")
Back to Journals » Journal of Inflammation Research » Volume 14
Vascular Adventitial Fibroblasts-Derived FGF10 Promotes Vascular Smooth Muscle Cells Proliferation and Migration in vitro and the Neointima Formation in vivo
Authors Chen Y, Chen Y, Jiang X, Shi M, Yang Z, Chen Z, Hua X , Chen J, Wang Y
Received 6 February 2021
Accepted for publication 21 April 2021
Published 25 May 2021 Volume 2021:14 Pages 2207—2223
DOI https://doi.org/10.2147/JIR.S305204
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Yuhan Chen,1,* Yuanyuan Chen,1,* Xueze Jiang,1 Mengkun Shi,2 Zhenwei Yang,1 Zhiyong Chen,1 Xuesheng Hua,1 Jie Chen,1 Yuepeng Wang1
1Department of Cardiology, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200092, People’s Republic of China; 2Department of Cardio-Thoracic Surgery, Shanghai Tongji Hospital, Tongji University School of Medicine, Shanghai, 200065, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuepeng Wang; Jie Chen
Department of Cardiology, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200092, People’s Republic of China
Tel +86 18217267289
; +86 15280380968
Email [email protected]; [email protected]
Background: Activation of vascular adventitial fibroblasts (VAFs) upon vascular injury contributes greatly to the medial vascular smooth muscle cells (VSMCs) proliferation, migration and the subsequent neointima formation. A number of factors including fibroblast growth factors (FGFs) have been shown to control VSMC growth, proliferation and phenotypic switching, suggesting that they may function as paracrine signals for VAFs to modulate VSMCs functions. However, little is known about the signaling molecule(s) and its mechanism of action. This study is set to identify which and how FGF family members are involved in VAFs mediated vascular remodeling.
Methods: We used qPCR, Western blot and Immunohistochemistry to observe the spatiotemporal expression of FGF10 and FGFR2 in injured vascular tissue. The proliferation and migration assays of VSMCs were performed in a co-culture system. The activation of signaling pathway was detected by Western blot, immunohistochemistry and immunofluorescence. Hematoxylin-eosin and immunofluorescence were used to assess the effects of exogenous FGF10 and siFGF10 on the neointima formation.
Results: The expression of FGF10 and FGFR2 were increased from day 3 through day 14 post injury. FGF10 was significantly upregulated in adventitia, and FGFR2 was detected in both media and neointima after injury. In vitro, FGF10 was most prominently expressed in VAFs and FGFR2 was significantly expressed in VSMCs. Both were regulated by PDGF. Co-culture of VAFs and VSMCs in vitro showed that VAF-derived FGF10 promoted the proliferation and migration of VSMCs. PDGF could synergistically enhance the process. VAF-derived FGF10 can significantly activate the FGFR2 in VSMCs and furthermore significantly activate the downstream MAPK/PI3K-AKT signaling pathways. Delivery of exogenous FGF10 potentiated the neointima formation, while siFGF10 attenuated the neointima formation.
Conclusion: VAFs-derived FGF10 promoted the proliferation and migration of VSMCs and neointima formation, and FGF10-FGFR2 signaling triggered the activation of MAPK/PI3K-AKT pathways in VSMCs and PDGF synergistically amplified FGF10 signaling.
Keywords: vascular adventitial fibroblasts, vascular smooth muscle cells, FGF10, proliferation, migration, neointima formation
Introduction
Percutaneous coronary intervention (PCI) is the most effective procedure for immediate treatment of symptomatic coronary disease.1 During the procedure, the vascular endothelial cells (VECs) are denuded and VSMCs of the medial layer are exposed to various circulatory stimulants, including platelet-derived growth factor (PDGF), thrombin, angiotensin-II and a variety of growth factors, and get activated. Subsequently, activated VSMCs undergo a phenotypic change to myofibroblast-like VSMCs and secret matrix metalloproteinases to remodel the extracellular matrix (ECM) and vascular basement membrane and then migrate to neointima, which finally cause restenosis.2,3 So far, it has been widely acknowledged that VSMCs play crucial roles in the neointima formation.3–8
However, other than VSMCs and VECs, the role of the remaining cell types in the vascular wall, especially the vascular adventitial fibroblasts (VAFs), in the neointima formation gained attention in recent years.9–11 A series of studies have shown that after vascular injury, similar to VSMCs, VAFs are also activated and undergo a phenotypic change into myofibroblasts to secrete multiple cytokines and growth factors, like IL6, CXCL1, OPN, TGF-β1, MCP-1, VEGF and reactive oxygen species (ROS). Some of them have been shown to promote proliferation of VSMCs in the medial layer.11–15 However, the key molecule mediating intercellular signaling transduction from VAFs to VSMCs is still unknown.
FGFs comprise a family of 22 distinct ligand proteins with pleiotropic signaling functions in organogenesis, development and homeostasis. Apart from four intracellular non-secretory members, the remaining 18 (FGF1~10 and FGF16~23) consist of four paracrine subfamilies (15 members; Figure 1) and one endocrine subfamily of three members.16 FGF10 belongs to the FGF7 paracrine subfamily and contains 215 amino acids. Previous studies showed that FGF10 plays a crucial role in the development of lungs, kidneys, salivary glands, adipose tissue and heart.17 However, as to its function in normal artery and in pathological vascular remodeling, there has been no report. Although it has been reported that FGF10 could promote VSMCs proliferation, it was an in vitro study.18
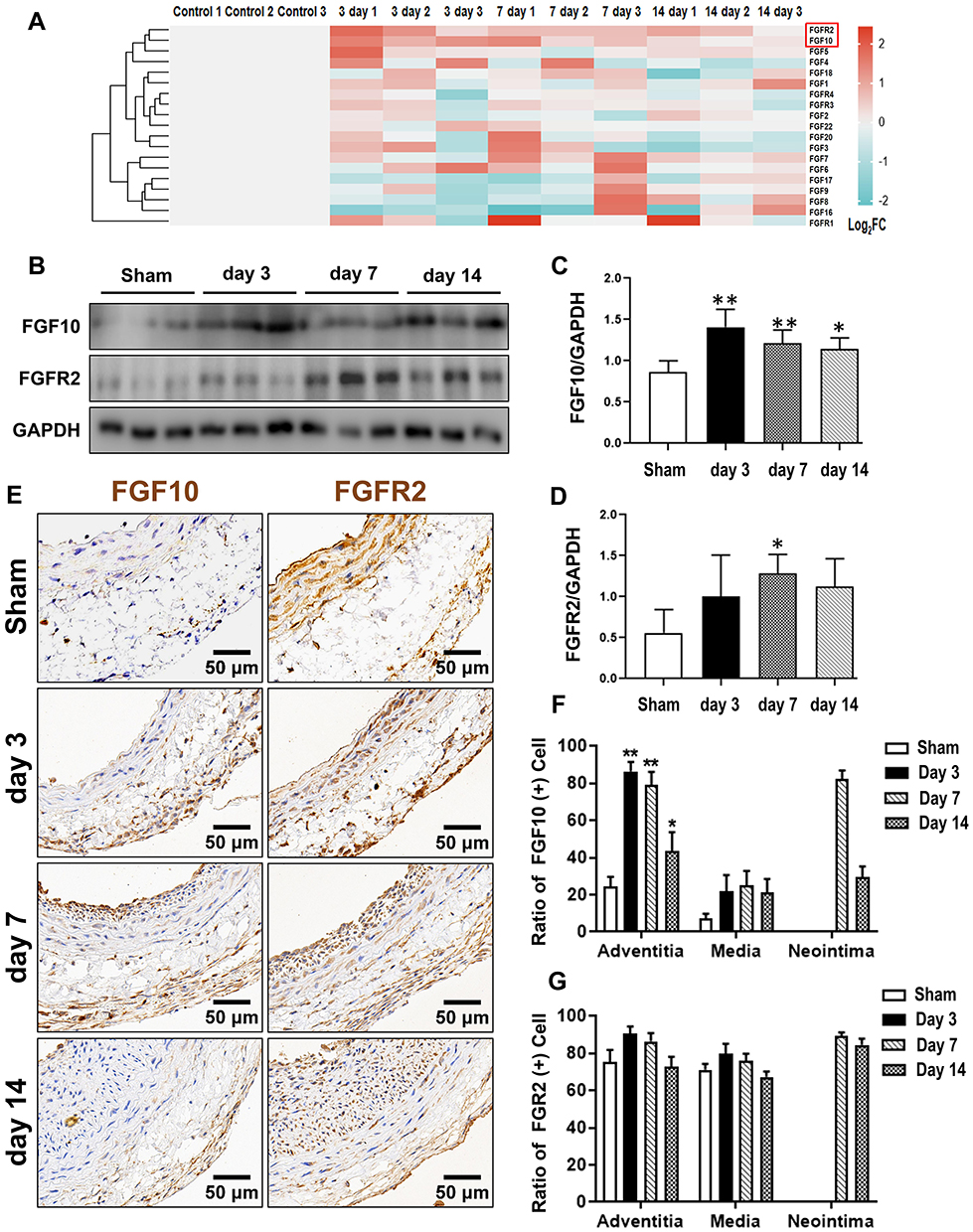 |
Figure 1 Secretory FGFs and their FGFRs expressions in balloon-injured rat left carotid artery. The balloon injuries were performed in rat LCA. The whole LCA were harvested at times as indicated post injury for qPCR (A), Western blotting (B) and IHC (E). (A) The mRNA fold change of 15 FGFs and 4 FGFRs were organized into a heat-map. Red box indexes the FGF10 and FGFR2. (B–D) Representative Western blotting (B) images and quantitative analyses of FGF10 and FGFR2 protein expression in the whole LCA (C and D) were shown. (E–G) Representative IHC images (E) and summaries of FGF10 positive cells and FGFR2 stained cells (E and G) were illustrated. Bar=50μm. n=6. *p<0.05 and **p<0.01 vs Ctl. |
Fibroblast growth factors (FGFs) ligands exert downstream biological effects by binding and activating their specific receptor FGFR.19 FGFR family contains four gene types (FGFR1-FGFR4). The structure of FGFRs is a typical one-way transmembrane protein, consisting of a cytoplasmic tyrosine kinase domain and an extracellular domain which are composed of three immunoglobulin-like repeats.16 Like other members of the FGF paracrine subfamilies, the functions of FGF10 are mediated principally by binding with its specific receptor, FGFR2, which is also known as keratinocyte growth factor receptor. FGFR2 is expressed in VSMCs and can be activated by four known ligands (FGF1, FGF3, FGF7 and FGF10) that are predominantly synthesized by mesenchymal cells. Binding of FGF10 to FGFR2 initiates autophosphorylation of its cytoplasmic tyrosine kinase domain and activates the downstream signaling.17,18
Methods and Materials
Antibodies and Reagents
The following seven antibodies were purchased from Cell Signaling: cyclin-D1 (55506), PCNA (13110), P-JNK (9255), PI3K (4249), p-PI3K (4228), P-AKT (4060) and AKT (4691). Antibodies purchased from Abclonal (USA) were FGF10 (A1201), FGFR2 (A12436), ERK1/2 (AP4782), p-ERK1/2 (AP0472), T-JNK (AP0631), P38 (AP11401) and p-P38 (AP0526). Antibodies purchased from Servicebio (China) were CD31 (GB11063), vimentin (GB11192), MMP-2 (GB11130), MMP-9 (GB11132) and GAPDH (GB11002). p-FGFR2 (Tyr653+Tyr654; A1201) antibodies were purchased from Affinity (USA). Normal rabbit IgG, HRP-conjugated goat anti-mouse and goat anti-rabbit secondary antibodies were purchased from Proteintech (Chicago, USA). The α-SMA antibody (F1804) and PDGF-BB (P4056) were from Sigma-Aldrich.
