")
Back to Journals » Veterinary Medicine: Research and Reports » Volume 14
Viral Protein 1 (VP1) Sequence-Based Genetic Diversity of SAT 2 FMDV Circulating in Ethiopia from 1990 to 2015
Authors Woldemariyam F , Paeshuyse J
Received 14 March 2023
Accepted for publication 19 May 2023
Published 25 May 2023 Volume 2023:14 Pages 91—101
DOI https://doi.org/10.2147/VMRR.S408352
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Young Lyoo
Fanos Woldemariyam,1,2 Jan Paeshuyse1
1Laboratory of Host-Pathogen Interaction, Division of Animal and Human Health Engineering, Department of Biosystems, KU Leuven, Leuven, Belgium; 2Department of Biomedical Sciences, College of Veterinary Medicine, Addis Ababa University, Bishoftu, Ethiopia
Correspondence: Jan Paeshuyse; Fanos Woldemariyam, Email [email protected]; [email protected]
Introduction: Pathogen molecular epidemiology determines the origin of specific outbreaks locality of foot-and-mouth disease virus serotype South African Territories-2 sequence-based analysis of highly variable Viral Protein 1 (VP1), which helps to identify the evolution of this virus through time and space. The objective of this study was to compare the differences between SAT-2 VP1 sequences of FMDV circulated in Ethiopia from 1990 to 2015 at the genetic level.
Methods: The nucleotide and amino acid sequences were analyzed using Basic Local Alignment Search Tools (BLAST), Multiple sequence alignment and sequence editing and Phylogenetic tree reconstruction. The nucleotide and amino acid sequences alignment, distance matrix, and phylogenetic tree constructions were done using the MEGA 6.0 software package.
Result and Discussion: In this analysis, we found 76% nucleotide identities and amino acid similarities among the sequences. The overall group mean distance at nucleotide level was 19% with a mean intra-population diversity of 2%. The lowest sequence variation was observed among sequences obtained from the years 2007/09/10, 2014/15, and 1990/91 which was less than 5% among them. This analysis revealed that in the last 25 years, four different topotypes of the FMDV SAT-2 were circulating in Ethiopia. The Arg-Gly-Asp (RGD) amino acid (AA) motif at AA position 144– 146 within the G-H loop of the VP1 protein of FMDV is conserved, but up- and downstream hyper-variable AA sequences are identified. In this study, it was observed that four topotypes (IV, XIV, XIII, and VII) were circulating in Ethiopia for 25 years. Further, compared with sequences from neighboring countries (Sudan, Kenya) confirmed the presence of these topotypes.
Conclusion: Pertinent to this genetic diversity control strategies in Ethiopia should be based on having regular antigenic and genetic vaccine matching tests with the circulating strain within a defined period, space, transboundary nature of the disease and applying biosecurity measures.
Keywords: amino acid, Ethiopia, FMDV, genetic diversity, nucleotide, SAT-2, viral protein
Introduction
The foot-and-mouth disease virus (FMDV) genome is a single-strand, positive-sense RNA of about 8.5kbp that codes for a single open reading frame (ORF) flanked by a 5′-untranslated region (UTR) and 3′-UTR. A single polyprotein is translated and further cleaved into a leader protein L, structural protein P1, non-structural protein P2, and P3. Leader protein (L) regulates host interferon responses, the structural proteins form the viral capsid, whereas replication and completion of the viral life cycle is the responsibility of the non-structural proteins.1 Immunologically, there are seven distinct serotypes with over 65 topotypes of the virus.2,3 The seven serotypes are O, A, and C circulated in the endemic area all over the world. Serotype Asia 1 is restricted to Asia and serotypes SAT-1, -2, and -3 are restricted to sub-Saharan Africa with spill over to Middle east.4 Out of the seven serotypes, only serotype C has not been encountered in any outbreak since 2004.5 Antigenic variation and new subtypes have been evolving due to mutations that happen in the capsid proteins encoding genes.6 This is a result of the lack of proof-reading ability of RNA-dependent RNA polymerase7 coupled with immune selection pressure by the host8 and recombination.9–12 This gives rise to immunological distinct variants that can escape the memory of the host’s immunological response.13 Considering the above genetic and antigenic variation of the virus, production of the protective vaccine, regular vaccination, post-vaccination sero-monitoring, and quarantine of newly introduced animals are very important in FMD control. Selection of an appropriate vaccine strain in the area where FMD is enzootic needs crucial attention. Genetic analysis of the highly variable capsid coding region, mainly the VP1 gene has paramount importance in selecting appropriate vaccine strain among the circulating strains.4 For this purpose, molecular epidemiology study of FMDV that links virus topotypes and lineages to specific geographical areas is crucial. Accordingly, the source and origin of the virus of a specific outbreak can be traced using homology phylogenetic analysis of sequences from the VP1 region of the viral genome.3,14–16 The viral protein P1 is one of the proteins produced from the FMDV genome that forms a structural component. It is cleaved by 3Cpro to produce two stable products, ie VP1 (1D), VP3 (1C), and a product with relatively intermediate stability, VP0 (1AB). The final cleavage of this VP0 is performed by RNA triggered auto cleavage of VP0 that results in mature VP4 (1A) and VP2 (1B).17 Based on the P1 polyprotein VP1 (1D) gene sequence, the timescale and population dynamics of FMDV can be studied.15–20 In addition, it is also a major component of the viral capsid and has major functional epitopes such as the G-H loop. Within the VP1 sequence, a region spanning from residue 135–146 is the G-H loop where the cell attachment site Arg-Gly-Asp (RGD) motive is located.
