")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Variability of the drug response to nonsteroidal anti-inflammatory drugs according to cyclooxygenase-2 genetic polymorphism
Authors Lee SJ , Park MK, Shin DS , Chun MH
Received 12 June 2017
Accepted for publication 8 August 2017
Published 13 September 2017 Volume 2017:11 Pages 2727—2736
DOI https://doi.org/10.2147/DDDT.S143807
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Sook Joung Lee,1,* Min Kyu Park,2,* Dong-Seong Shin,3 Min Ho Chun4
1Department of Physical Medicine and Rehabilitation, Dong-A University College of Medicine, Dong-A University Hospital, 2Department of Pharmacology and Clinical Pharmacology, Dong-A University College of Medicine, Dong-A University Hospital, Busan, 3Clinical Trials Center, Gachon University Gil Medical Center, Incheon, 4Department of Rehabilitation Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea
*These authors contributed equally to this work
Purpose: Cyclooxygenase (COX) is the main pharmacodynamic target of nonsteroidal anti-inflammatory drugs (NSAIDs). We investigated the inhibitory effects on COX-2 after NSAIDs administration using a lipopolysaccharide (LPS)-derived COX-2 induction model in whole blood, according to the genotypes of COX-2 single-nucleotide polymorphisms (SNPs).
Patients and methods: Seven genotypes of COX-2 SNPs were selected from public databases and analyzed in 324 healthy subjects. Two genotypes showing a high percentage of variability were selected. A clinical trial examining pharmacodynamics according to the genotype of two SNPs (rs5275 and rs689466) was conducted. Twenty subjects were administered a single oral dose of 200 mg of celecoxib, and pharmacokinetic and pharmacodynamic analyses were performed.
Results: In the analysis of the pharmacokinetic parameters, significant differences in drug exposure were not investigated for each SNP genotype. The pharmacodynamic analysis revealed that the maximum effect of COX-2 inhibition was achieved at 2.0 hours for all genotypes of COX-2 SNPs after a single oral administration of 200 mg celecoxib. The inhibitory effects according to the genotype of COX-2 SNPs were investigated, and the area under the effect curve of the rs689466 GG genotype was significantly lower than that for the AA or AG genotype.
Conclusion: Our results demonstrated that inhibitory effects of celecoxib on COX-2 induction were different according to the genotype of COX-2 SNPs. In the present study, rs689466 is responsible for the variability of the response to celecoxib, suggesting that a subject with the GG genotype of rs689466 would be more responsive to celecoxib in terms of COX-2 inhibition.
Keywords: nonsteroidal anti-inflammatory agent, cyclooxygenase-2, single-nucleotide polymorphism, drug response, pharmacodynamics, clinical trial
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the most widely prescribed pharmaceutical agents worldwide and have broad clinical utility in the management of pain, fever, inflammation and inflammatory disease. Cyclooxygenase (COX) is the main pharmacodynamic target of NSAIDs. COX, also known as prostaglandin H synthase, is a major enzymatic source of prostaglandin (PG) production from arachidonic acid and comprises two isoforms, COX-1 and COX-2. Although COX-1 and COX-2 exhibit a high degree of homologous genetic information, their structural stability, enzymatic mechanism and functions differ.1,2 COX-1 isoform is constitutively expressed in most tissues and cells throughout the body and produces PGs involved in regulating physiological homeostatic processes.3–5 In contrast, COX-2 is an inducible isoform that is upregulated by various mitogenic and inflammatory stimuli, such as pro-inflammatory cytokines, growth factors and tumor promoters.6
Although NSAIDs show various beneficial effects, such as antipyretic, analgesic and anti-inflammatory effects, response variability and unwanted adverse drug effects are also observed in patients using NSAIDs. Because the mechanisms of inflammation and pain are complex and vary between individuals, there is interindividual variability in the effectiveness and safety of NSAIDs.
