")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 14
Viral Nephropathies, Adding SARS-CoV-2 to the List
Authors Pramod S , Kheetan M, Ogu I, Alsanani A, Khitan Z
Received 11 February 2021
Accepted for publication 7 May 2021
Published 3 June 2021 Volume 2021:14 Pages 157—164
DOI https://doi.org/10.2147/IJNRD.S303080
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Sheena Pramod, Murad Kheetan, Iheanyichukwu Ogu, Ahlim Alsanani, Zeid Khitan
Division of Nephrology, Department of Internal Medicine, Joan C Edwards Marshall University School of Medicine, Huntington, WV, USA
Correspondence: Sheena Pramod
Joan C Edwards Marshall University School of Medicine, 1690 Medical Center Drive, Huntington, WV, 25701, USA
Tel + 1 3045262532
Email [email protected]
Abstract: Viral infections in the immunocompetent host can cause both acute and chronic kidney disease either as a direct damage to the infected kidney cells or as a consequence of systemic immune responses that impact kidney function. Since identifying these entities in the 1970s and 80s, major breakthroughs in the understanding of the viral mechanisms have occurred. Viruses have evolved mechanisms to hijack signaling pathways of infected cells to evade antiviral immune responses by the host. Over time, the clinical presentations and management of these diseases have evolved along with our in-depth understanding of the various pathophysiological mechanisms causing these conditions. Similarly, both at the cellular and systemic levels, the host has evolved mechanisms to counter viral subversion strategies for mutual survival. Since the start of the current COVID-19 pandemic, numerous cases of acute kidney injury have been reported in the literature with various possible pathophysiological mechanisms. In this review, we summarize lessons learned from prior viral pandemics related to viral mechanisms utilized in the pathogenesis of numerous renal manifestations to attempt to utilize this knowledge in predicting post-COVID-19 kidney disease.
Keywords: viral nephropathy, COVID-19, viral immunogenicity, hepatitis B, hepatitis C, HIV, SARS-CoV-2
Introduction
Virus-related nephropathies have been well established in several viral infections like hepatitis B, C, HIV, parvovirus-B resulting in a spectrum of renal lesions in an immunocompetent host. The evolutionary race between infectious agents and the human immune system over time has created elaborate viral mechanisms to escape immune clearing and equally complex host response to counter viral evasion strategies.1 Several mechanisms are involved in the pathogenesis of virus-related nephropathies, including tropism of the virus in the kidney, induction of abnormal immune complexes, direct cytopathogenic effects, and multiorgan failure.2 Since the start of the global pandemic due to the novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) manifesting as COVID-19, an increase in cases of acute kidney injury (AKI) ranging from 3 −43% has been observed with significant number exhibiting proteinuria (42%) and a smaller fraction developing hematuria (11.3–40.9%).3 Kidney biopsies showed various pathologies including ATN, collapsing FSGS and acute endothelial injury.4
In this review, we explore the affinity of various viruses to the renal tissue along with pathological manifestations and discuss the potential mechanisms that can explain current renal syndromes with respect to COVID-19 pandemic.
Epidemics of Hepatitis B, C, and Pandemic of HIV with New Roadmap of Glomerular Disease in the Past Century