Balloon Injury of Rat Left Carotid Artery
All procedures were conducted in compliance with both the Guide for the Animal Care and Use Committee of Xinhua Hospital at SJTUSM and the guidance for the care and use of experimental animals published by NIH (the eighth Edition, NRC 2011). The experiments were approved by the Animal Care and Use Committee of Xinhua Hospital at SJTUSM. Male Sprague-Dawley rats in 10~12 weeks-old (300~350g) were anesthetized and heparinized by intraperitoneal injections of 2% Phenobarbital (40 mg/kg) and heparin (100~200 U/kg). After the bifurcation of LCA was surgically exposed, a 2-F Fogarty balloon catheter (diameter of balloon 2 mm, length 15 mm, Medtronic Company, USA) was inserted into LCA up to the aortic arch bifurcation from the external carotid artery. A pressure about 0.5–0.7 atm was given to inflate balloon and the same segment of common carotid artery was injured three times. After ligation of the external carotid artery, blood flow between the common and the internal carotid artery was confirmed to be recovered. For some of the rats, the injured carotids were surrounded by 300 μL of 30% Pluronic F-127 gel (Sigma) with or without 200ug/mL FGF10 or 10μM siFGF10 before closure of skin. All rats were housed in the Xinhua Hospital animal facilities. Four, seven, and fourteen days later, the injured carotids were excised for analyses.
Quantitative Real-Time PCR
Total RNA, extracted from the whole injured rat LCA using TRIzol (Takara, Cat# RR036A), was reversely transcribed into cDNA using the Prime-ScriptTMRT reagent kit (Takara, RR036A). qRT-PCR was performed using specific primers (Supplementary Table 1), Premix Ex Taq™ (Probe qPCR) and normalized to GAPDH expression. ABI 7500 detector (Applied Biosystems) with standard PCR conditions (95°C for 30s, followed by 40 cycles of 95°C for 5s and 60°C for 34s) was used to run the samples.
Immunohistochemistry (IHC) and Cellular Immunofluorescence Staining
Injured arteries were harvested on day 3, 7 and 14 after the procedure and the contralateral uninjured carotid arteries were also harvested as controls. Common carotid arteries were harvested after rats were euthanized with sodium pentobarbital (70mg/kg) in combination with isoflurane inhalation. Then, we fixed them in 4% paraformaldehyde for 24 h and embedded them in paraffin thereafter. Briefly, 4µm-thick slides were heated to 100°C, incubated with 5% Donkey serum and then incubated with primary antibodies. After rinsing, the secondary antibody was incubated. Visualization was accomplished using DAB (Yeasen, China).
For cellular immunofluorescence analysis, cells cultured on coverslips were fixed, permeabilized, incubated with primary antibodies and then incubated with FITC-conjugated or Rhodamine-conjugated secondary antibodies (Servicebio, China) before being counterstained with 4′6-diamidino-2-phenylindole (DAPI) (Beyotime). Both IHC and immunofluorescence were imaged under Olympus BX51 microscope (Olympus, Japan).
Hematoxylin-Eosin (HE) Staining and Immunofluorescence Staining of Tissue
Injured arteries with or without FGF10 and siFGF10 were harvested on day 14 after the procedure and the contralateral uninjured carotid arteries were also harvested as Sham group. Harvested vascular tissues were immersed in 4% paraformaldehyde overnight and then embedded in paraffin. Paraffin section (5μm), after being dewaxed and rehydrated, were used for immunofluorescence and HE staining. Primary antibodies used were anti-rat FGF10 antibody (Abconal, A1201), anti-rat PCNA antibody (Servicebio,13110) and anti-rat MMP9 antibody (Servicebio, GB12132). Secondary antibodies used were goat anti-rabbit antibody Alexa 647 (Beyotime), goat anti-rabbit antibody Alexa 488 (Beyotime) and goat anti-mouse antibody Alexa-488 (Beyotime). DAPI (Beyotime) was used to counter stain the nuclei. HE staining was performed with a HE Staining kit (Beyotime) according to the manufacturer’s instructions.
Isolation, Culture and Identification of Cells
VAFs, VSMCs and vascular endothelial cells (VECs) were isolated from thoracic aortas of 6~8-week-old male SD rats (150–200g) purchased from JSJ company (Shanghai, China). Briefly, VECs were first isolated by collagenase-II digestion method as previously described.20 Subsequently, the medial layer was scraped off and left the remaining adventitia. The medial layer was sliced into 1 mm2 piece explants and seeded to the culture dish containing DMEM with 20% fetal bovine serum (FBS; Yeasen, China). VSMCs were obtained in 7 days and were further sub-cultured. The adventitia were digested by 0.1% collagenase-II (Sigma, USA) in 37°C for 2h, filtered, centrifuged and re-suspended in the medium for culture in DMEM with 10% FBS at 5% CO2 and 37°C. Three types of cells were characterized by immunofluorescence staining using antibodies specific against CD31, α-SMA and vimentin.21 Cells in passage 2~5 were used for experiments.
RNA Interference
Small interfering RNA (siRNA) for FGF10 and scrambled siRNA were synthesized by Biotend (China). For transfection, VAFs were cultured to 60~80% confluence. 50nM siRNA was transfected into VAFs by using Lipofectamine RNAiMAX reagent (Life Technologies) according to the manufacturer’s protocol. The efficiencies of knocking down were evaluated by Western blotting.
Co-Culture System
Co-culture of VAFs and VSMCs was performed in 6-well plates with insets (3412, pore size 0.4 μm; Corning Costar, USA). VAFs were seeded in the upper wells and cultured in DMEM supplemented with 10% FBS for 24 h and then incubated with scramble siRNA or siRNA against FGF10, as mentioned above. After 48 h, medium was changed to fresh DMEM supplemented with or without 20ng/mL PDGF for 8h. VSMCs were seeded in the lower wells and cultured in DMEM supplemented with 10% FBS for 24 h. Before co-cultured with VAFs, VSMCs were cultured in DMEM supplemented with or without 20ng/mL PDGF for 8h, and then cultures in serum-free medium for 12h. VSMCs co-cultured with VAFs were used for cell cycle analysis, scratch-wound assay, cellular immunofluorescence staining and Western blotting.
Assessment of FGF10 Concentration
The concentration of FGF10 in co-culture system was measured using a commercial ELISA kit (Lengton Bioscience, China). The media were added to each anti-FGF10-coated well and then incubated with anti-Rat FGF10 primary antibody. After washing three times, streptavidin-enzyme conjugates were added into each well, followed by quantification on a spectrophotometer at 450nm.
Cell Proliferation Assays
Cell proliferation was assessed by CCK-8 assay according to the manufacturer’s protocol (Yeasen, China). For cell cycle analysis, VSMCs were harvested from the co-culture system by trypsinization, followed by washed twice with PBS and fixed by 70% ethanol in 4°C for 2h. The cells were then centrifuged (800 rpm, 3 min), washed once with PBS, and re-suspended in a staining buffer solution containing propidium iodide (PI) and RNAase for 30min in the dark at 37°C. The minimal cell numbers counted per specimen is 20,000. Cell cycles were determined using a flow cytometer (Beckman, USA) and analyzed using the software CytExpert 2.1.