The genetic diversity between topotypes of SAT-2 has been generally defined by Knowles and Samuel as having 20% or more nucleotide differences in the VP1 (1D) region. South African Territories (SAT) serotypes are distinct in their genetic, immunologic, and geographic location aspects.3 Of the three SAT serotypes, SAT-2 is the dominant in its presence as well as efficient cross-species transmission. African buffalo (Syncerus caffer) particularly in Southern Africa acts as a maintenance host as well as a primary host for epidemiological purposes. South African Territory-2 (SAT-2) has 14 topotypes (I to XIV) based on VP1(1D) gene analysis with all limited to the African continent except for some spillover to the Middle East.4
Amino acid substitution of the VP1 region of SAT-2 FMDV from Egypt was found to have a 25–26% amino acid change as compared to the reference strain.18 To accurately determine the origin of regional and continental distinct viruses, viral protein 1 sequence-based identification has paramount importance.19,20 Therefore, the aim of this study was viral protein 1 region nucleotide sequence and amino acid residues comparison of SAT-2 FMDV that was causing outbreaks of FMD in Ethiopia in 25 years (1990–2015) to see genetic variability among sequences of different years and geographic location.
Materials and Methods
Sequence Search
The retrieval for publicly available/published Ethiopian SAT2 VP1 FMDV sequences that was published between 1990 and 2015 using the following keywords: ¨Foot and Mouth disease virus SAT-2 sequence, Ethiopia¨ from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). All available sequences of SAT-2 isolated from Ethiopia were included. Besides the actual sequences, also the following meta data were collected: year of outbreak, accession number, virus name, and geographical origin (Table 1). The total number of sequences retrieved was 25 that were from outbreaks in 1990 (n=2), 1991 (n=3), 2007 (n=1), 2009 (n=7), 2010 (n=3), 2014 (n=2), and 2015 (n=7) (Table 1).
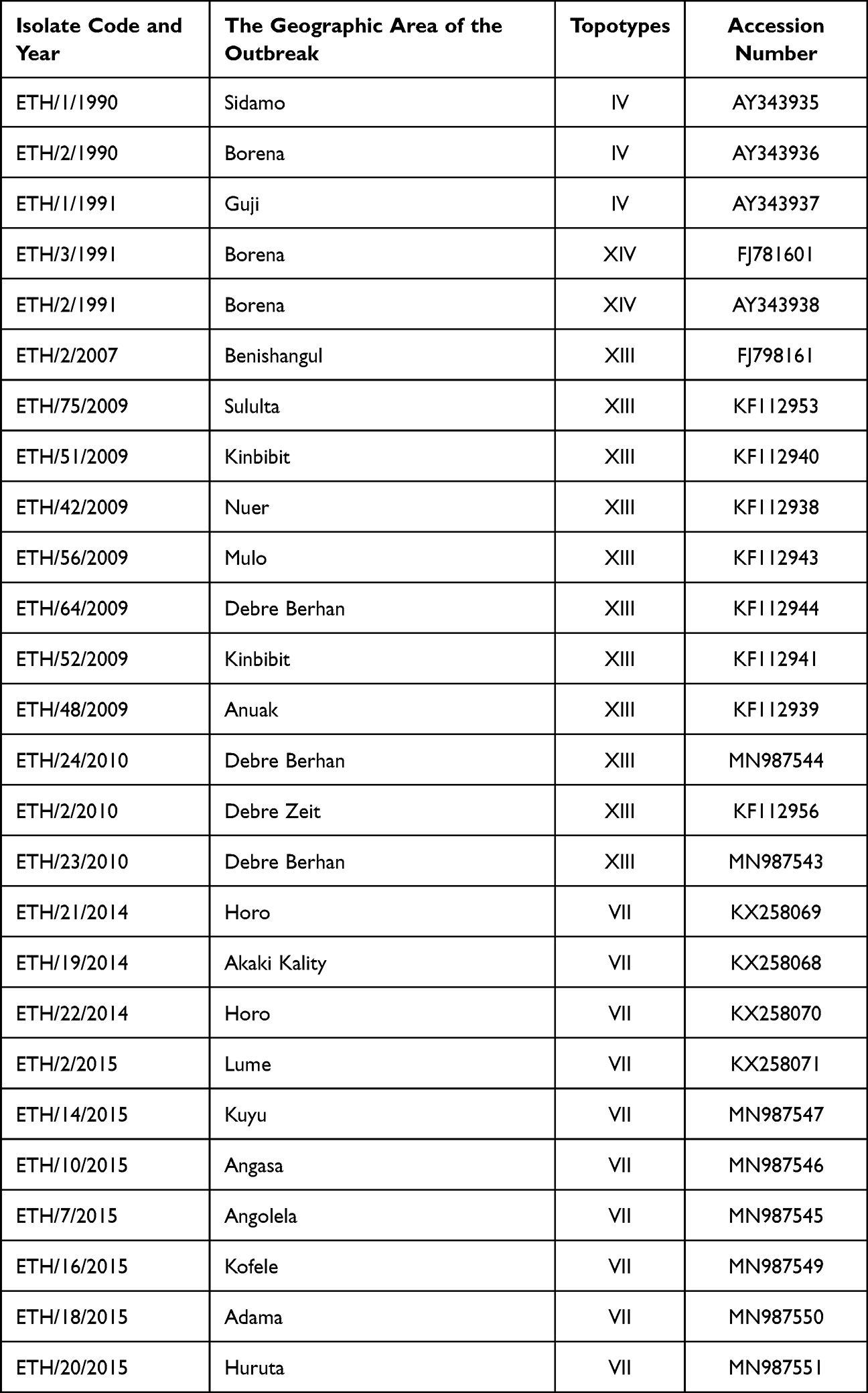 |
Table 1 The Sequences Retrieved from the Gene Bank with Geographic Area, Year Code, Topotypes, and Accession Number |
Basic Local Alignment Search Tools (BLAST)