Approximately one-third of the world’s population have been infected with hepatitis B (HBV) with 5% of this population being chronic carriers.5 HBV involves both viral and host factors in pathogenesis with an association of HLA-II genes indicating genetic predisposition. Although there is evidence of direct infectivity of glomerular cells resulting in mesangial proliferation and expressing extracellular matrix proteins, manifested as IgA nephropathy and FSGS,6 the main pathogenic mechanism in HBV-related renal disease is immune-complex deposition in the glomeruli (Table 1). In HBV, the immune response is directed towards the viral antigens surface (s), core (c) and extracellular (e), that forms different immune complexes that deposit in the glomeruli according to their size and charge. This results in various glomerular patterns of injury as seen in the classical membranous nephropathy (MN) or membranoproliferative glomerulonephritis (MPGN) pathology, where the smaller cationic HBeAg deposit in the subepithelial region as in MN and the larger anionic HBcAg and HBsAg deposit in the subendothelial and mesangial regions causing MPGN with or without type III cryoglobulinemia.7,8 In HBV-associated polyarteritis nodosa (PAN), HBsAg immune complexes are deposited in medium-sized blood vessels. On kidney biopsy, the mesangial hypercellularity and subendothelial deposits may suggest a secondary MN with endothelial cell tubuloreticular inclusions.9 Cryoglobulinemia (type III) can be seen in HBV compared to type II in HCV-MPGN.9 Since the first reported cases in 1980s, the HIV pandemic has continued, with 37 million active carriers in the world and two million newly infected cases per year.10 The manifestations of renal disease in HIV infection depends on the treatment status of the patient and by their coinfection status with other viruses such as with HBV (5–10%) or with HCV (25–30%).11 HIV-associated nephropathy (HIVAN), a form of collapsing FSGS was first reported in 1984.11 On the other hand, HIV-associated immune complex kidney disease (HIVICK) is a result of immune complexes deposition in the glomerulus, unlike HIVAN, treatment with ART does not show major effect on its progression to ESRD.12,13 Over the past 30 years, the epidemiology of HIV- related renal disease has changed from HIVAN to more cases of HIVICK and ART-induced nephrotoxicity.14
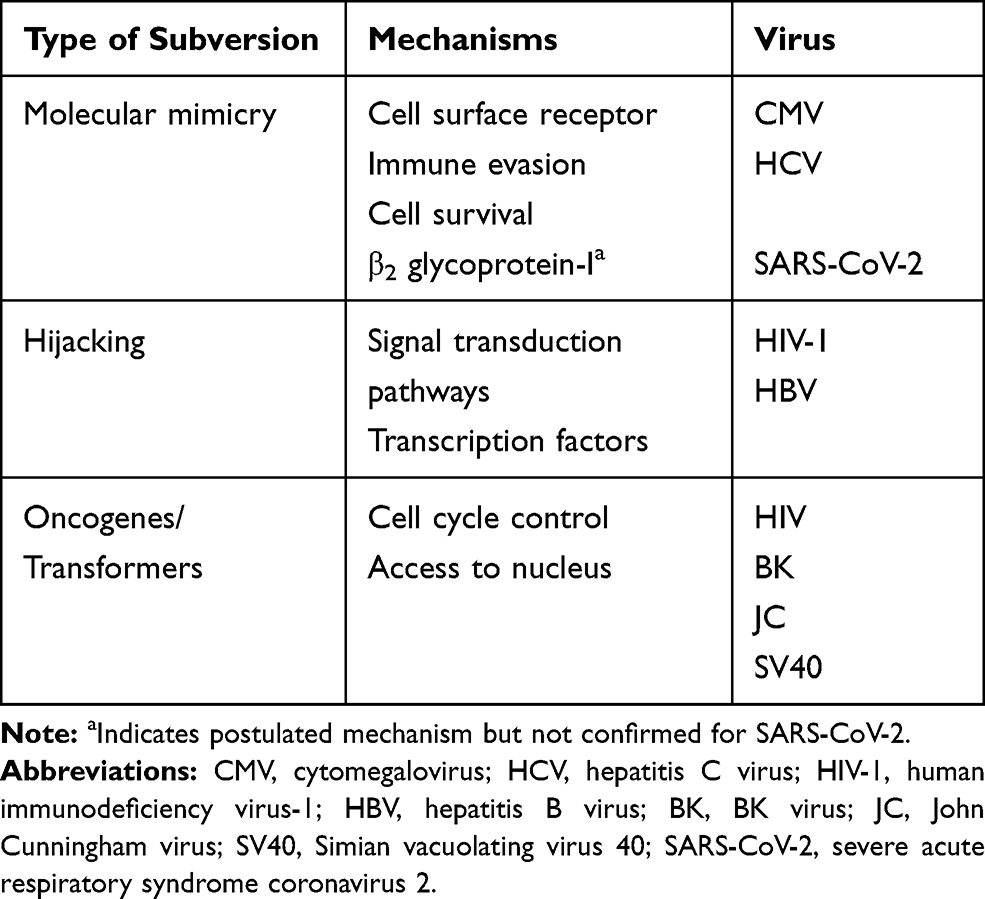 |
Table 1 Type of Virus and Mechanism of Subversion |
Kidney disease in HIV is mediated by factors related to the virus itself, whether it is direct infectivity of the podocytes and tubular epithelial cells (tropism) or cytopathic effects on renal epithelial cells in the presence of accessory proteins (Nef and Vpr) resulting in podocyte dysregulation giving the FSGS phenotype. Factors related to the host as in APOL1 variants make HIVAN predominantly a disease of those of African heritage, while circulating immune complexes depositing in the glomerulus can result in a variety of glomerulonephritides including IgA nephropathy, MPGN, MN, lupus-like GN, immunotactoid GN and fibrillary GN (Table 1). 15,16 In HIVAN, renal biopsy usually shows FSGS with the collapsing variant, tubulointerstitial disease with microcystic disease of renal tubules, interstitial nephritis and the presence of tubuloreticular inclusions.17,18 In HIVICK, deposits vary based on the site of immune-complex deposition, with some shared features with HIVAN, including podocyte hyperplasia, tubular dilation, interstitial inflammation and tubuloreticular inclusion bodies.15