Migration Assay
Transwell assays were performed as described previously.22 Scratch-wound assays were performed in the co-culture system. VSMCs were seeded in the lower layer and VAFs were seeded in the upper layer of the system. VAFs and VSMCs were co-cultured after the VSMCs were scratched.
Western Blot
Cells were washed with cold PBS triplicate and homogenized in RIPA buffer (Biotime, China) mixed with Phenylmethylsulfonyl fluoride (PMSF, Biotime, China) and proteinase inhibitors (Sigma) before being left on ice for 30 min. The lysate was centrifuged (4°C, 12000rpm, 15min) to eliminate debris. A BCA Protein Assay Kit (Yeasen, China) was used to measure protein concentration of the lysate. Samples solubilized in a loading buffer were boiled for 10 min, separated by SDS-PAGE, transferred to the PVDF membrane (Sigma, USA), blocked by 5% non-fat milk in TBST and incubated with primary antibodies in 4°C overnight. The membranes were incubated with secondary antibodies at room temperature for 1h and washed with TBST 3 times before being applied to Gel Imaging System (Tanon 5200) for visualization and quantified by densitometry software (Image J).
Statistical Analysis
Data were represented as mean ± SD or percentage of at least three independent experiments. Two-tailed Student’s tests were used for two-group comparisons, and one-way ANOVA respectively tests were used for multiple group comparisons. A value of p<0.05 was considered statistically significant. Data were analyzed using GraphPad prism 8.0 software.
Results
Spatiotemporal Expression of FGF10 and FGFR2 in Injured Vascular Tissue
To explore the differential expression patterns of each FGF member and their cognate receptors, we extracted the total RNA from the whole injured rat LCA, and determined the expression of 15 secretory FGF family members (1–10, 16~18, 20, 22) and their receptors (FGFR1~4) by qPT-PCR. As shown in Figure 1A, FGF10 of the 15 FGFs and FGFR2 of the 4 FGFRs have the most typical and similar changes in time courses: an initial high peak-phase, followed by a gradually declining plateau-phase. In detail, FGF10 was significantly upregulated, with peak on day 3. Then, the upregulation was slightly attenuated on day 7 and further attenuated on day 14. FGFR2 was also significantly upregulated, with peak at third day. Then, the upregulation was slightly attenuated on both day 7 and day 14.
Therefore, we next verified the upregulated FGF10 and FGFR2 in the whole LCA on protein levels by WB (Figure 1B). Similar to cDNA levels, FGF10’s upregulation peaked on day 3, gradually declined on day 7 and further on day 14 (Figure 1C). However, on day 14 FGF10 still remained in high levels. FGFR2’s expression increased during day 3~7 with peak on day 7 and declined slightly on day 14 (Figure 1C and D).
The spatiotemporal expression patterns of both FGF10 and FGFR2 in injured LCA were evaluated using IHC (Figure 1E). In the adventitia, FGF10 was significantly upregulated on day 3 and 7, 4 days earlier than its remarkable increase in the neointima on day 7 (Figure 1F). FGFR2 maintains similar expression patterns in both adventitia and media. In the neointima, it was significantly upregulated along with its formation on day 7, and kept a high level on day 14 (Figure 1G).
The Expression of FgF10 and FGFR2 Were Regulated by PDGF in vitro
The LCA contains three major vascular cell types, naming VECs, VSMCs and VAFs. To determine which cell type is responsible for the observed increase in FGF10 and FGFR2 expression shown in Figure 1, we isolated and cultured three cell types from rat thoracic aorta (Figure 2A). The characteristics of each isolated cell population were confirmed by immunofluorescence staining with antibodies against VECs marker CD31, VSMCs marker α-SMA and mesenchymal origin cell marker vimentin, respectively (Figure 2B).
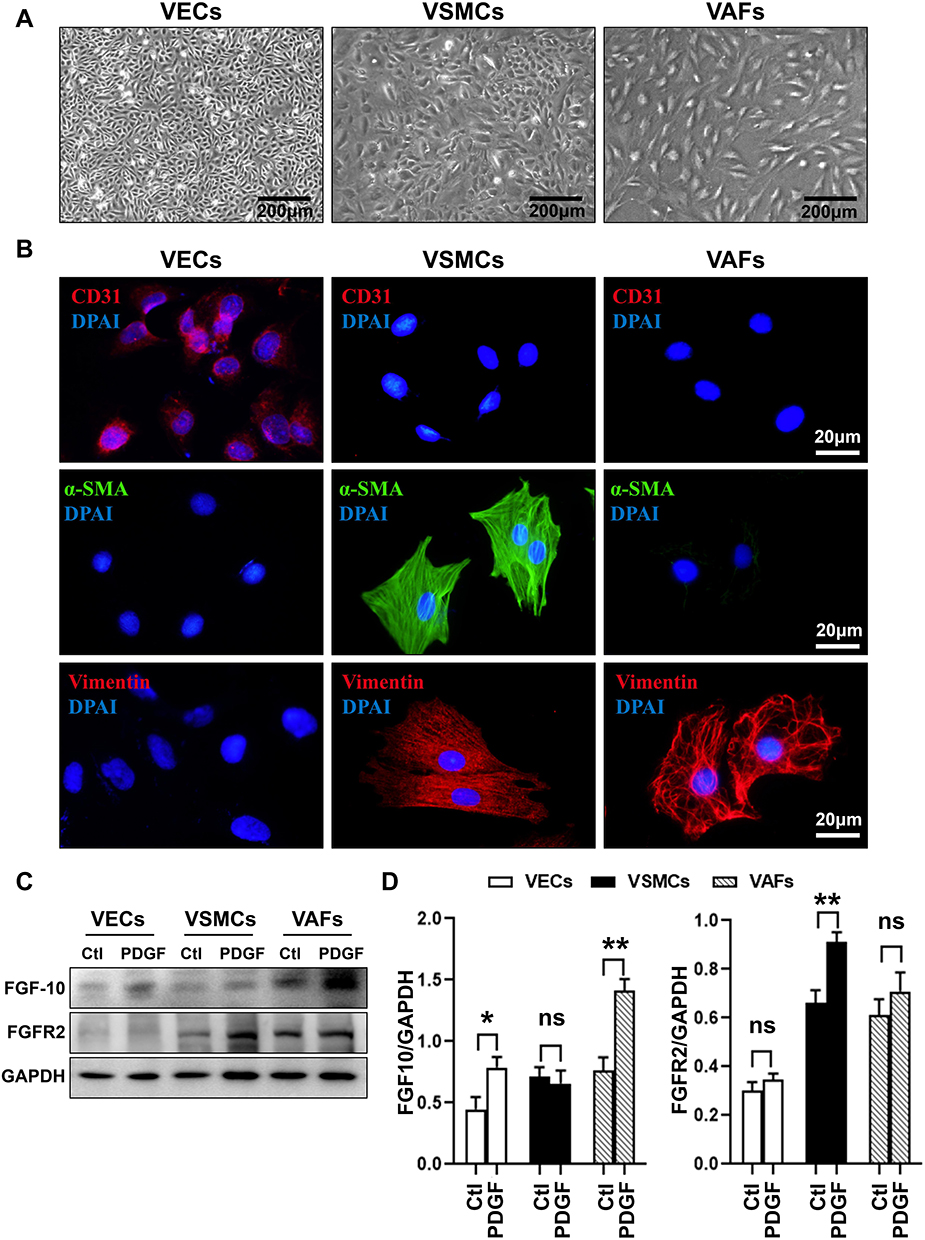 |
Figure 2 PDGF differentially regulates expression of FGF10 and FGFR2 in VECs, VSMCs and VAFs. VECs, VSMCs and VAFs were isolated from rat aorta and cultured. (A) Morphological features of VECs, VSMCs and VAFs in primary cultures. Bar=200μm. (B) Immunofluorescence staining of CD31, α-SMA and Vimentin in three types of cells. Nuclei were stained with DAPI. Bar=20μm. (C and D) Representative WB images (C) and summaries (D) of FGF10 and FGFR2 protein expressions in three types of cells with or with put treatment of PDGF (20ng/mL). n=3. *p<0.05 and **p<0.01. Abbreviation: ns, no significance. |
The expression levels of FGF-10 and FGFR2 in VEC, VSMC and VAFs were determined by Western blot analysis using cell lysates from each isolated culture cell. As a result, at basal level, FGF10 expression levels were highest in VAFs while the basal expression levels of FGFR2 were higher in both VAFs and VSMCs as compared with VECs (Figure 2C and D).
Previous studies have shown that PDGF is a crucial stimulator to activate VAFs and to induce the neointima formation.8,12 We tested whether PDGF treatment increased FGF10 and FGFR2 expression in three types of cells. As shown in Figure 2C and D, the FGF10 expressions were remarkably upregulated by PDGF treatment in VAFs and VECs, but not in VSMCs. FGFR2 expression, on the other hand, was upregulated in VSMCs, but not in VECs and VAFs after PDGF treatment. These results confirmed the expression of FGF10 and FGFR2 in both VAFs and VSMCs. FGF10 was expressed higher in VAFs and lower in VSMCs, while FGFR2 was expressed higher in VSMCs; PDGF can stimulate FGF10 expression in VAFs while FGFR2 expression in VSMCs can be stimulated by PDGF.