All sequences identified were analyzed using BLASTn21 using the default parameter settings. Except for the organism name that was set to Aphthovirus SAT-2 (tax id: 12,109). Then, very closely related sequences, including their accession number and year of isolation were copied to Microsoft notepad and saved in FASTA format for further analysis. This was done until no new sequences matching the search criteria could be recovered from GenBank.
Multiple Sequence Alignment and Sequence Editing
Multiple sequence alignment was done by the MEGA 6.0 package using Muscle22,23 with the default parameter setting. Editing was performed if needed using the Bio edit 7.053 software.24 Next, the aligned sequences were exported from the MEGA 6.0 package and saved in MEGA format.
Phylogenetic Tree Reconstruction
The aligned nucleotide sequences were used to construct a phylogenetic tree in the MEGA 6.0 program with the following parameters: midpoint-rooted neighbor-joining tree, Kimura 2-parameter, nucleotide substitution model, distances matrices. The 1000 bootstrap replicates were used to assess the tree topology and its robustness.
Data Analysis
The nucleotide and amino acid sequences alignment, distance matrix, and phylogenetic tree constructions were done using the MEGA 6.0 software package.22 The location of the topotypes identified were indicated on a map using QGIS2.18.14 software (2014).25
Results
Nucleotide Identity and Amino Acid Similarities
The geographic distribution of the FMDV outbreak caused by SAT-2 serotype in Ethiopia for 25 years covered the southern, western central, and to some extent, the northwestern part of Ethiopia. This area is known mainly for high cattle densities and is linked with extensive infrastructure for livestock marketing26 (Figure 1).
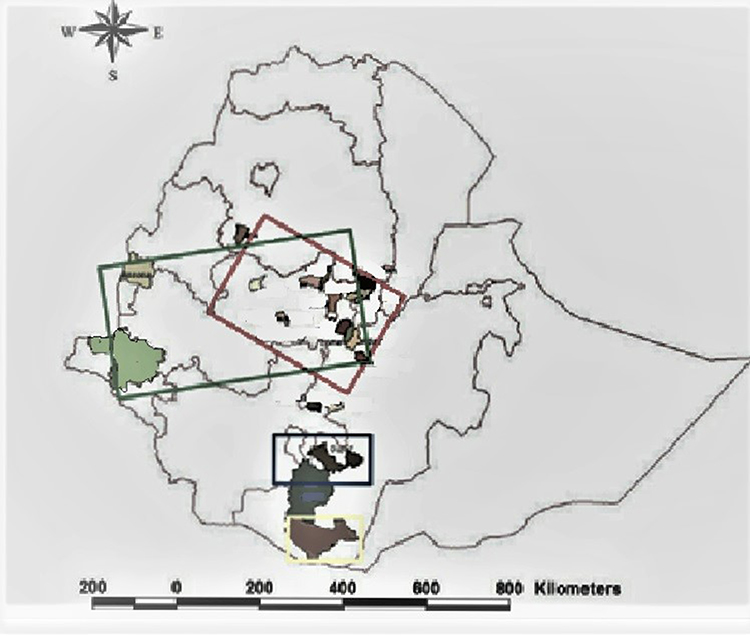 |
Figure 1 This is a map of the federal democratic republic of Ethiopia as of February 2022. The grey line on the map shows regional states’ administrative boundaries. The geographic location of outbreaks from the year 1990 to 2015 were indicated as follows: Red rectangle − 2014, 2015 outbreak (Central Ethiopia), Green rectangle − 2007, 2009, 2010 outbreak (Gambella, Benishangul gumuz and central Ethiopia), Blue rectangle − 1990, 1991 outbreaks (Sidamo, and Arsi), Yellow rectangle −1991 outbreaks (Borena). |
This study used 25 FMDV serotype SAT-2 (Table 1) complete VP1 sequences (648 bp) including gaps. These sequences could be translated into 216 AA sequences. Among these 25 FMDV serotype SAT-2 sequences from Ethiopia studied, 76% nucleotide identity and AA similarity were observed.