As of the 2015 census, an estimated 71.1 million individuals are chronically infected with HCV.19 Occult HCV has been detected in 30–50% of patients with idiopathic MN, IgA, FSGS, ANCA positive vasculitis and MPGN.17 It is estimated that only 20% of these individuals are aware of the diagnosis of HCV and 15% of those known receive treatment.19 HCV causes upregulation of the humoral immune response, with specific viral proteins, like the HCV E2 envelope protein, demonstrating molecular mimicry resulting in B-lymphocyte activation causing most of the immune mediated disorders associated with HCV such as type II cryoglobulinemia. Other histopathological presentations that are immune complex mediated include MN, PAN, IgA nephropathy, immunotactoid GN, fibrillary GN and thrombotic microangiopathy (TMA), where FSGS is speculated to be secondary to direct podocyte injury (Table 1).20 With the current availability of direct-acting antiviral therapy (DAA) which can achieve a viral remission of 95% for most HCV genotypes, the prevalence of glomerular disease in this population should progressively decline in the coming decade.21
Virulence of Viruses and Immunogenicity
Virulence can dramatically increase when pathogens shift to infect new host species resulting in new and devastating outbreaks like Ebola, HIV and the current COVID-19 pandemic.22 Pathogenetic links between viral infection and renal disease are often difficult to establish.23 In order to prove causality, besides recognition of the clinical syndrome, serological diagnosis, identification of specific viral antigenemia and detection in glomerular structures of viral antigen by polymerase chain reaction (PCR) or immune complexes either by in-situ PCR, culture or the presence of inclusion bodies intracellularly are needed.2,17,23 Resolution or regression of the glomerular lesion with host immune clearance of viremia or with antiviral therapy also supports this.17 There have been some reported cases of renal hepatitis C manifestations where viremia was completely absent.24,25
Viruses are obligate parasites that cannot complete their lifecycle without a host to synthesize new protein.1 The first function is to provide for the structural proteins of the coat, second is to ensure their replication of the genetic material and the third is to modify the host cellular machinery to facilitate the first two functions. The last function which subverts the normal cellular activities is unique to individual viruses and leads to pathogenesis and disease.26
There are several subversion mechanisms that viruses use to replicate in the host (Table 2). Immune evasion strategies like molecular mimicry used by cytomegalovirus manipulate the immune system by producing proteins that mimic host cytokines and chemokines thereby altering several key immunologic functions like lymphocyte recruitment and B cell functions. RNA viruses like hepatitis C virus have immune evasion strategies which cause inactivation of Toll-like receptor (TLR) signaling complex thereby blocking the interferon antiviral mechanism leading to persistent infection.26 Latency is another immune evasion strategy to maintain viral persistence in the host by silencing replication as in the case of EBV using miR-BART2 miRNA.1
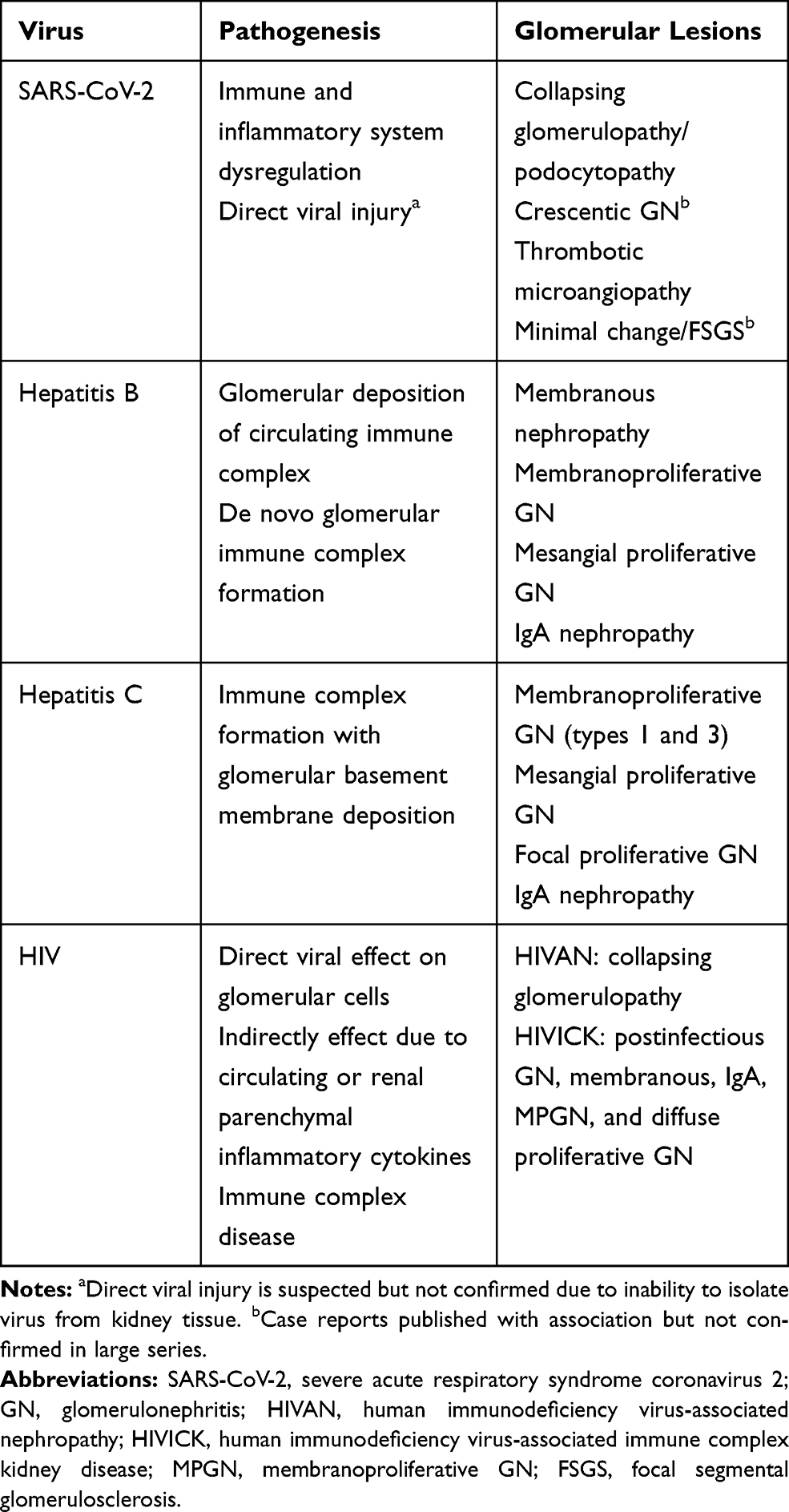 |
Table 2 Viruses, Pathogenic Mechanism and Associated Glomerulopathy |
Another strategy viruses use is cellular hijacking HIV-1 by hijacking Src-signaling.26 In HIVAN, HIV genes Nef induces podocyte dysfunction and proliferation along with Vpr induced apoptosis of renal epithelial cells playing a key role in the pathogenesis.27 Studies have shown that retroviruses, polyomaviruses and herpesviruses all encode proteins to manipulate mTOR signaling to facilitate viral protein production.1