VAFs-Derived FGF10 Cooperates with PDGF to Promote VSMCs Proliferation
Based on the above results, we hypothesized that VAFs-derived FGF10 is a major factor and acts in a paracrine way on VSMCs to promote VSMCs proliferation, migration, and the neointima formation upon vascular injury.
To verify this hypothesis and to determine the effect of VAFs-secreted FGF10 on VSMCs proliferation and migration, we first used FGF10-specific siRNA to knock-down FGF10 in cultured VAFs. Compared with scramble, FGF10 expression was reduced 60%~70% when VAFs were transfected with indicated siFGF10s (Figure 3A). Among those siFGF10s, siFGF10-2 was the most effective.
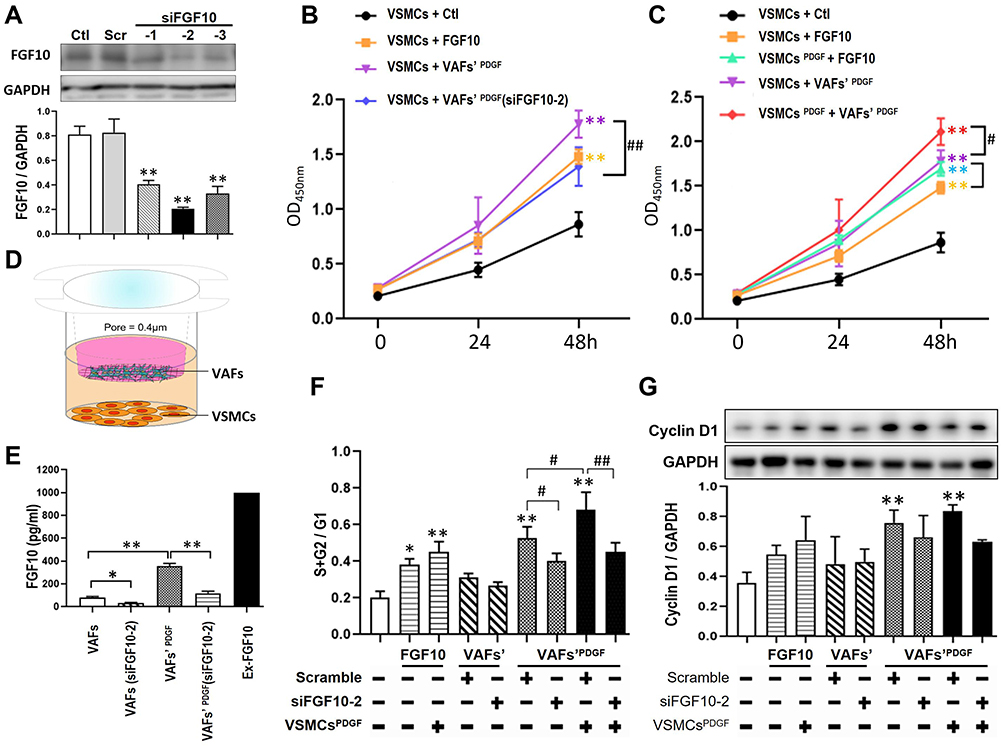 |
Figure 3 Both exogenous and VAFs-derived FGF10 promote the proliferation and cell cycle of VSMCs. (A) In cultured VAFs, knocking-down efficiency of FGF10 by siRNA was determined by WB. (B and C) Relative VSMCs numbers under different experimental conditions were analyzed by CCK-8 proliferation assay. (D) The schematic illustration of VAFs-VSMCs co-culture system. The co-cultured VSMCs were used for flow cytometry and Western blotting test. (E) In this co-cultured system, the concentrations of VAF-derived FGF10 in different experimental conditions as indicated were measured by Elisa. (F) Cell cycle progression in VSMCs under different experimental conditions were evaluated by using propidium iodide staining (flow cytometry). (G) Cyclin D1 expressions in co-cultured VSMCs were analyzed by WB and its quantitative analysis. n=3. *p<0.05, **p<0.01 compared with Ctl. #p<0.05, ##p<0.01 between two groups as indicated. |
Next, we tested the effects of VAF-derived FGF10 on VSMC proliferation by CCK-8 cell proliferation assay. As shown in Figure 3B, compared with VSMCs cultured in serum-free media [VSMCs+Ctl], addition of exogenous FGF10 [1ng/mL; VSMCs+FGF10] to the culture media significantly enhanced VSMCs’ proliferation. To explore the effect of VAFs-derived FGF10 on VSMCs’ proliferation, we used PDGF to stimulate VAFs for 8h to increase FGF10 secretion with or without siFGF10 transfection. Following the washout of PDGF in the media and further cultured for 16h, we collected the conditional medium to culture VSMCs. As compared with VSMCs [VSMCs + VAFs’ PDGF], the proliferation of VSMCs with VAFsPDGF(siFGF10-2) conditional media [VSMCs + VAFs’ PDGF (siFGF10)] was significantly reduced. These results indicated that both exogenous and endogenous FGF10 play a role in regulating VSMCs proliferation.
As shown in Figures 1 and 2, FGF-10 was mainly secreted from VAFs whereas FGFR2 was highly expressed in VSMCs and was further up-regulated by PDGF only in VSMCs. We next examined whether PDGF could amplify VAFs-derived FGF10’s effects on VSMC proliferation by CCK-8 assaying (Figure 3C). As compared with VSMCs stimulated by FGF10 [VSMCs + FGF10], the VSMCs with pretreatment of PDGF for 8 h [VSMCs PDGF + FGF10] (followed by medium change for removal of remaining PDGF and further culture for 16 h) illustrated indifferent cell proliferation. Similarly, as compared to VSMCs stimulated by conditional medium [VSMCs + VAFs’ PDGF], the VSMCs pretreated with PDGF [VSMCs PDGF + VAFs’ PDGF] also show an enhanced proliferation. These results indicated that PDGF pretreatment to VSMCs could amplify both exogenous and endogenous FGF10-mediated signaling on promoting VSMCs proliferation.
We used a co-culture system to determine the effects of the VAFs-derived FGF10 on cell cycles of VSMCs. In this system, VAFs were seeded in the upper chamber (pore=0.4um) and VSMCs were seeded in the lower chamber (Figure 3D). The concentration of VAFs-derived FGF10 in co-culture system was assayed by Elisa. We first verified the inhibitory efficiency of siFGF10-2 in this co-culture system. As shown in Figure 3E, siFGF10 efficiently blocked FGF10 secretion from VAFs. Consistent with the above findings, we observed that PDGF (20ng/mL) treatment significantly upregulated FGF10 secretion. siFGF10 reduced 67% of the PDGF-induced FGF10 secretion.
As shown in Figure 3F and Supplementary Figure 1A, we first assayed the impact of exogenous FGF10 on cell cycles. Compared with VSMCs [VSMCs+Ctl], exogenous FGF10 [1ng/mL; VSMCs+FGF10] significantly increased S+G2 phase. When compared with VSMCs [VSMCs + FGF10], the VSMCs pretreated with PDGF for 8h, cultured in serum-free medium for 12h and followed by FGF10 treatment [VSMCPDGF+FGF10] further increased the S+G2 fractions. These results indicated that exogenous FGF10 could accelerate cell cycles and PDGF could amplify this effect.
Next we performed co-culture experiments for assaying cell cycles (Figure 3F and Supplementary Figure 1A). Before co-culture, VAFs had been stimulated with or without PDGF for 8h and VSMCs had been starved for 12h and then had been pretreated with PDGF for 8h. After co-culture for 24h, the VSMCs were harvested for cell cycle analysis by flow cytometry and for Cyclin D1 expression by Western blotting. As shown in Figure 3F and Supplementary Figure 1B, compared with VSMCs [VSMCs+VAFs], the cells [VSMCs+VAFs’ (si-FGF10)] showed unchanged cell cycle progression. Compared to VSMCs co-cultured with PDGF pretreated VAFs [VSMCs+VAFsPDGF], the cells [VSMCs+VAFsPDGF(si-FGF10)] demonstrated an attenuated cell cycle progression. Similarly, as compared with VSMCs [VSMCsPDGF+VAFsPDGF], the cells [VSMCsPDGF+VAFsPDGF(si-FGF10)] also showed a slowdown in cell cycle progression. Consistently, cyclin D1 expression levels in the co-cultured VSMCs altered in similar tendencies (Figure 3G). These results further supported that endogenous FGF10 increased VSMCs proliferation and PDGF synergistically potentiated FGF10’s effect.
VAFs-Derived FGF10 Cooperates with PDGF to Promote VSMCs Migration
In order to investigate the effect of VAFs-derived FGF10 on VSMC migration, we first used transwell chamber (pore=8μm) to perform experiments. Compared to VSMCs [VSMCs-Ctl], both exogenous FGF10 [VSMCs+FGF10] and exogenous FGF10 with VSMCs pretreated by PDGF [VSMCPDGF+FGF10] significantly increased VSMCs migration. The effects were mildly amplified in cells [VSMCPDGF+FGF10] (Figure 4A and D).