Regarding the pharmacokinetic characteristics of NSAIDs, several studies have investigated the variability of adverse events and the effectiveness of NSAIDs according to the genetic polymorphism of hepatic cytochrome P450 isoenzymes, which are involved in drug metabolism. Indeed, CYP2C subfamilies exhibit many single-nucleotide polymorphisms (SNPs) with different frequencies, and CYP2C9 metabolizes many NSAIDs including ibuprofen, diclofenac, naproxen, piroxicam and tenoxicam.7,8 However, these previous studies have not investigated the relationship between NSAIDs and COX-2 inhibition. Because the effect of NSAIDs in individual patients is based on pharmacodynamic activity related to the induction and continued participation of COX-2 in the inflammatory process, the investigation of various inhibitory effects on COX-2 activity and its duration is necessary to explain the variability of NSAID response.
Regarding the pharmacodynamic target molecule, COX-2 genetic polymorphisms have been reported in various cancers and can change COX-2 expression and alter susceptibility to carcinomas.9 In breast cancer, a reduced risk of hormone receptor-positive breast cancer has been reported in subjects with the COX-2 SNP rs5275 using NSAIDs. These findings suggest that the effects of NSAIDs could be modulated by the characteristics of COX-2, such as genetic polymorphisms.
Previous studies have demonstrated a relationship between interindividual variability in COX-2 gene expression and the variable response to celecoxib treatment.10–12 However, no comparisons of COX-2 expression associated with genetic polymorphisms have yet been conducted using objective pharmacodynamic parameters.
In the present study, we investigated the characteristics of the inhibitory effects of COX-2 after NSAID administration according to the genotypes of COX-2 SNPs. To objectively evaluate the inhibitory effects of COX-2, we used a previously investigated lipopolysaccharide (LPS)-derived COX-2 induction model in whole blood. Pharmacokinetic and pharmacodynamic analyses were conducted to exclude causative factors other than COX-2 characteristics. Prior to clinical trials, the genotypes of the seven most common COX-2 SNPs were selected and analyzed in a larger population to identify the COX-2 SNP types to be used in the present clinical study. Celecoxib, a type of NSAID designed for COX-2 inhibition that is used clinically to treat inflammatory disease, was employed in this study.
Patients and methods
Analysis of COX-2 SNP
Study population
The present study, involving a COX-2 SNP analysis, was approved by the institutional review board of our institution, and written informed consent for participation was obtained from all subjects after providing a full explanation of the study procedures (Dong-A University Hospital IRB No: 14-071). In total, 324 subjects were screened and participated in the study without exclusion. All subjects were older than 19 years of age and had no genetic disease according to their medical history and physical examination. Although disease states other than genetic disease were not included in the exclusion criteria, all subjects were healthy.
Selection of COX-2 SNPs and genotyping
The human COX-2 gene contains 10 exons, and its molecular weight is approximately 8.3 kilobases.13 In the present study, data on COX-2 polymorphisms were collected from available public databases, including dbSNP, and several types of COX-2 SNPs were selected for analysis according to the following criteria: 1) a reported minor allele frequency is 5% or greater and 2) previously reported data or evidence indicating an association with COX-2 function. Based on these criteria, a total of seven SNPs of COX-2 (rs20417, rs5275, rs689466, rs4648276, rs2745557, rs689470 and rs2066826) were selected. After the selection of COX-2 SNPs, genomic DNA was extracted from peripheral blood leukocytes using a commercial QIAamp® DNA blood mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Genotyping was conducted via a single-base primer extension assay using the ABI PRISM SNaPShot Multiplex kit (Thermo Fisher Scientific, Waltham, MA, USA). The genomic DNA sequences flanking the SNPs of COX-2 were amplified through polymerase chain reaction (PCR) using forward and reverse primer pairs. Information on the primers is presented in Table 1. After amplification, the PCR products were purified and added to a SNaPshot Multiplex Ready reaction mixture for the primer extension reaction. Following the removal of excess fluorescent dye terminators with shrimp alkaline phosphatase, the sequence analysis was performed with GeneMapper software (version 4.0; Thermo Fisher Scientific).