Several viruses use oncogenic transformation to help survival. For example, increase proliferation of epithelial cells in HIVAN is different from common forms of FSGS where there is loss of renal cell mass.
Some nephropathies occur due to systemic (cytokine storm as in Dengue) or local antigen deposition as in HB-MN while others occur due to viral tropism in the kidney as in polyomaviruses BK, JC, herpesvirus CMV and hanta virus nephropathy.1,26
One of the earlier observations by Su et al described coronavirus like particles in EM in proximal tubular epithelium with a positive immunofluorescence staining for the virus in tubular epithelium and podocytes using an antibody targeting SARS-CoV-2 virus.28 There have also been reports describing isolation of viral elements within endothelial cell and accumulation of inflammatory cells from postmortem renal tissue29 These findings may suggest the induction of endotheliitis in organs as a consequence of viral involvement and of host inflammatory response leading to systemic impaired microcirculatory dysfunction.29 In contrast, several reports failed to identify viral particles in renal cells which possibly disapproves viral tropism.30–32
Multiple studies have also demonstrated virus encoded products that can be recognized by immune and nonimmune cells as pathogen-associated molecular patterns (PAMP) which are structural motifs that serve as danger signals to the host indicating the presence of the virus. This causes activation of immune cells within the podocyte or glomerular basement membrane inducing bystander injury to the renal parenchyma in renal disease that may not be conventionally considered to be of viral origin.26 Organ cross talk mediated by cytokines, growth factors and release of damage associated molecular patterns (DAMPS) have been suggested to mediate AKI in the setting of ARDS.3
SARS-CoV-2: Characteristics, Immunogenicity Targets in the Kidney
SARS-CoV-2 is an enveloped RNA virus that belongs to the beta coronavirus genus, sharing about 79% genetic similarity with SARS-CoV, the agent responsible for the SARS from 2002–2003 pandemic and 51.8% similarity with the Middle East respiratory syndrome coronavirus (MERS-CoV).33 Human angiotensin-converting enzyme 2 (ACE2) has been identified as the functional receptor of SARS-COV-2 in humans.33
The spike (S) protein of SARS-CoV-2 has two main subunits, the N-terminal S1 subunit which contains the receptor binding domain (RBD) and the C-terminal S2 subunit which is important in viral cell fusion.33 The binding of the RBD to ACE2 initiates endocytosis of the virus followed by cleavage of the S protein by the endosomal proteases and exposes it to the endosomal proteases which cleaves the S protein, a step called priming, exposing the fusion peptide which in turn mediates the fusion to the target cell membrane and subsequent virus entry.33 SARS-CoV-2, like other coronaviruses, also depends on transmembrane protease serine 2 (TMPRSS2) for its entry inside the host cell.33 Interestingly, similar to MERS-CoV and unlike SARS-CoV, SARS-CoV-2 has a furin cleavage site at the border of S1/S2 that can contribute to its broad tissue tropism and infectivity.33 Another potential virus entry route that has been shown in in vitro is of host cell receptor CD147 which is widely expressed including in the proximal convoluted tubules and SARS-CoV-2 spike protein.34 The authors showed that loss of CD147 or blocking CD147 in Vero E6 and BEAS-2B cell lines by anti-CD147 antibody, meplazumab, inhibits SARS-CoV-2 amplification which supports this receptor being an entry route.34 Further research exploring this avenue in human kidneys is ongoing.