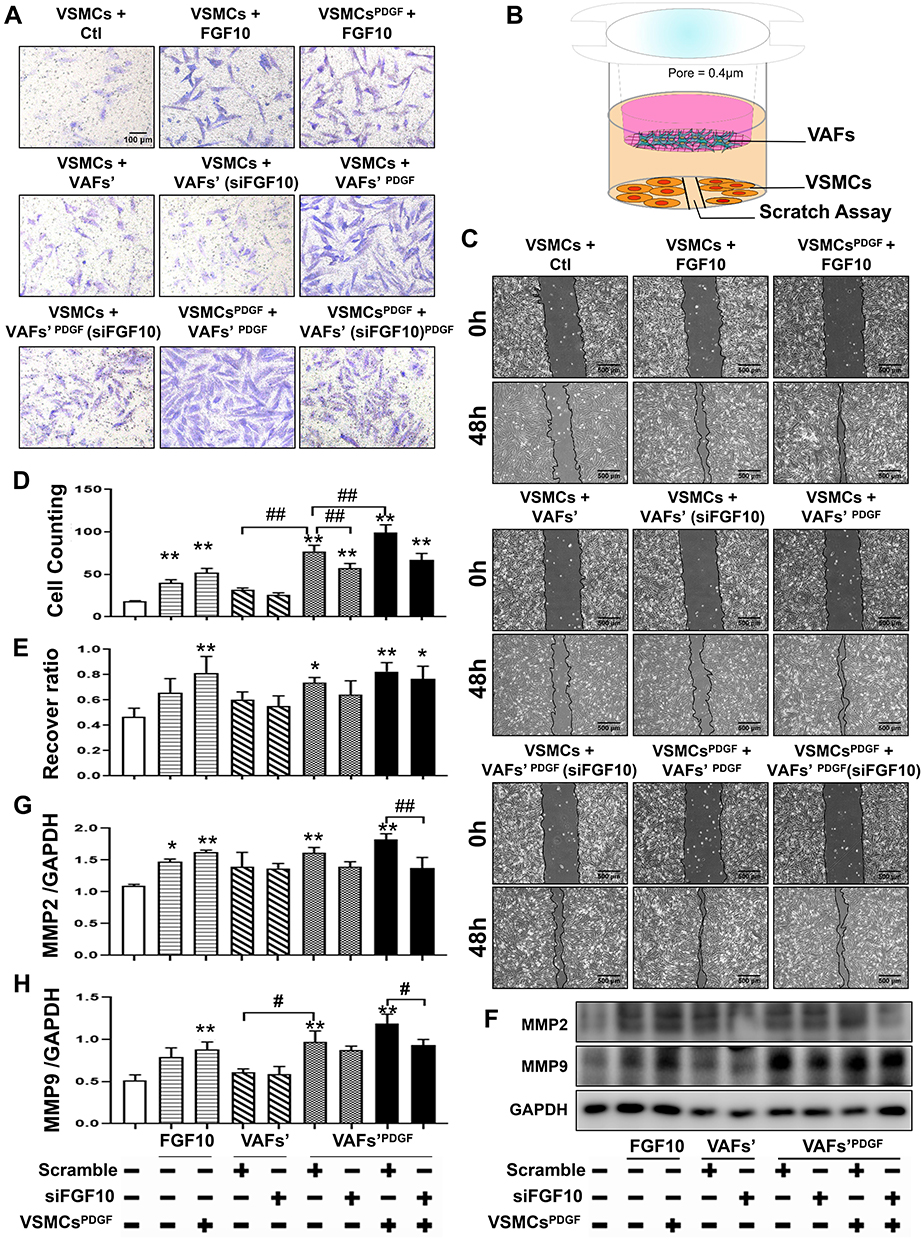 |
Figure 4 Both exogenous FGF10 and VAFs-derived FGF10 promote VSMCs migration. (A and C) Evaluation of VSMCs migration by Transwell assay (A) and scratching assay (C) and quantitative analyses (D and E). (B) Schematic illustration of VAFs-VSMCs co-cultured system in which co-cultured VSMCs were used for scratching assay and WB analyses of MMP2 and MMP9. (F–H) Representative WB images (F) and their quantitative analyses (G and H) of MMP2 and MMP9 protein expressions in co-cultured VSMCs under different conditions as indicated. n=3. *p<0.05, **p<0.01 compared with Ctl. #p<0.05, ##p<0.01 between two groups as indicated. |
To determine the effect of endogenous VAFs-derived FGF10 on VSMC cell migration, we collected conditional media from PDGF pretreated VAFs cultured with or without FGF10 knockout for the transwell experiment.
Next, the conditional media were obtained from VAF culture in serum-free medium for 12h or had been pretreated with PDGF for 8h. VSMCs, serum-starved for 12h, were seeded in the upper layer of the chamber. The VSMCs were then treated with or without recombinant FGF10 or the conditional medium and were further incubated for 24h, followed by counting the number of cells that migrated.
As shown in Figure 4A and D, as compared with VSMCs [VSMCs+VAFs], the cells [VSMCs+VAFs’ (si-FGF10)] illustrated a mildly decreased migration. While as compared with VSMCs [VSMCs+VAFsPDGF], the cells [VSMCs+VAFsPDGF(si-FGF10)] demonstrated an attenuated cell migration. Compared with VSMCs [VSMCsPDGF+VAFsPDGF], the cells [VSMCsPDGF+VAFsPDGF(si-FGF10)] also showed a decreased cell migration.
The FGF10 effect on VSMCs migration was also assessed by using scratching assay in co-culture system (Figure 4B). As shown in Figure 4C and E, compared with control VSMCs, co-culturing with PDGF-pretreated VAFs significantly enhanced the wound healing. Pretreatment with PDGF in VSMCs facilitates the healing process, whereas siFGF10 knockdown of FGF10 in VAFs reduces the healing speed.
MMP2 (Figure 4F and G) and MMP9 (Figure 4F and H) expressions in the co-cultured VSMCs demonstrated similar changes. These results indicated that endogenous FGF10 promoted VSMCs migration and PDGF could intensify FGF10’s effect.
VAFs-Derived FGF10 Cooperates with PDGF to Activate FGFR2 in VSMCs
Previous studies have shown that FGF10 binding increased FGFR2 phosphorylation leading to activate its downstream cascades to exert its biological effects.17,19 FGFR2 phosphorylation is a key indicator for FGF-FGFR signaling activity. We used IHC to determine if there were changes in spatiotemporal FGFR2 phosphorylation in the LCA upon injury (Figure 5A). In the medial layer, the phosphor-FGFR2 protein was significantly increased on day 3 after injury, peaked on day 7 and declined on day 14. Even on day 14, the percentage of phosphor-FGFR2+ cells was still higher than that in Sham group. In the neointima, whose formation started to be obvious on day 7, showed a high phosphorylation level of FGFR2 that nearly 70% of cells were positive for the staining. The percentage was slightly decreased on day 14 after injury, but still higher than that in the medial layer (Figure 5A and C).
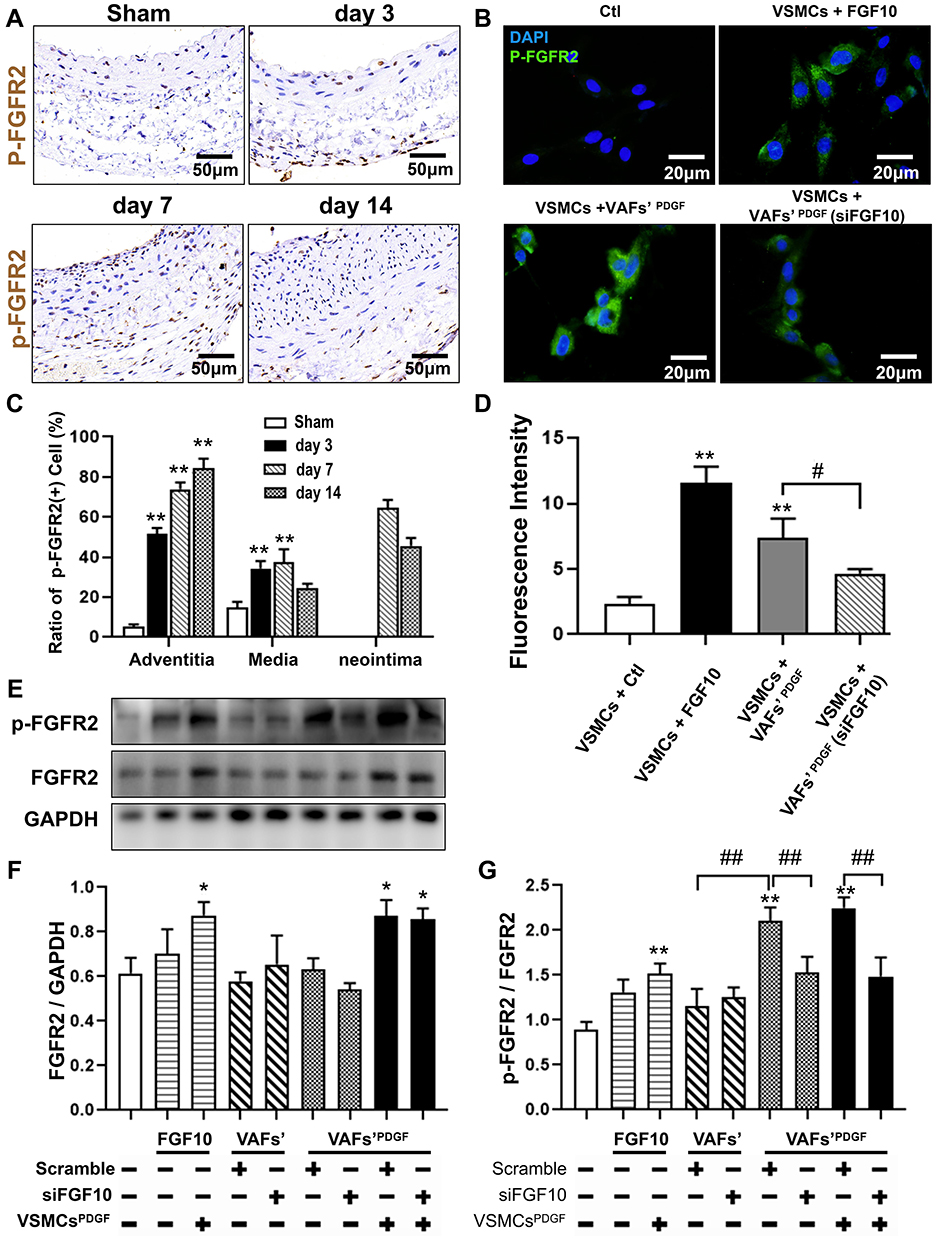 |
Figure 5 The activities of FGFR2 in VSMCs in vivo and in vitro. (A and C) Representative IHC images of P-FGFR2 staining (A) and its quantitative analysis (C) in rat LCA sections harvested at indicated times after balloon-injury. Scale bar=50μm, n=6. (B and D) Representative immunofluorescence images of P-FGFR2 staining (B) and its quantitative analysis (D) in cultured VSMCs under conditions as indicated. Scale bar=20μm, n=3. (E–G) The protein expression of P-FGFR2 in co-cultured VSMCs under conditions described in Figure 3D were analyzed by WB (E) and its quantitative analyses (F and G), n=3. *p<0.05; **p<0.01 vs Ctl. #p<0.05; ##p<0.01 between two groups as marked. |
To further explore the effect of VAFs-derived FGF10 on the activity of FGFR2 in VSMCs, we use exogenous FGF10 and the co-culture system depicted in Figure 3D to stimulate VSMCs, followed by immunofluorescence staining of p-FGFR2 in VSMCs. The results showed that exogenous FGF10 and PDGF-pretreated co-culture system can significantly increase the phosphorylation levels of FGFR2 in VSMCs, and such an increase can be significantly reduced by knocking down the expression of FGF10 in the co-cultured VAFs (Figure 5B and D).