The FMDV SAT-2 VP1 sequences identified in Ethiopia in the year 1990 were 97% identical with Yemeni sequences identified in the same year. Similarly, this sequences were compared with sequences identified in Kenya in the years of 2009, 2007, 2006, 2005, 2002, 1999, 1994, 1984, 1983, 1982, and 1981 with 88% identity. The Ugandan sequence identified in 2013 were also seen to have 88% identity. The sequences obtained from outbreaks during 1991, ie, ETH/2/91 and ETH/3/91, were grouped together as a separate branches. The VP1 sequences of 2007, 2009, and 2010 were identical among themselves and share 98% identity to sequences from Sudan (2008, 1977). The 2015 VP1 sequences were 98% identical to those of 2014. The sequences from these 2 years were 95% identical to sequences from Sudan obtained in 2010, from Egypt in 2012, and from Eritrea in 1998. The sequences obtained from isolates from 1990 to 1991 showed 26 to 28.2% nucleotide differences from sequences 2007, 2009, 2010, 2014, and 2015 (Table 2 and Figure 2).
 |
Table 2 Nucleotide Differences Among the Sequences Using the P-Distance Model |
 |
Figure 2 Neighbor-joining tree indicating genetic relationships of serotype SAT-2 FMD viruses from Ethiopia (1990–2015) and neighboring countries Sudan, Eritrea, Egypt, Kenya, and Uganda from 1970 to 2012. Bootstrap values were estimated based on 1000 replications. The roman numerals indicate the topotypes. The scale represents 2% differences at the nucleotide level, Red triangles: outbreak year 2014, and 2015, red squares: the outbreak year 2007, 2009, and 2010, red diamonds: the outbreak year 1990, and red circles: the outbreak year 1991. The red square indicates topotype XIII, red circle indicates topotype VII, red triangle indicates topotype XIV, and red triangle with a hyphen bar indicates topotype IV. |
Diversity of the Sequences
The overall mean distance (number of sites at which two compared sequences differ) was 19% nucleotide diversity (nucleotide substitution), and with a mean intra-population diversity of 2% between 24 SAT-2 sequences over 25 years. The mean nucleotide distance between sequences of different years, as shown in Table 1, indicates that sequences from 2007, 2009 and 2010 among each other and sequence pairs from 2014 and 2015 were very closely related to each other with only less than 3% difference. The remaining sequences were distant from each other with a minimum of 25 and a maximum of 29.8% nucleotide difference between group means. Except for isolates ETH/2/91, ETH/1/91, ETH/2/90 and ETH/1/90 had a 14.6% group mean nucleotide difference for each other, see Table 2.
Phylogenetic Tree Analysis
The complete VP1 gene sequences (648bp) were used to compare phylogenetic differences between the SAT-2 virus sequences collected over 25 years from 1990 to 2015, and made publicly available in GenBank. The neighbor-joining (NJ) tree revealed two clusters of viral sequences. Cluster I grouped sequences from 2007, 2009, and 2010, as well as sequences from the year 1991 (ETH/2/91, ETH/3/1991), cluster II comprises sequences from 1991 (ETH/1//1991), 1990, 2014, and 2015. The identified clusters with the corresponding topotypes are illustrated in the phylogenetic tree depicted in Figure 3. In this phylogenetic tree, sequences from previous authors such as Sahle et al,27 Ayelet et al,28 and Suleyman et al,29 were used to construct the phylogenetic tree. Furthermore, the sequences from 25 years were compared among African sequences and a phylogenetic tree was constructed. Accordingly, the 2015 SAT-2 VP1 nucleotide sequences of FMDV were grouped as topotype (VII) with sequences from Sudan (SUD/4/2010), and Egypt (ie, EGY/1/2012, EGY/2/2012 EGY/3/2012, EGY/4/2012,EGY/5/2012, EGY/6/2012, EGY/9/2012, EGY/10/2012, EGY/13/2012 EGY/14/2012 and EGY/15/2012) and Eritrea (ie, ERI/12/98). The sequences of 2007, 2009, and 2010 were grouped as topotype (XIII) with sequences from Sudan (ie, SUD/2/2008, SUD/1/2008, SUD/6/77 and SUD/9/77). The sequences of 1990 were grouped as a topotype (IV) together with sequences from Kenya (ie, KEN/11/2009 KEN/12/2009 and KEN/122/2009) as well as other Kenyan sequences from 2007, 2006, 2005, 2002, 1999, 1994, 1984, 1983, 1982 and 1981) and sequences from Uganda from 2013. The sequences of 1991 (ETH/2/91, and ETH/3/91) are grouped in a separate branch on the tree as topotype (XIV), see Figure 2.