In the kidney, ACE2 and TMPRSS2 are colocalized in the proximal tubules and collecting ducts and to a lesser extent in podocytes, mesangial cells and parietal epithelium, making these cells a target for SARS-CoV-2 infection.33 ACE1 is characterized by a genetic deletion/insertion (D/I) polymorphism in intron 16, which is associated with alterations in circulating and tissue concentrations of ACE.35 ACE polymorphism and susceptibility to SARS-CoV-2 may be a possible hypothesis to why the African American population develops more severe forms of COVID-19 compared to Western populations.35 A study showed the presence of viral RNA in the kidneys preferentially targeting the glomerular cells which goes to support the possibility of renal cell infection.33,36 Renal pathology studies of COVID-19 patients with AKI have reported microscopic findings of possible viral inclusion bodies in tubular epithelial cells and podocytes suggesting possible direct cytopathic effect of infected cells by SARS-CoV-2.33,37 However, there has been caution on the conclusion of these finding as viral inclusion bodies since they can be normal substructural organelles (clathrin-coated vesicles) mimicking viral pathogens or microvesicular bodies (MVBs) representing podocyte endocytosis especially since viral RNAs were not demonstrated in the renal tissues.30,33,37 In two studies looking at 2738 and 6339 autopsy specimens respectively, a detectable viral load using SARS-CoV-2 screening test RT-PCR was shown in homogenized kidney tissue after incubating the tissue with primate epithelial (Vero) cells.36 There exists a strong plausibility of direct renal cell infection by SARS-CoV-2 given its properties and the physiology of the potentially susceptible renal cells, however, demonstration of viral RNAs will lend more credence to it and thus the need for more studies.
The absence of identifiable viral particles in kidney tissues have led to an alternate theory of indirect viral cytopathic mechanism in the observed renal pathology. Cases of collapsing glomerulopathy has been observed in COVID-19 patients presenting with heavy proteinuria, mainly in African American patients with high-risk APOL1 genes have been reported.33,37,40 Interestingly none of these reports demonstrated viral particles in the kidney tissue. It is possible, as postulated, that this may have resulted from upregulation of APOL1 expression by the viral infection and other inflammatory response leading to podocyte dysregulation and injury that culminated in collapsing glomerulopathy.41
Cytopathic effect of SARS-CoV-2 can lead to pyroptosis leading to severe inflammation with release of proinflammatory cytokines including IL-6, IFNy, MCP1. This may trigger dysfunctional immune response that may potentially result in a cytokine storm manifesting with multi-organ damage including AKI, proteinuria and glomerulopathy.42
SARS-CoV-2 Associated Renal Disease
The incidence of AKI associated with COVID-19 is variable depending on the geographic regions.43 AKI occurs in approximately 25–30% of patients admitted to hospitals and higher for ICU COVID-19 patients.43 The most common injury observed in autopsy and biopsy findings is acute tubular injury (Figure 1).35 In a recently published study, authors reported SARS-CoV-2 causing specific manifestations of proximal tubule dysfunction.44 In a cohort of 49 patients the authors found evidence of low molecular weight proteinuria (70–80%), neutral aminoaciduria (46%) and defective handling of uric acid (46%) or phosphate (19%).46 Histopathology showed proximal tubular injury, acute tubular necrosis, intraluminal debris, marked decrease in megalin expression in the brush border and electron microscopy evidence of particles resembling coronaviruses in cisternae of the endoplasmic reticulum in proximal tubule cells.44 Reports also indicate that rhabdomyolysis occurs in 7–20% of patients with COVID-19 AKI.3
