJ, Long LM, Yang N, et al. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol Sin. 2013;34(5):681–690. doi:10.1038/aps.2013.22
28. Hayman TJ, Wahba A, Rath BH, et al. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin Cancer Res. 2014;20(1):110–119. doi:10.1158/1078-0432.CCR-13-2136
29. Shapiro GI, Bell-McGuinn KM, Molina JR, et al. First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin Cancer Res. 2015;21(8):1888–1895. doi:10.1158/1078-0432.CCR-14-1306
30. Lee W, Ortwine DF, Bergeron P, et al. A hit to lead discovery of novel N-methylated imidazolo-, pyrrolo-, and pyrazolo-pyrimidines as potent and selective mTOR inhibitors. Bioorg Med Chem Lett. 2013;23:5097–5104. doi:10.1016/j.bmcl.2013.07.027
31. Song SH, Jeong WK, Choi D, et al. Evaluation of early treatment response to radiotherapy for HCC using pre- and post-treatment MRI. Acta Radiol. 2019;60(7):826–835. doi:10.1177/0284185118805253
32. Yu JI, Choi GS, Lim DH, et al. Treatment of naive HCC combined with segmental or subsegmental portal vein tumor thrombosis: liver resection versus TACE followed by radiotherapy. Anticancer Res. 2018;38(8):4919–4925. doi:10.21873/anticanres.12808
33. Suri OP, Satti NK, Suri KA. Microwave induced acetoacetylation of hetaryl and aryl amines. Synth Commun. 2000;30(20):3709–3718. doi:10.1080/00397910008086998
34. Hafiz ISA, Ramiz MMM, Sarhan AAM. Activated Anilide in heterocyclic synthesis: synthesis of new dihydropyridines, dihydropyridazines and thiourea derivatives. Chin J Chem. 2011;29(6):1154–1162. doi:10.1002/cjoc.201190216
35. Caballero J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J Mol Graph Model. 2010;29(3):363–371. doi:10.1016/j.jmgm.2010.08.005
36. Schrodinger LLC The PyMOL molecular graphics system, version 1.8. 2015.
37. Sun N, Li B, Shao J, et al. A general and facile one-pot process of isothiocyanates from amines under aqueous conditions. Beilstein J Org Chem. 2012;8:61–70. doi:10.3762/bjoc.8.6
38. Chen Y, Feng F, Gao X, et al. MiRNA153 reduces effects of chemotherapeutic agents or small molecular kinase inhibitor in HCC cells. Curr Cancer Drug Targets. 2015;15(3):176–187. doi:10.2174/1568009615666150225122635
39. Feng F, Jiang Q, Cao S, et al. Pregnane X receptor mediates sorafenib resistance in advanced hepatocellular carcinoma. Biochim Biophys Acta Gen Subj. 2018;1862(4):1017–1030. doi:10.1016/j.bbagen.2018.01.011
40. Zhang Y, Li D, Jiang Q, et al. Novel ADAM-17 inhibitor ZLDI-8 enhances the in vitro and in vivo chemotherapeutic effects of Sorafenib on hepatocellular carcinoma cells. Cell Death Dis. 2018;9(7):743–756. doi:10.1038/s41419-018-0804-6
41. Shao Z, Li Y, Dai W, et al. ETS-1 induces Sorafenib-resistance in hepatocellular carcinoma cells via regulating transcription factor activity of PXR. Pharmacol Res. 2018;135:188–200. doi:10.1016/j.phrs.2018.08.003
42. Feng Y, Xu X, Zhang Y, et al. HPIP is upregulated in colorectal cancer and regulates colorectal cancer cell proliferation, apoptosis and invasion. Sci Rep. 2015;5:9429–9440. doi:10.1038/srep09429
43. Liu G, Wang W, Wan Y, et al. Application of 3D-QSAR, pharmacophore, and molecular docking in the molecular design of diarylpyrimidine derivatives as HIV-1 nonnucleoside reverse transcriptase inhibitors. Int J Mol Sci. 2018;19(5):1436–1452. doi:10.3390/ijms19051436
44. Liu G, Wan Y, Wang W, et al. Docking-based 3D-QSAR and pharmacophore studies on diarylpyrimidines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Mol Divers. 2019;23(1):107–121. doi:10.1007/s11030-018-9860-1
45. Pei Z, Blackwood E, Liu L, et al. Discovery and biological profiling of potent and selective mTOR inhibitor GDC-0349. ACS Med Chem Lett. 2013;4(1):103–107. doi:10.1021/ml3003132
46. Bhagwat SV, Gokhale PC, Crew AP, et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther. 2011;10(8):1394–1406. doi:10.1158/1535-7163.MCT-10-1099
47. Quemener A, Maillasson M, Arzel L, et al. Discovery of a small-molecule inhibitor of interleukin 15: pharmacophore-based virtual screening and hit optimization. J Med Chem. 2017;60(14):6249–6272. doi:10.1021/acs.jmedchem.7b00485
48. Verheijen JC, Richard DJ, Curran K, et al. Discovery of 4-morpholino-6-aryl-1H-pyrazolo[3,4−d]pyrimidines as highly potent and selective ATP-competitive inhibitors of the Mammalian Target of Rapamycin (mTOR): optimization of the 6-Aryl substituent. J Med Chem. 2009;52(24):8010–8024. doi:10.1021/jm9013828
49. Tong Y, Zhu W, Huang X, et al. PI3K inhibitor LY294002 inhibits activation of the Akt/mTOR pathway induced by an oncolytic adenovirus expressing TRAIL and sensitizes multiple myeloma cells to the oncolytic virus. Oncol Rep. 2014;31(4):1581–1588. doi:10.3892/or.2014.3020
50. Yang X, Yang JA, Liu BH, et al. TGX-221 inhibits proliferation and induces apoptosis in human glioblastoma cells. Oncol Rep. 2017;38(5):2836–2842. doi:10.3892/or.2017.5991
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.