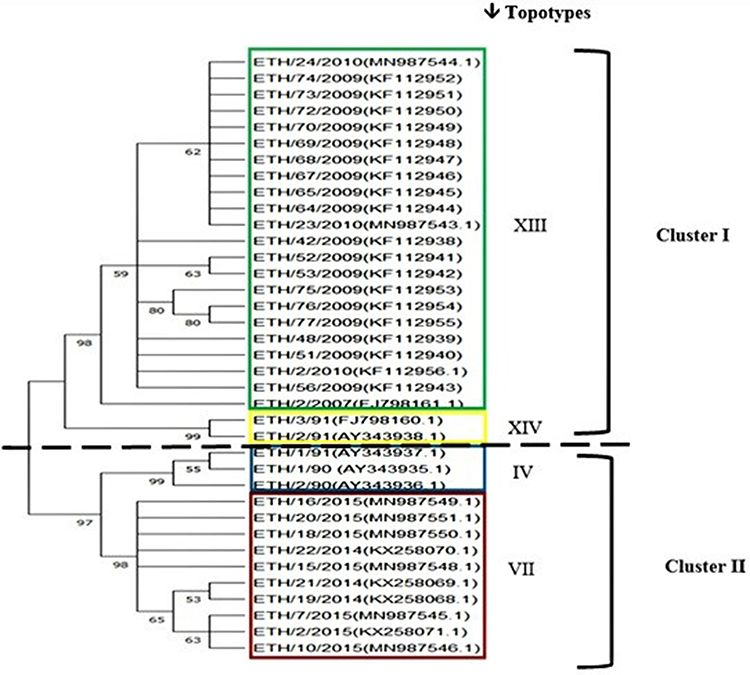 |
Figure 3 Neighbor-joining tree indicating genetic relationships of serotype SAT-2, FMD viruses from Ethiopia (from 1990 to 2015). Bootstrap values were estimated based on 1000 replications. The roman numbers I and II correspond to clusters, indicating grouping into two as associated by year of the outbreak. The colored boxes indicate the year of an outbreak and the topotypes. The green box is the outbreak year 2007, 2009 and 2010, topotype: (XIII), the yellow box: outbreak year 1991 and topotype (XIV), the blue box: the outbreak year 1990 and 1991 has a topotype (IV), the red box: outbreak year 2014 and 2015 that belongs to topotypes (VII). The scale represents 2% differences at the nucleotide level. |
Distribution of Mutations
The nucleotide sequences and amino acid residues of the full-length VP1 encoding gene of 24 isolates of Ethiopia between 1990 and 2015 showed that 24% nucleotide (155/648) and amino acid (52/216) variability. The hyper-variable regions were located at amino acid positions 21–28, 43–51, 81–101, 135–142, 155–160, whereas complete conservation was also observed at amino acid positions ranging from 175 to 191, and positions beyond have variation with some exceptions (Figure 2). The cell-attachment site Arg-Gly-Asparagine (“RGD”) within the G-H loop (134–161) and the cysteine residue at the base of the loop (residue position 135), as well as residues at −1 and +4 of RGD, were conserved. The amino acid present at the +1 position was a positively charged arginine in all Ethiopian SAT-2 sequences. Position +2, +3, +10, and +12 were also checked for their variability as they are reported previously as monoclonal antibody neutralization sites for SAT-2 FMDV15,30 within the GH-loop. It was found in this study that +3 (A/V), +10 (G/D/N) and +12 (N/K/R/G) have two, three, and four amino acid differences, respectively. Whereas at the +2 position, the amino acid is conserved throughout the sequences (A). The cleavage site (VP1/2A) (residue position between 214/215) contained amino acid sequences KQ/LC and RQ/TL. Here, the reference sequence was ETH/24/2010 (MN987544). This sequence belongs to the same outbreak of 2009 from which the NVI vaccine strain for SAT-2 was identified. Accordingly, 2007, 2009, and 2010 isolates were seen to have 97% amino acid similarities, whereas 2015, 2014, 1991, and 1990 have variable regions as indicated in the alignment (Figure 4).