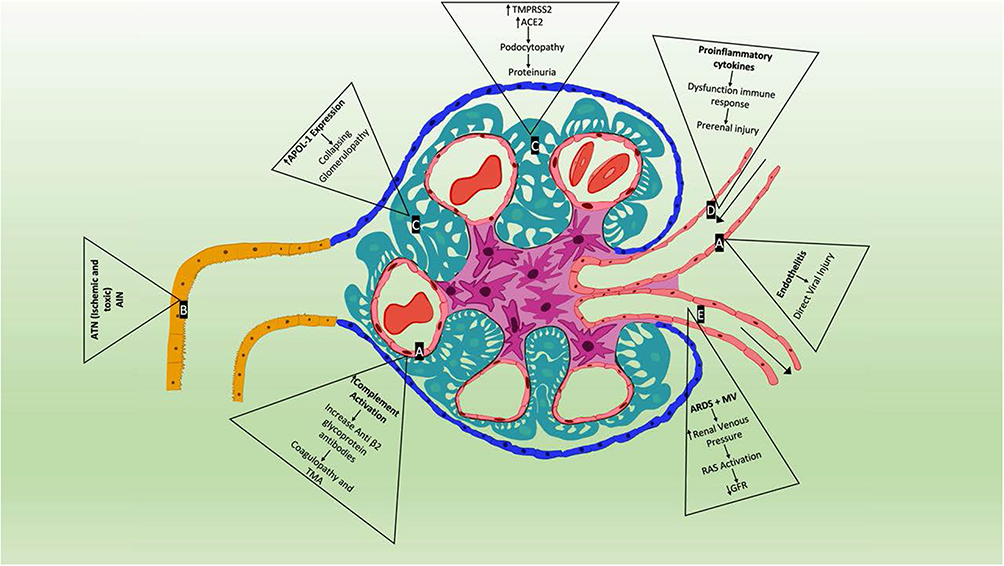 |
Figure 1 Proposed mechanisms of SARS-CoV-2 associated renal disease. (A) Endothelial cell—following infection in the lungs, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly infects endothelial cells owing to their high level of angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) expression triggering inflammatory and thrombotic processes leading to endotheliitis and complement activation causing increase in anti-β2 glycoprotein antibodies leading to coagulopathy and TMA. (B) Proximal tubular cell—due to systemic shock state, use of multiple drugs including antivirals, antibiotics and rhabdomyolysis commonly seen in SARS-CoV-2 infections can lead to acute tubular necrosis (ATN) and acute interstitial nephritis (AIN). (C) Podocyte – another site of increased ACE2 and TMPRSS2 expression may cause podocyte dysfunction leading to proteinuria and hematuria by altering glomerular capillary hemostasis. Also, presence of APOL1 genotype in some patients in the setting of SARS-CoV-2 infection induces a collapsing glomerulopathy. (D) Afferent arteriole—pro-inflammatory cytokines including IL-6, IFNy and MCP1 cause systemic cytokine storm causing hypoperfusion injury. Other factors such as sepsis, cardiac dysfunction, ARDS and mechanical ventilation, hypovolemia all contribute to renin-angiotensin system activation causing reduction in glomerular filtration rate and prerenal injury. (E) Efferent arteriole. |
Though data are still sparse and early, SARS-CoV-2 is emerging as a possible cause of virus-associated glomerulopathy (VAGP). Kissling et al described a case of a 63-year-old man with African heritage with COVID-19 homozygous for APOL1 gene who developed AKI, massive proteinuria. Kidney biopsy showed collapsing glomerulopathy with spherical particles in the podocytes on electron microscopy concerning for viral inclusion bodies but was negative for SARS-CoV-2 RNA.45 In another report of a 44-year-old African American female with high risk APOL1 gene, severe COVID-19, AKI on chronic kidney disease and proteinuria was noted to have collapsing glomerulopathy, tubular reticular inclusion in glomerular endothelial cells but negative for SARS-CoV-2 by in situ hybridization.46 In a study of six patients of African descent with COVID-19 and mild respiratory symptoms, AKI and new-onset nephrotic syndrome, collapsing glomerulopathy with microcystic tubular changes were seen in all six while endothelial reticular aggregates were seen in three. All six-patient had high-risk variants in the gene encoding apo L1 (APOL1). However, in situ hybridization failed to show SARS-CoV-2 RNA in the tissues.47 Another evidence of COVID-19 mediated podocytopathy may be seen in a case reported by Chueh et al of a patient with repeat renal biopsy showed transition from minimal change disease to collapsing focal segmental glomerulosclerosis (FSGS).48
Endothelial dysfunction as evidenced by high D-dimer levels and microvascular damage may be a key factor in COVID-19 associated coagulopathy.4 In a report by Jhaveri et al kidney biopsy done on a patient with COVID-19 revealed severe acute thrombotic microangiopathy with cortical necrosis.49 Detailed complement testing revealed a low factor H complement antigen, elevated plasma CBb complement and plasma SC5b-9 levels suggesting an activation of the alternative complement pathway.49 Zhang et al noted the presence of anticardiolipin immunoglobulin A (IgA) antibodies, anti-β2 glycoprotein I IgA and IgG antibodies in patients with COVID-19 who had hypoxemic respiratory failure.50 In a prospective single center observational study, elevated prevalence of anticardiolipin/anti-β2-glycoprotein-I antibodies were found in 12% of the COVID-19 patients.51 Researchers have suggested that IgG in patients with COVID-19 could recruit neutrophilic aggregation with similar ability to that of antiphospholipid antibody.