 | Table 1 Primer sets and Tm for the SNaPshot assay |
Clinical trial
Subjects
Based on the results regarding the COX-2 SNP genotypes, a clinical trial examining pharmacodynamics according to the genotypes of two SNPs (rs5275 and rs689466) was conducted. The subjects carrying either rs5275 (T>C) or rs689466 (A>G) were selected for screening, and the 20 participants who passed the screening evaluation were enrolled. The following inclusion criteria were used: age 19–50 years, body mass index (BMI) 18.0–30.0 kg/m2 and a lack of clinically significant medical history, physical examination findings or routine laboratory tests (12-lead electrocardiogram [ECG], blood chemistry, hematology, infectious serology and urine analysis). Subjects with gastrointestinal disease that may affect the absorption of the investigational product and those who had ingested any prescribed medication within 2 weeks prior to study initiation were excluded. After a full explanation of the objectives and study procedures, written informed consent was obtained from all subjects. The study protocol was approved by the institutional review board, and the study was conducted according to the Declaration of Helsinki and Korean Good Clinical Practice (Dong-A University Hospital IRB No: 15-148, CRIS No: KCT0002094).
Study design
This clinical trial was an open-label, single-oral dose, one-treatment study conducted in Korean male subjects at the Clinical Trials Center of our institution. On Day −1, the enrolled subjects were hospitalized at 6 pm, and each of the subjects was provided with an identical, controlled meal after undergoing baseline laboratory tests (eg, clinical laboratory tests, ECG). The subjects were assigned to each group according to their genotype (Table 2). On Day 1, the subjects received a single oral dose of 200 mg celecoxib with 150 mL of water in the fasting state. Subsequently, the participants remained in a sitting position and continued to fast for 4 hours. Blood sampling for pharmacokinetic and pharmacodynamic analyses was performed over the following 24 hours according to a schedule.
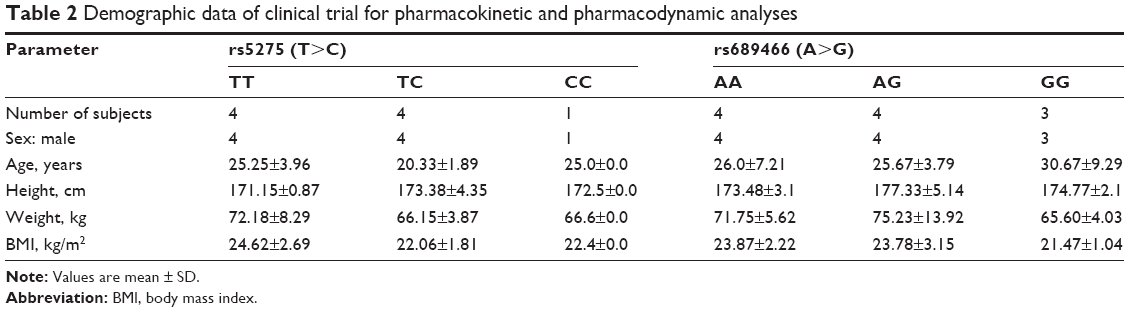 | Table 2 Demographic data of clinical trial for pharmacokinetic and pharmacodynamic analyses |
Pharmacokinetic and pharmacodynamic assessment
Venous blood samples for the pharmacokinetic evaluation were collected before dosing (0 hour) and at 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 hours after administration of the study drug to determine the plasma concentration of celecoxib. At the indicated time points, blood was drawn into heparinized tubes and immediately centrifuged at 3,000 rpm for 10 minutes at 4°C. The separated plasma samples were transferred to sterile cryotube vials and stored at −70°C until further analysis.
For the pharmacodynamic evaluation of prostaglandin E2 (PGE2), which represents COX-2 activity, venous blood samples were collected before dosing (0 hour) and at 2, 4, 8, 12 and 24 hours after study drug administration. Samples of 10 mL blood were drawn into K2 EDTA tubes and stored in a 36°C incubator after treatment with 10 ng/mL LPS for 24 hours. After the appropriate incubation time for COX-2 induction, plasma was separated by centrifugation at 3,000 rpm for 10 minutes at 4°C and frozen at −70°C until PGE2 measurement.