 |
Figure 4 Amino acid sequence alignment of the VP1 (1D) gene sequence of 24 SAT-2 FMD viruses circulating for 25 years in Ethiopia. Indicates an amino acid site identical to the sequence of ETH/24/2010 and “?”Indicates ambiguous sites. The cell attachment site coded by AA Arg-Gly-Asp (“RGD”) is highlighted in blue and the green bar indicates the G-H loop that is the major immunodominant site of the FMDV capsid, a red bar indicates the variable sites of the G-H loop, the yellow bar indicates an alpha helix (A, B, Z), a black bar a beta-sheet (B, C, D, E, F, G1, G2, H, I), light yellow bar the C-terminus of the major immunodominant site, an arrow indicates the 2A cleavage site. |
Amino acid variations were predominantly at the G-H loop (AA positions 134–161) and the C-terminus AA positions (191–216) position of VP1. The RGD cell attachment sites within the G-H loop of the gene at positions 144–146 were conserved in all sequences and reference strains used in the analysis (ETH/24/2010) from the same outbreak from which the NVI vaccine for SAT-2 is prepared.
Discussions
In Ethiopia, FMD is an endemic disease described 60 years ago. Circulating FMDV come as different serotypes, ie, O, A, SAT-2, SAT-1, and C (no longer reported after 1984).28,31,32 There has been an attempt to investigate this disease throughout Ethiopia based on outbreak reports. In the period 1981–2007, two topotypes of serotype O (EA-3 and EA-4), one genotype of serotype A (Africa), and three topotypes of serotype SAT-2 (IV, XII, and XIV) were identified28 and recently by Sulayeman et al.29 The temporal distribution of the outbreaks occurrence as indicated by their years of outbreak indicates that at least 2 years’ gap is observed in Ethiopia before they were succeeded by other topotype outbreak (ie, circulated in 1990, and 1991, 2007, 2009, and 2010, 2014, and 2015). This is in agreement with El-Nahas and Salem which identified the genetic diversity of SAT-2 viruses between different outbreak years in Egypt where FMD is endemic.18 In addition, previous genetic diversity of serotypes in successive years has been reported for other serotypes (O, A, SAT-1) in different parts of Africa.19,33
According to their sequence identity, the topotypes (IV) (1990) were found to have higher nucleotide identity with Kenyan and Ugandan sequences. Topotype (XIV) of 1991 (ETH/2/91 and ETH/3/91) did not show significant nucleotide identity with neighboring countries’ sequences. Topotype (VII) sequence of 2014 and 2015 has nucleotide identity with Sudan sequences. On the other hand, 2007, 2009, and 2010 outbreaks caused by topotype (XIII) have also nucleotide identity with 1977 and 2008 Sudan sequences. Viral incursions from the neighboring countries are possible because these countries share common borders. In addition, animal trading across these borders is a common practice34 that increased the risk of incursion. In the presence of a huge livestock population, no or little attempt at disease control or prevention strategy, vulnerable production systems, illegal trading of animal and animal products easily maintain cycles of FMD epizootics in one country or might become a source for outbreaks in neighboring countries.35
In this study, two phylogenetic clusters of viruses corresponding to four topotypes were identified, in agreement with two topotypes of Sahle et al,36 identified as (IV) (ETH/1/91, ETH/1/90, ETH/2/90) and (XIV) (ETH/2/91) by sequence-based analysis. The additional topotypes (VII) was also in agreement with.29 The other topotype (XIII) identified in 2007, 2009, and 2010 sequences were in agreement with the finding of Ayelet et al.28 Therefore, we can conclude that a total of four topotypes from 1990 to 2015 were identified. Based on the date of isolation and the risky husbandry practices, the possible route of incursion into Ethiopia was from the west border with Sudan moving to the east consisting of topotype (VII) (2014–2015) and (XIII) (2007, 2009, 2010) and from the south to north direction of the neighboring countries from Kenya topotype (IV) (1990 and 1991). Topotype (XIV), which caused an outbreak in 1991, might be extinct. Therefore, the appearance of four topotypes (IV,VII, XIII, and XIV) of SAT-2 FMDV in Ethiopia over 25 years, based on VP1 sequence analysis, warns that multiple area-specific vaccines may have to be developed to deal with the heterogeneity of the circulating virus.37
Conclusions
In this particular study, it is worth to conclude that sequences of serotype SAT-2 FMD viruses circulating in Ethiopia during 25 years (1990 to 2015) cluster phylogenetically according to their prevalence in four distinct geographical areas (topotypes) in neighboring countries like Sudan, Eritrea, and Kenya in the same timeframe but also with isolates found in Egypt. In chronological order, the identified topotypes were IV, XIV, XIII, and VII, based on phylogenetic classifications available in scientific literature. In all of the SAT-2 FMDV sequences studied, the cell attachment site RGD of the VP1 protein was conserved, although significant amino acid changes were observed in the flanking regions. This might affect the efficacy of neutralizing antibodies induced by vaccination or infection. Further studies should indicate whether the SAT-2 FMD vaccine currently in use in Ethiopia provides significant protection to all of the topotypes observed. The current vaccine is based on an outbreak strain dating back to 2009. It should however be noted that FMD control should not only rely on using a well-matched vaccine but should also include strict biosecurity measures at the herd level and regulation and control of animal movements and trade at a national and international level. Furthermore, the re-emergence of topotypes and information on the status of new variants and their regional prevalence pattern is inadequate. Hence, more research on virus diversity and effective control policy design should be effectuated including all circulating topotypes and taking into account the specific husbandry practices for each region.