Patients with severe COVID-19-associated pneumonia/ARDS are often treated with increasing positive end-expiratory pressure (PEEP) which leads to increased intrathoracic pressure and increased sympathetic tone leading to secondary activation of renin-angiotensin system leading to reduced filtration in the kidney.3
Infection with SARS-CoV-2 has been associated with cytokine release syndrome, a cytokine storm which contributes to hypoperfusion related injury of renal tubules.35 Viral infection in alveolar cells, leads to massive recruitment of immune cells causing cytokine-mediated AKI.37
Conclusion
Over the past several decades various pandemics have highlighted the different mechanisms by which viruses cause kidney disease. With the novel coronavirus pandemic which has so far affected 96 million people as of January 2021 a new cohort of patients with kidney injury has been brought to light. The high infectivity causing rapid spread of this virus among the global population can have a significant burden of long-term kidney complications than prior pandemics. Though there have been several publications over the past few months with attempts to identify the exact mechanism of kidney injury in these patients, the exact pathophysiology of COVID-19 AKI in the kidney remains unclear. There exists a strong plausibility of direct renal cell infection by SARS-CoV-2 given its properties and the physiology of the potentially susceptible renal cells, however, demonstration of viral RNAs will lend more credence to viral tropism. One plausible explanation for lack of viral particles on the kidney biopsy may be attributed to biopsies being performed at a later phase of the infection. The absence of viral particles demonstrated may also indicate an alternate mechanism for renal injury explained in part by a dysfunctional immune response leading to a cytokine storm causing a cascade of renal injury including ATI, interstitial inflammation, microangiopathy, proteinuria, and possible glomerulopathy. As in the case of the hepatitis viruses and HIV-associated kidney disease, we may be seeing the start of a new disease entity as recently described of COVID-19-associated nephropathy (COVAN) in the African descent population along with increased CKD in patients with COVID-associated AKI in the future. Targeting the pathophysiologic mechanism of SARS-CoV-2 infection will be a pragmatic way of managing its host effects and developing therapies to regulate immune dysfunction in COVAN and maybe help limit a potential CKD pandemic until disease eradication is achieved by widely adopting effective vaccines.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bruggeman LA. Common mechanisms of viral injury to the kidney. Adv Chronic Kidney Dis. 2019;26(3):164–170. doi:10.1053/j.ackd.2018.12.002
2. Lai AS, Lai KN. Viral nephropathy. Nat Clin Pract Nephrol. 2006;2(5):254–262.
3. Nadim MK, Forni LG, Mehta RL, et al. COVID-19-associated acute kidney injury: consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat Rev Nephrol. 2020;16(12):747–764.
4. Akilesh S, Nast CC, Yamashita M, et al. Multicenter clinicopathologic correlation of kidney biopsies performed in COVID-19 patients presenting with acute kidney injury or proteinuria. Am J Kidney Dis. 2020.
5. Sharma SK, Saini N, Chwla Y. Hepatitis B virus: inactive carriers. Virol J. 2005;2:82.
6. Diao Z, Ding J, Yin C, Wang L, Liu W. Purified hepatitis B virus induces human mesangial cell proliferation and extracellular matrix expression in vitro. Virol J. 2013;10:300.
7. Gupta A, Quigg RJ. Glomerular diseases associated with Hepatitis B and C. Adv Chronic Kidney Dis. 2015;22(5):343–351.
8. Chan TM. Hepatitis B and Renal Disease. Curr Hepat Rep. 2010;9(2):99–105.
9. Kupin WL. Viral-associated GN: hepatitis B and other viral infections. Clin J Am Soc Nephrol. 2017;12(9):1529–1533. doi:10.2215/CJN.09180816
10. Piot P, Abdool Karim SS, Hecht R, et al. Defeating AIDS–advancing global health. Lancet. 2015;386(9989):171–218.
11. Fabian J, Naicker S, Goetsch S, Venter WD. The clinical and histological response of HIV-associated kidney disease to antiretroviral therapy in South Africans. Nephrol Dial Transplant. 2013;28(6):1543–1554.
12. Kalyesubula R, Perazella MA. Nephrotoxicity of HAART. AIDS Res Treat. 2011;2011:562790.
13. Booth JW, Hamzah L, Jose S, et al. Clinical characteristics and outcomes of HIV-associated immune complex kidney disease. Nephrol Dial Transplant. 2016;31(12):2099–2107.
14. Mallipattu SK, Salem F, Wyatt CM. The changing epidemiology of HIV-related chronic kidney disease in the era of antiretroviral therapy. Kidney Int. 2014;86(2):259–265.
15. Cohen SD, Kimmel PL. Immune complex renal disease and human immunodeficiency virus infection. Semin Nephrol. 2008;28(6):535–544.
16. Ross MJ. Advances in the pathogenesis of HIV-associated kidney diseases. Kidney Int. 2014;86(2):266–274.
17. Kupin WL. Viral-Associated GN: hepatitis C and HIV. Clin J Am Soc Nephrol. 2017;12(8):1337–1342.
18. Post FA, Campbell LJ, Hamzah L, et al. Predictors of renal outcome in HIV-associated nephropathy. Clin Infect Dis. 2008;46(8):1282–1289.