Assay of celecoxib plasma concentrations
The plasma concentration of celecoxib was measured using high-performance liquid chromatography–tandem mass spectrometry (LC–MS/MS) in human plasma. Method validation was conducted with celecoxib (98% purity) and celecoxib-d7, as an internal standard (IS), which were purchased from Toronto Research Chemicals (North York, ON, Canada) and CDN Isotopes, respectively. For solvent and sample delivery, an Agilent 1,200 system consisting of a G1312A quaternary pump, G1379B degasser, 1367B auto-sampler, G1316A thermostat and G1316A column and an oven (TTC) compartment (Agilent, Santa Clara, CA, USA) was used. An API 4,000 triple-quadrupole mass spectrometer equipped with a TurboIonSpray (EIS; Thermo Fisher Scientific) source was used for mass analysis and detection.
A 50 μL aliquot of 0.2 M zinc sulfate was added to 100 μL of plasma sample, followed by vortex mixing for 10 seconds. This plasma sample was combined with 1 mL of acetonitrile (in celecoxib-d7, 100 ng/mL), and the contents of the tube were vortexed for 30 seconds, followed by centrifugation at 12,000 rpm for 5 minutes. The supernatant was finally transferred to appropriate auto-sampler vials, and an aliquot (5 μL) was injected into an LC–MS/MS instrument for analysis. The mobile phase consisted of acetonitrile–0.1% formic acid (85:15, v/v) at a flow rate of 0.2 mL/min, and the used column was Gemini C18® (50×2.0 mm, 5 μm; Phenomenex, Torrance, CA, USA). The column temperature was maintained at 35°C.
A stock solution of 10 mg of celecoxib in 10 mL of acetonitrile was used to produce seven standard working solutions of 100, 500, 1,000, 2,500, 5,000, 10,000 and 30,000 ng/mL of celecoxib. These solutions were generated by further dilution of the stock solution with acetonitrile–water (50:50, v/v).
The calibration standards were prepared at concentrations of 10, 50, 100, 250, 500, 1,000 and 3,000 ng/mL of celecoxib in plasma, and the corresponding QC samples were prepared at 30, 800 and 2,400 ng/mL. The performance of the proposed method, as the % coefficient of variation (CV) of QC samples, was less than 97.47% and 100.21%. For celecoxib, the calibration curves were linear within the range of 10–3,000 ng/mL.
Assay of COX-2 activity
COX-2 activity was estimated by measuring the blood PGE2 level according to a previously reported method. Plasma PGE2 levels were determined using commercially available enzyme immunoassay kits (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer’s instructions. The standard and experimental samples were assayed in parallel, and the detection limit of PGE2 quantification was 15 pg/mL. The reaction solution, including 50 μL of human plasma in each well with 50 μL of enzyme immunoassay reagent, PGE2 and a PGE2 monoclonal antibody, was incubated for 1.5 hours. The incubated samples were measured in 96-well plates using a spectrophotometer at 405 nm (SpectraMax 340; Molecular Devices Corp., Sunnyvale, CA, USA).
Pharmacokinetic and pharmacodynamic analyses
The pharmacokinetic parameters of celecoxib were estimated using the non-compartmental analysis method of WinNonlin® 6.4 (Pharsight Co, Mountain View, CA, USA), based on the plasma concentrations of celecoxib and the actual sampling time. The Cmax and the time to Cmax (Tmax) were estimated directly from the plasma concentration–time profiles. The individual area under the concentration–time curve (AUC0–t) from dosing to 24 hours or the AUClast was calculated using the linear-up and log-down trapezoidal method. The elimination rate constant (λz) was estimated from the slope of the terminal log-linear phase, and the terminal elimination half-life (t1/2) was calculated as ln(2)/λz. The apparent oral clearance (CL/F) was calculated as dose/AUC.
For objective pharmacodynamic analysis, the pharmacodynamic parameters, including the Imax, Tmax and the area under the effect curve (AUEC), were estimated. Therefore, in the present study, the individual AUEC was estimated from the plasma PGE2 level–time profiles using the linear trapezoidal approximation method to compare the genotypes of COX-2 SNPs. Additionally, the maximum inhibitory effect on PGE2 production and the Tmax were estimated from the time course of PGE2 production.