The similar result was also verified by WB results (Figure 5E). Moreover, as compared with VSMCs [VSMCs-Ctl], both expression levels of FGFR2 in PDGF-pretreated VSMCs [VSMCsPDGF+VAFsPDGF; VSMCsPDGF+VAFsPDGF(si-FGF10)] were significantly upregulated nearly 1.5 folds but the cells [VSMCsPDGF+VAFsPDGF(si-FGF10)] showed significantly decreased levels of FGFR2’s phosphorylation.(Figure 5F and G). In summary, these data indicated that VAFs-derived FGF10 cooperates with PDGF to induce FGFR2 phosphorylation in VSMCs.
VAFs-Derived FGF10 Cooperate with PDGF to Activate the MAPK/PI3K-AKT Pathways
Previous studies have shown that mitogen activated protein kinase (MAPK) and phosphoinositide 3 kinase-protein kinase B (PI3K-AKT) signaling are two classical pathways for FGFR2-mediated proliferation and migration in cells other than VSMCs.17,23,24 To determine whether these two pathways can be activated in VSMCs by exogenous or VAFs-derived FGF10, we performed WB to analyze the protein levels of key components in MAPK and PI3K-AKT pathways and their phosphorylation status in VSMCs under the same experimental conditions of Figure 3D. As shown in Figure 6A and B, both exogenous FGF10 or FGF10 secreted from PDGF-pretreated VAFs, could significantly increase the phosphorylation levels of MEK1, ER1/2, JNK, P-38, PI3K and AKT (Figure 6A and B). These effects could be significantly decreased by knocking-down FGF10 in PDGF-pretreated VAFs. In addition, we also found that under these experimental conditions, PDGF-pretreatment of VSMCs could further amplify the phosphorylation levels of ERK1/2, JNK, PI3K and AKT (Figure 6A and B). These results indicated that VAFs-derived FGF10 regulated the activation of MAPK and PI3K pathways in VSMCs.
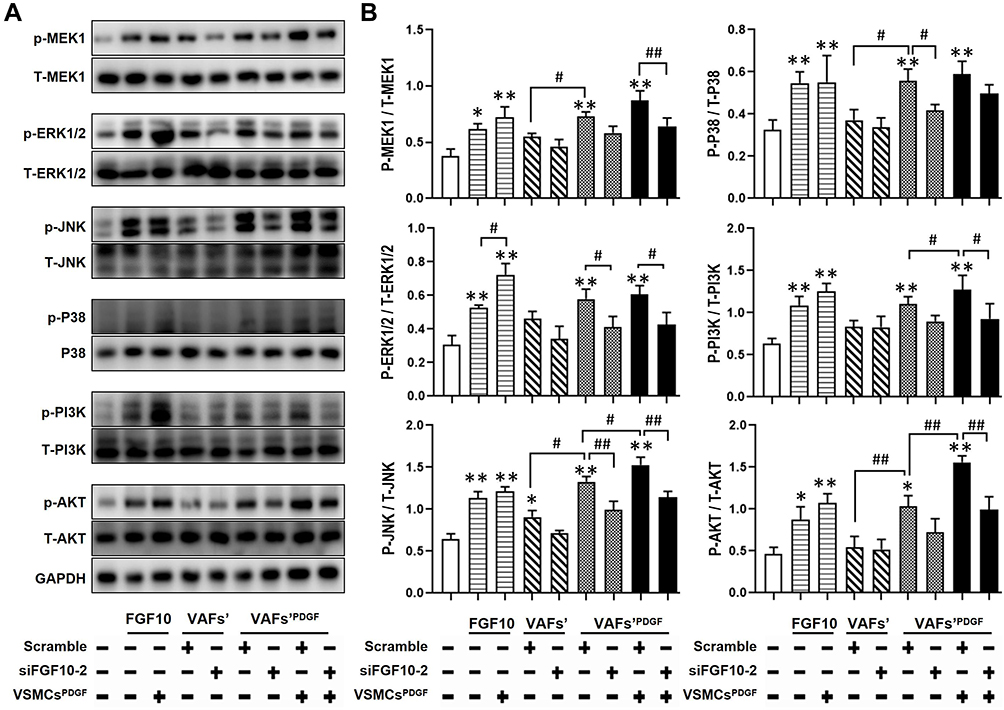 |
Figure 6 FGF10 regulates MAPK and PI3K-AKT pathways in VSMCs. MAPK and PI3K signaling molecules in co-cultured VSMCs under conditions exactly same to Figure 3D were analyzed by WB (A) and their quantitative analyses (B), n=3. *p<0.05; **p<0.01 vs Ctl. #p<0.05; ##p<0.01 between two groups as marked. |
Effects of Delivery of Exogenous FGF10 or Knocking-Down of Endogenous FGF10 on Expressions of MMP9 and PCNA and on Neointima Formation
In order to investigate the effects of FGF10 on VSMCs’ proliferation, migration and neointima formation in vivo, we performed standard balloon injury in rat LCA, with or without F127 gel-mediated delivery of FGF10 or siFGF10. As compared with Control, exogenous FGF10 promoted the expression of PCNA (Figure 7), MMP9 (Figure 8), and neointima formation upon injury (Figure 9), while siFGF10 reduced PCNA (Figure 7), MMP9 (Figure 8) expression and reduced neointima formation (Figure 9A and B) upon balloon injury. These results indicated that VAFs-mediated FGF10 plays a key role in neointima formation by regulating VSMCs proliferation and migration in vivo.