19. Spearman CW, Dusheiko GM, Hellard M, Sonderup M. Hepatitis C. Lancet. 2019;394(10207):1451–1466.
20. Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int. 1999;56(6):2203–2213.
21. Jadoul M, Martin P. Hepatitis C treatment in chronic kidney disease patients: the kidney disease improving global outcomes perspective. Blood Purif. 2017;43(1–3):206–209.
22. Longdon B, Hadfield JD, Day JP, et al. The causes and consequences of changes in virulence following pathogen host shifts. PLoS Pathog. 2015;11(3):e1004728.
23. Wenderfer SE. Viral-associated glomerulopathies in children. Pediatr Nephrol. 2015;30(11):1929–1938.
24. Fowell AJ, Sheron N, Rosenberg WM. Renal hepatitis C in the absence of detectable serum or hepatic virus. Liver Int. 2008;28(6):889–891.
25. Bruggeman LA, Ross MD, Tanji N, et al. Renal epithelium is a previously unrecognized site of HIV-1 infection. J Am Soc Nephrol. 2000.
26. Berns JS, Cohen AH. Viruses and diseases of the kidneys. Clin J Am Soc Nephrol. 2007;2(Suppl 1):S1.
27. Medapalli RK, He JC, Klotman PE. HIV-associated nephropathy: pathogenesis. Curr Opin Nephrol Hypertens. 2011;20(3):306–311.
28. Su H, Yang M, Wan C, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98(1):219–227.
29. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418.
30. Calomeni E, Satoskar A, Ayoub I, Brodsky S, Rovin BH, Nadasdy T. Multivesicular bodies mimicking SARS-CoV-2 in patients without COVID-19. Kidney Int. 2020;98(1):233–234.
31. Kudose S, Batal I, Santoriello D, et al. Kidney Biopsy Findings in Patients with COVID-19. J Am Soc Nephrol. 2020;31(9):1959–1968.
32. Rossi GM, Delsante M, Pilato FP, et al. Kidney biopsy findings in a critically ill COVID-19 patient with dialysis-dependent acute kidney injury: a case against “SARS-CoV-2 nephropathy”. Kidney Int Rep. 2020;5(7):1100–1105.
33. Park SE. Epidemiology, virology, and clinical features of severe acute respiratory syndrome -coronavirus-2 (SARS-CoV-2; Coronavirus Disease-19). Clin Exp Pediatr. 2020;63(4):119–124.
34. Wang K, Chen W, Zhang Z, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020;5(1):283.
35. Izzedine H, Jhaveri KD. Acute kidney injury in patients with COVID-19: an update on the pathophysiology. Nephrol Dial Transplant. 2020.
36. Parmar MS. Acute Kidney Injury associated with COVID-19 - Cumulative evidence and rationale supporting against direct kidney injury (Infection). Nephrology (Carlton). 2020.
37. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23.
38. Puelles VG, Lutgehetmann M, Lindenmeyer MT, et al. Multiorgan and Renal Tropism of SARS-CoV-2. N Engl J Med. 2020;383(6):590–592.
39. Braun F, Lutgehetmann M, Pfefferle S, et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet. 2020;396(10251):597–598.
40. Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003;77(16):8801–8811.
41. Nasr SH, Kopp JB. COVID-19-associated collapsing glomerulopathy: an emerging entity. Kidney Int Rep. 2020;5(6):759–761.
42. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–374.
43. Kellum JA, Nadim MK, Forni LG. Sepsis-associated acute kidney injury: is COVID-19 different? Kidney Int. 2020;98(6):1370–1372.
44. Werion A, Belkhir L, Perrot M, et al. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 2020;98(5):1296–1307.
45. Kissling S, Rotman S, Gerber C, et al. Collapsing glomerulopathy in a COVID-19 patient. Kidney Int. 2020;98(1):228–231.
46. Larsen CP, Bourne TD, Wilson JD, Saqqa O, Sharshir MA. Collapsing glomerulopathy in a patient with COVID-19. Kidney Int Rep. 2020;5(6):935–939.
47. Wu H, Larsen CP, Hernandez-Arroyo CF, et al. AKI and collapsing glomerulopathy associated with COVID-19 and APOL 1 high-risk genotype. J Am Soc Nephrol. 2020;31(8):1688–1695.
48. Chueh TI, Zheng CM, Hou YC, Lu KC. Novel evidence of acute kidney injury in COVID-19. J Clin Med. 2020;9:11.
49. Jhaveri KD, Meir LR, Flores Chang BS, et al. Thrombotic microangiopathy in a patient with COVID-19. Kidney Int. 2020;98(2):509–512.
50. Zhang Y, Xiao M, Zhang S, et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N Engl J Med. 2020;382(17):e38.
51. Siguret V, Voicu S, Neuwirth M, et al. Are antiphospholipid antibodies associated with thrombotic complications in critically ill COVID-19 patients? Thromb Res. 2020;195:74–76.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.