Ethics Statement
Ethical clearance regarding this research was obtained from ethical review committee of Addis Ababa University, College of Veterinary Medicine (Ref. number VM/ERC/03/12/2016).
Acknowledgments
We would like to thank KU LEUVEN Belgium (HPI), and the sequences obtained were from the publicly available GenBank database and accordingly the authors would like to acknowledge the source.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was financially supported by Global Minds PhD scholarship, KU Leuven (Project number: 4520196970).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Domingo E, Baranowski E, Escarmís C, Sobrino F. Foot-and-mouth disease virus. Comp Immunol Microbiol Infect Dis. 2002;25:297–308. doi:10.1016/s0147-9571(02)00027-9
2. Carrillo C, Tulman ER, Delhon G, et al. Comparative genomics of foot-and-mouth disease virus. J Virol. 2005;79:6487–6504. doi:10.1128/JVI.79.10.6487
3. Knowles NJ, Samuel AR. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 2003;91:65–80. doi:10.1016/s0168-1702(02)00260-5
4. Rweyemamu M, Roeder P, MacKay D, et al. Epidemiological patterns of foot-and-mouth disease worldwide. Transbound Emerg Dis. 2008;55:57–72. doi:10.1111/j.1865-1682.2007.01013.x
5. Knight-Jones TJD, Robinson L, Charleston B, et al. Global foot-and-mouth disease research update and gap analysis: 1 - overview of global status and research needs. Transbound Emerg Dis. 2016;63(1):3–13. doi:10.1111/tbed.12528
6. Haydon DT, Samuel AR, Knowles NJ. The generation and persistence of genetic variation in foot-and-mouth disease virus. Prev Vet Med. 2001;51:111–124. doi:10.1016/s0167-5877(01)00210-0
7. Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, VandePol S. Rapid evolution of RNA genomes. Science. 1982;215:1577–1585. doi:10.1126/science.7041255
8. Domingo E, Mateu MG, Martinez MA, Dopazo J, Moya A, Sorbino F. Genetic variability and antigenic diversity of foot and mouth disease virus. Appl Virol Res. 1990;2:233–266.
9. Balinda SN, Siegismund HR, Muwanika VB, et al. Phylogenetic analyses of the polyprotein coding sequences of serotype O foot-and-mouth disease viruses in East Africa: evidence for interserotypic recombination. Virol J. 2010;7:1–9. doi:10.1186/1743-422X-7-199
10. Ferretti L, Pérez-Martín E, Zhang F, et al. Pervasive within-host recombination and epistasis as major determinants of the molecular evolution of the foot-and-mouth disease virus capsid. Phil Trans R Soc B. 2020;374:1–23. doi:10.1371/journal.ppat.1008235
11. Ferretti L, Di Nardo A, Singer B, et al. Within-host recombination in the foot-and-mouth disease virus genome. Viruses. 2018;10:1–14. doi:10.3390/v10050221
12. Jamal SM, Ferrari G, Ahmed S, Normann P, Belsham GJ. Molecular characterization of serotype Asia-1 foot-and-mouth disease viruses in Pakistan and Afghanistan; emergence of a new genetic Group and evidence for a novel recombinant virus. Infect Genet Evol. 2011;11:2049–2062. doi:10.1016/j.meegid.2011.09.015
13. OIE. Manual of diagnostic tests and vaccines for terrestrial animals. Collect, submiss storage diagnostic specimens; 2009. Available from: https://www.fao.org/fileadmin/templates/rap/files/meetings/2014/140318-reference.pdf.