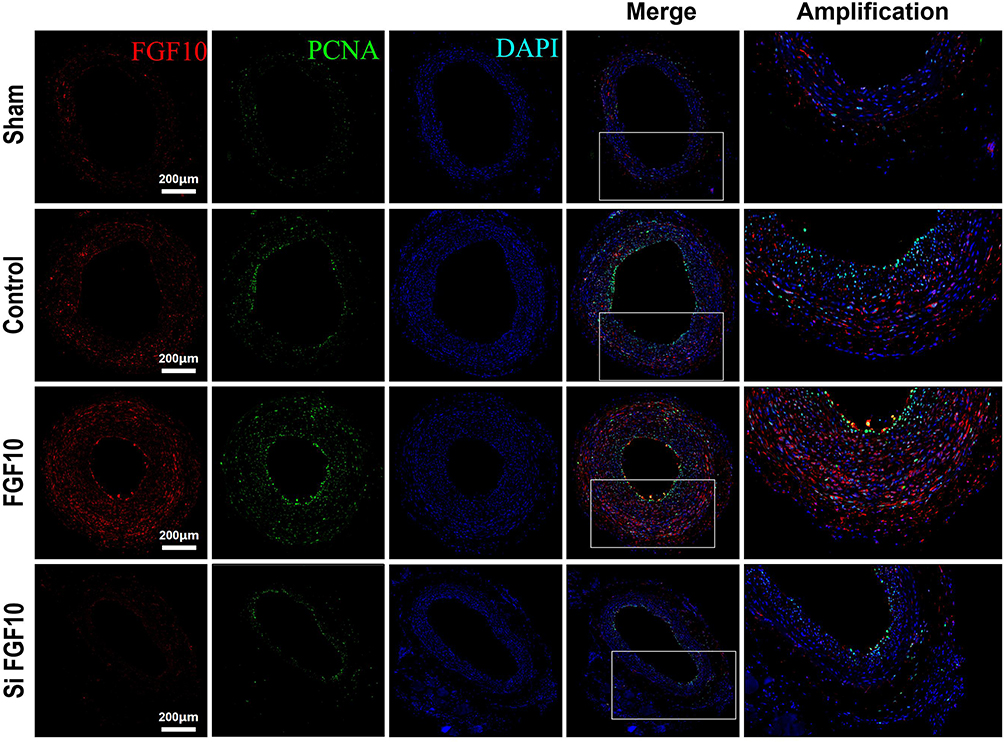 |
Figure 7 PCNA expressions were upregulated by FGF10 in vivo. The balloon injuries were performed in rat LCA with or without deliveries of exogenous FGF10 or siFGF10-2 and the whole LCA were harvested on day 14 post the procedure for staining of FGF10, PCNA and DAPI. Bar=200μm. n=5. |
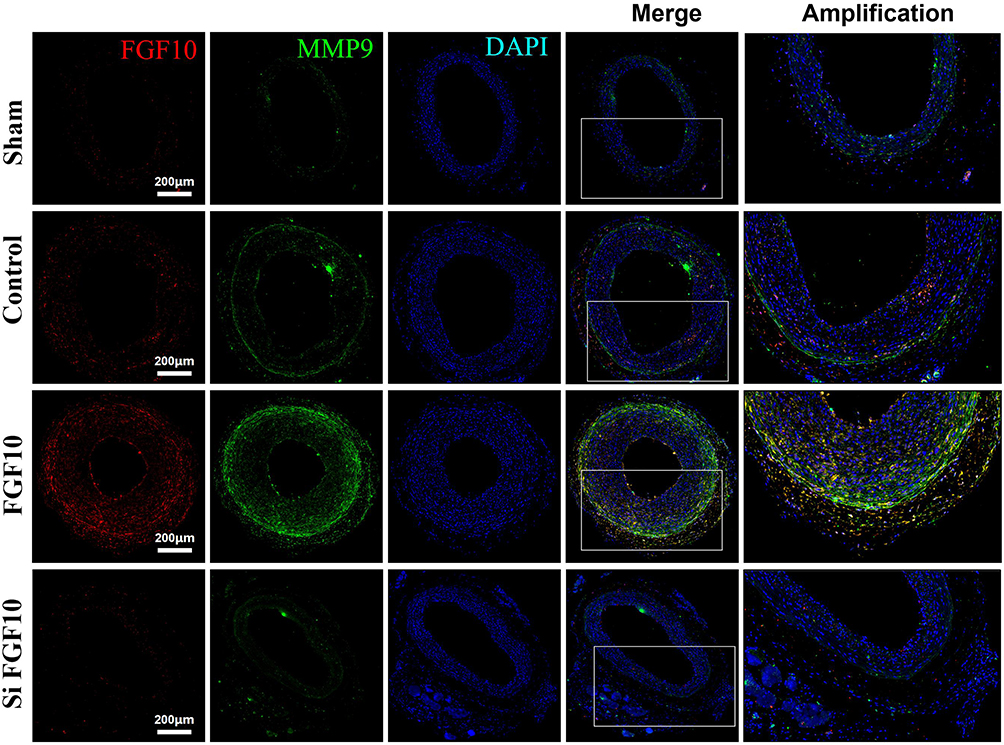 |
Figure 8 MMP9 expressions were upregulated by FGF10 in vivo. The balloon injuries were performed in rat LCA with or without deliveries of exogenous FGF10 or siFGF10-2 and the whole LCA were harvested on day 14 post the procedure for staining of FGF10, MMP9 and DAPI. Bar=200μm, n=5. |
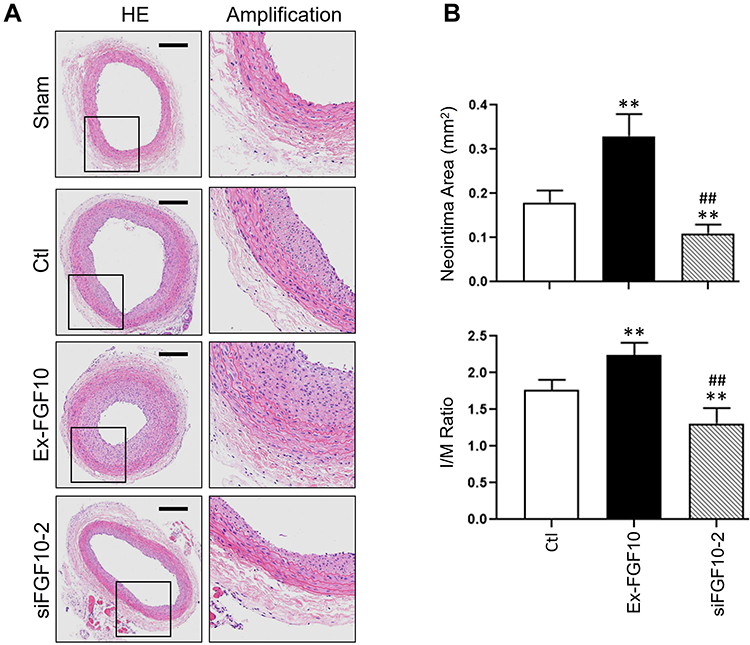 |
Figure 9 FGF10 potentiated balloon injury-induced neointima formation. The balloon injuries were performed in rat LCA with or without deliveries of exogenous FGF10 or siFGF10-2 and the whole LCA were harvested on day 14 post the procedure for HE staining (A) and quantitative analyses (B) of the neointima. Bar=200μm. n=6. **p<0.01 vs Ctl; ##p<0.01 vs FGF10. |
Discussion
Previous studies identified that the activation of VAFs induced by vascular injury has a significant promoting effect on VSMC proliferation and migration and neointima formation.11,12,25,26 However, all studies focused on the identification of this pathological phenomenon itself. The underlying molecular mechanisms remain elusive. In this study, we identified for the first time the importance of FGF10-FGFR2 axis in regulating VSMC growth and neointima formation. Our major findings are the following: 1) FGF10 was highly expressed in adventitia from day 3 after injury and maintained high through day 7 and day 14 post injury. 2) Coincidentally, the expression of FGFR2 in the medial layer VSMCs was upregulated. 3) Both FGF10 in VAFs and FGFR2 in VSMCs were significantly upregulated by PDGF in vitro. 4) Both exogenous FGF10 and VAFs-derived FGF10 promoted VSMCs proliferation and migration in an in vitro VAF-VSMC co-culture system. PDGF synergistically promoted these effects. 5) Both exogenous FGF10 and VAFs-derived FGF10 significantly activated the FGFR2 in VSMCs in vitro. 6) The phosphorylation levels, but not the expression levels, of FGFR2 in VSMCs of both media and neointima were significantly increased after balloon injury in vivo. 7) FGF10-FGFR2 axis activated both MAPK and PI3K-AKT pathways. 8) Administration of exogenous FGF10 upregulated PCNA and MMP9 gene expression and promoted the neointima formation post injury, while siFGF10 downregulated PCNA and MMP9 gene expression and attenuated neointima formation post injury in a rat balloon injury model.
In this study, we compared FGF10 spatiotemporal expression and FGFR2 expression and activities in LCA samples. As demonstrated by IHC, the phosphorylation levels of FGFR2 in VSMCs of both media and neointima were significantly increased after injury, while the number of FGFR2 expression-positive cells among the adventitia, media and neointima did not change significantly. Therefore, we deduced that the regulation by FGF10-FGFR2 axis of VSMCs proliferation and migration in the media and neointima formation is mainly achieved through both up-regulating FGF10 ligand and enhancing FGFR2 activity, rather than the expression levels of FGFR2. Our in vitro and in vivo WB results also supported this deduction. Although the percentages of FGFR2(+) VSMCs in the media illustrated no obvious changes either between w/wo injury or among day 3-7-14th post-injury, the percentages of FGFR2(+) VSMCs in the neointima were significantly higher than that in media from 7–14th day. These results reflected a higher proliferative status of VSMCs in neointima than media.
Meanwhile, PDGF significantly upregulated the expression level of FGFR2 in VSMCs, and in this way, the enhanced phosphorylation levels of FGFR2 caused by FGF10 were further potentiated. These results indicated that PDGF and FGF10 have a synergistic effect on activation of FGFR2.
FGF10-FGFR2 axis has been reported to mediate multiple downstream pathways, such as MEK1-ERK1/2, PI3K-AKT, Wnt and Shh.17 Since MAPK and PI3K-AKT are two major classic pathways that govern cell proliferation, migration and survival, we therefore verified their activities in VSMCs in vivo and in VAFs-VSMCs co-culture system. Our results showed that FGF10-FGFR2 axis significantly activated the pathways and upregulated cyclin-D1, MMP2 and MMP9. Pretreatment of either VSMCs or VAFs by PDGF further amplified these effects.
There are two limitations in this study. Firstly, this study lacked rescuing experiments in vivo. Although we used pluronic gel as a medium to apply FGF10 or siFGF10-2 to the surrounding of the injured LCA, the target was not specifically to VAFs. To circumvent this, we adopted a VAFs-VSMCs co-culture system and successfully identified the role of VAFs-derived FGF10 as a cytokine for intercellular signaling communication to VSMCs after injury. However, this was not in vivo experiment. A better explanation for the effects of VAFs-derived FGF10 on neointima formation would be if the expression of FGF10 in the adventitia could be conditional knocked down in VAFs and further followed by exogenous delivery of FGF10 to the local vessels to observe subsequent changes in neointima formation upon injury. Currently, this scheme is technically limited because VAFs have no specific lineage characteristics,27 leading to without specific Cre for use. Secondly, we only identified MAPK and PI3K-AKT pathways that are mediated by FGF10-FGFR2 axis. Whether other pathways, such as Wnt, Shh and other few mechanisms were involved was still unknown, although they has been reported to mediate the cell proliferation and migration.17,28 To make up for this limitation, we scheduled an analysis of the role of FGF10-FGFR2 axis in VSMCs with phosphoproteomics.
In conclusion, as shown in Figure 10, this study showed that after balloon injury, activated VAFs secreted FGF10 to act on VSMCs’ FGFR2. The activated FGFR2 mediated VSMCs proliferation and migration through MAPK and PI3K-AKT pathways, and promoted the neointima formation. PDGF upregulated the expression of FGFR2 in VSMCs and amplified the effects of FGF10-FGFR2 axis. Our results indicate that FGF10 is a potential therapeutic target for fighting the restenosis in patients post-PCI.
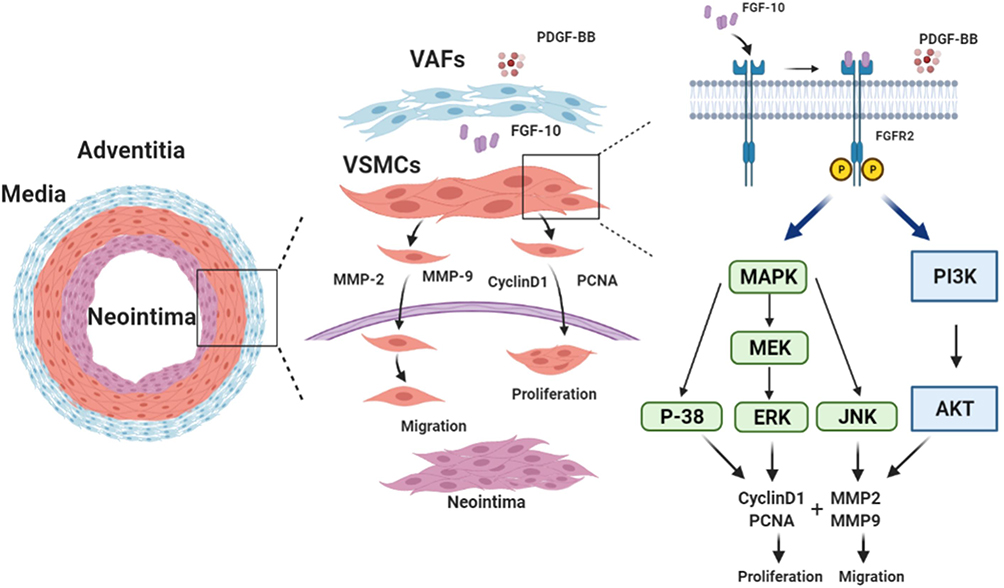 |
Figure 10 Schematic summary of VAFs-derived FGF10 and FGF10-FGFR2 signaling axis promotes VSMCs proliferation and migration in vitro and the neointima formation in vivo. |
Acknowledgments
We thank Hua Liu of Shanghai Zhongshan hospital for assistance to Elisa assay and HE staining.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Natural Science Foundation of China (NSFC) Grant No.81974041 (to Y. W.), No.81670414 (to Y. W.), No.82070443 (to J. C.), No.81470497 (to J. C.).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jukema JW, Verschuren JJW, Ahmed TAN, et al. Restenosis after PCI. Part 1: pathophysiology and risk factors. Nat Rev Cardiol. 2012;9(1):53–62. doi:10.1038/nrcardio.2011.132
2. Sorokin V, Vickneson K, Kofidis T, et al. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front Immunol. 2020;11:599415. doi:10.3389/fimmu.2020.599415
3. Abedi H, Zachary I. Signalling mechanisms in the regulation of vascular cell migration. Cardiovasc Res. 1995;30(4):544–556. doi:10.1016/S0008-6363(95)00092-5
4. Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372(6503):231–236. doi:10.1038/372231a0
5. Christen T, Verin V, Bochaton-Piallat ML, et al. Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation. 2001;103(6):882–888. doi:10.1161/01.CIR.103.6.882
6. Groves PH, Banning AP, Penny WJ, et al. Kinetics of smooth muscle cell proliferation and intimal thickening in a pig carotid model of balloon injury. Atherosclerosis. 1995;117(1):83–96. doi:10.1016/0021-9150(95)05562-B
7. Southgate KM, Fisher M, Banning AP, et al. Upregulation of basement membrane-degrading metalloproteinase secretion after balloon injury of pig carotid arteries. Circ Res. 1996;79(6):1177–1187. doi:10.1161/01.RES.79.6.1177
8. Rectenwald JE, Moldawer LL, Huber TS, et al. Direct evidence for cytokine involvement in neointimal hyperplasia. Circulation. 2000;102(14):1697–1702.
9. Li G, Chen SJ, Oparil S, et al. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101(12):1362–1365. doi:10.1161/01.CIR.101.12.1362
10. Han X, Wu A, Wang J, et al. Activation and migration of adventitial fibroblasts contributes to vascular remodeling. Anat Rec. 2018;301(7):1216–1223. doi:10.1002/ar.23793
11. Li XD, Chen J, Ruan CC, et al. Vascular endothelial growth factor-induced osteopontin expression mediates vascular inflammation and neointima formation via Flt-1 in adventitial fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32(9):2250–2258. doi:10.1161/ATVBAHA.112.255216
12. Dutzmann J, Koch A, Weisheit S, et al. Sonic hedgehog-dependent activation of adventitial fibroblasts promotes neointima formation. Cardiovasc Res. 2017;113(13):1653–1663. doi:10.1093/cvr/cvx158
13. Xu F, Liu Y, Hu W. Adventitial fibroblasts from apoE(-/-) mice exhibit the characteristics of transdifferentiation into myofibroblasts. Cell Biol Int. 2013;37(2):160–166. doi:10.1002/cbin.10027
14. Shi Y, Niculescu R, Wang D, et al. Increased NAD(P)H oxidase and reactive oxygen species in coronary arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2001;21(5):739–745. doi:10.1161/01.ATV.21.5.739
15. Li XD, Hong MN, Chen J, et al. Adventitial fibroblast-derived vascular endothelial growth factor promotes vasa vasorum-associated neointima formation and macrophage recruitment. Cardiovasc Res. 2020;116(3):708–720. doi:10.1093/cvr/cvz159
16. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4(3):215–266.
17. Watson J, Francavilla C. Regulation of FGF10 signaling in development and disease. Front Genet. 2018;9:500. doi:10.3389/fgene.2018.00500
18. Onda M, Naito Z, Wang R, et al. Expression of keratinocyte growth factor receptor (KGFR/FGFR2 IIIb) in vascular smooth muscle cells. Pathol Int. 2003;53(3):127–132. doi:10.1046/j.1440-1827.2003.01445.x
19. Zhang X, Ibrahimi OA, Olsen SK, et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281(23):15694–15700. doi:10.1074/jbc.M601252200
20. Xu X, Qimuge A, Wang H, et al. IRE1α/XBP1s branch of UPR links HIF1α activation to mediate ANGII-dependent endothelial dysfunction under particulate matter (PM) 2.5 exposure. Sci Rep. 2017;7(1):13507. doi:10.1038/s41598-017-13156-y
21. Huang G, Cong Z, Wang X, et al. Targeting HSP90 attenuates angiotensin II-induced adventitial remodelling via suppression of mitochondrial fission. Cardiovasc Res. 2020;116(5):1071–1084. doi:10.1093/cvr/cvz194
22. Song S, Liu L, Yu Y, et al. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis. J Mol Cell Cardiol. 2019;127:83–96. doi:10.1016/j.yjmcc.2018.12.002
23. Francavilla C, Rigbolt KG, Emdal K, et al. Functional proteomics defines the molecular switch underlying FGF receptor trafficking and cellular outputs. Mol Cell. 2013;51(6):707–722. doi:10.1016/j.molcel.2013.08.002
24. Mavila N, James D, Utley S, et al. Fibroblast growth factor receptor-mediated activation of AKT-β-catenin-CBP pathway regulates survival and proliferation of murine hepatoblasts and hepatic tumor initiating stem cells. PLoS One. 2012;7(11):e50401. doi:10.1371/journal.pone.0050401
25. Stenmark KR, Yeager ME, El Kasmi KC, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75(1):23–47. doi:10.1146/annurev-physiol-030212-183802
26. Haurani MJ, Pagano PJ. Adventitial fibroblast reactive oxygen species as autocrine and paracrine mediators of remodeling: bellwether for vascular disease? Cardiovasc Res. 2007;75(4):679–689. doi:10.1016/j.cardiores.2007.06.016
27. Kuwabara JT, Tallquist MD. Tracking adventitial fibroblast contribution to disease: a review of current methods to identify resident fibroblasts. Arterioscler Thromb Vasc Biol. 2017;37(9):1598–1607. doi:10.1161/ATVBAHA.117.308199
28. Wongsurawat T, Woo CC, Giannakakis A, et al. Distinctive molecular signature and activated signaling pathways in aortic smooth muscle cells of patients with myocardial infarction. Atherosclerosis. 2018;271:237–244. doi:10.1016/j.atherosclerosis.2018.01.024
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.