14. Vosloo W, Bastos ADS, Sangare O, Hargreaves SK, Thomson GR. Review of the status and control of foot and mouth disease in sub-Saharan Africa. Rev Sci Tech. 2002;21:437–449. doi:10.20506/rst.21.3.1349
15. Bastos ADS, Haydon DT, Sangaré O, Boshoff CI, Edrich JL, Thomson GR. The implications of virus diversity within the SAT 2 serotype for control of foot-and-mouth disease in sub-Saharan Africa. J Gen Virol. 2003;84:1595–1606. doi:10.1099/vir.0.18859-0
16. Sangare O, Bastos ADS, Venter EH, Vosloo W. Retrospective genetic analysis of SAT-1 type foot-and-mouth disease outbreaks in West Africa (1975–1981). Vet Microbiol. 2003;93:279–289. doi:10.1016/s0378-1135(02)00439-x
17. Han S-C, Guo H-C, Sun S-Q. Three-dimensional structure of foot-and-mouth disease virus and its biological functions. Arch Virol. 2015;160:1–16. doi:10.1007/s00705-014-2278-x
18. El Nahas AF, Salem SAH. Meta-analysis of genetic diversity of the VP1 gene among the circulating O, A, and SAT2 serotypes and vaccine strains of FMD Virus in Egypt. J Vet Res. 2020;64:487–493. doi:10.2478/jvetres-2020-0069
19. Sangare O, Bastos ADS, Marquardt O, Venter EH, Vosloo W, Thomson GR. Molecular Epidemiology of Serotype O foot-and-mouth disease virus with Emphasis on West and South Africa. Virus Genes. 2001;22:345–351. doi:10.1023/A:1011178626292
20. Sangare O. Molecular Epidemiology of foot-and-mouth-disease virus in West Africa. University of Pretoria; 2002. Available from: https://repository.up.ac.za/handle/2263/23010.
21. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi:10.1016/S0022-2836(05)80360-2
22. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi:10.1093/molbev/mst197
23. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi:10.1093/nar/25.24.4876
24. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In: Nucleic Acids Symposium Series. Information Retrieval Ltd; 1999:95–98. doi:10.14601/Phytopathol_Mediterr-14998u1.29
25. QGIS Developement Team. QGIS geographic information system. Open Source Geospatial Foundation; 2009. Available from: http://qgis.osgeo.org.
26. Leta S, Mesele F. Spatial analysis of cattle and shoat population in Ethiopia: growth trend, distribution and market access. Springerplus. 2014;3:1–10. doi:10.1186/2193-1801-3-310
27. Sahle M, Dwarka RM, Venter EH, Vosloo W. Study of the genetic heterogeneity of SAT-2 foot-and-mouth disease virus in sub-Saharan Africa with specific focus on East Africa. Onderstepoort J Vet Res. 2007;74:289–299. doi:10.4102/ojvr.v74i4.115
28. Ayelet G, Mahapatra M, Gelaye E, et al. Genetic characterization of foot-and-mouth disease viruses, Ethiopia, 1981–2007. Emerg Infect Dis. 2009;15:1409–1417. doi:10.3201/eid1509.090091
29. Sulayeman M, Dawo F, Mammo B, Gizaw D, Shegu D. Isolation, molecular characterization and sero-prevalence study of foot-and-mouth disease virus circulating in central Ethiopia. BMC Vet Res. 2018;14:1–10. doi:10.1186/s12917-018-1429-9
30. Crowther JR, Rowe CA, Butcher R. Characterization of monoclonal antibodies against a type SAT 2 foot-and-mouth disease virus. Epidemiol Infect. 1993;111:391–406. doi:10.1017/s0950268800057083
31. Martel JL. Foot-and-mouth disease in Ethiopia. Distribution of serotypes of foot-and-mouth disease virus. Rev Elev Med Vet Pays Trop. 1974;27:169–175. doi:10.19182/remvt.7961
32. Gizaw D, Tesfaye Y, Wood BA, et al. Molecular characterization of foot-and-mouth disease viruses circulating in Ethiopia between 2008 and 2019. Transbound Emerg Dis. 2020;67:2983–2992. doi:10.1111/tbed.13675
33. Slager-bastos AD. Molecular epidemiology and diagnosis of SAT-type foot-and-mouth disease in Southern Africa; 2001.
34. Duchatel F, Bronsvoort M de C, Lycett S. Phylogeographic analysis and identification of factors impacting the diffusion of foot-and-mouth disease virus in Africa. Front Ecol Evol. 2019;7:1–12. doi:10.3389/fevo.2019.00371
35. McDermott JJ, Arimi SM. Brucellosis in sub-Saharan Africa: epidemiology, control and impact. Vet Microbiol. 2002;90(1–4):111–134. doi:10.1016/s0378-1135(02)00249-3
36. Sahle M, Venter EH, Dwarka RM, Vosloo W. Molecular epidemiology of serotype O foot-and-mouth disease virus isolated from cattle in Ethiopia between 1979–2001. Onderstepoort J Vet Res. 2004;71:129–138. doi:10.4102/ojvr.v71i2.275
37. Bastos ADS, Anderson EC, Bengis RG, Keet DF, Winterbach HK, Thomson GR. Molecular epidemiology of SAT3-type foot-and-mouth disease. Virus Genes. 2003;27:283–290. doi:10.1023/a:1026352000959
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.