Statistical analysis
For the statistical analyses conducted in the present study, SPSS 22.0 (SPSS Korea, Seoul, Republic of Korea) was used. The analysis of pharmacokinetic parameters was performed for the 20 subjects in whom all procedures for the pharmacokinetic analysis were conducted. The analysis of variance (ANOVA) was performed to compare the AUC0–t and Cmax pharmacokinetic values between COX-2 SNP types.
For the pharmacodynamic analysis, the AUEC for the inhibition of PGE2 production was used to compare COX-2 SNP types, and the Kruskal–Wallis test, followed by Bonferroni’s correction, was employed for statistical analysis. The differences between COX-2 SNP groups were considered significant at the probability level of 0.05.
Results
COX-2 genotyping
For COX-2 genotyping, a total of 324 subjects (292 males) were enrolled, and seven COX-2 SNPs were selected and analyzed in whole blood. The mean age of the subjects was 26.47±4.49 years, and their mean body weight was 69.45±10.20 kg. The mean height and BMI of the subjects were 173.16±6.99 cm and 23.09±1.87 kg/m2, respectively.
The results for the seven SNPs of COX-2 are presented in Table 3. With the exception of the COX-2 SNP rs4648276, the other SNPs were not analyzed in some subjects. The numbers of rs689466, rs20417, rs2745557, rs2066826, rs4648276, rs5275 and rs689470 polymorphisms were 323, 323, 318, 321, 324, 322 and 322, respectively. Based on the seven COX-2 SNPs analyzed, the minor allele frequencies of the rs689466 and rs5275 genotypes were remarkably high, at 46% and 18%, respectively. The minor allele frequencies of the other five genotypes were very low, at approximately 1.1%–4.5%; thus, rs689466 and rs5275 were selected for subsequent clinical trials.
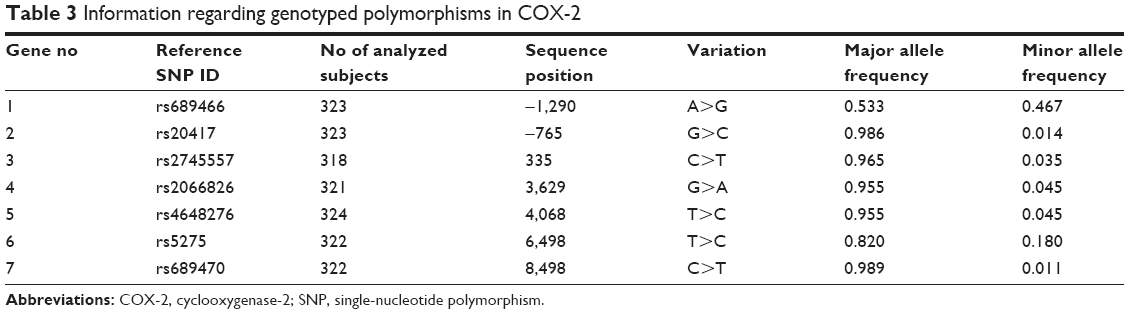 | Table 3 Information regarding genotyped polymorphisms in COX-2 |
Clinical trial
In the clinical trial, a total of 20 healthy male subjects were enrolled and completed the study. Twelve subjects were assigned to each allele type of rs5275, and eight subjects were assigned to each allele type of rs689466. The demographic data for each allele type of rs5275 and rs689466 are presented in Table 2.
Pharmacokinetic parameters of celecoxib
The plasma concentration–time profiles of celecoxib were obtained via high-performance LC–MS/MS. The mean plasma concentration–time profiles of celecoxib are presented in Figure 1, and the corresponding pharmacokinetic parameters (Cmax, AUC, etc) are presented in Table 4. For rs5275 (T>C), the maximum observed plasma concentration (Cmax) was 434.80±118.33, 350.54±96.19 and 627.88±0.0 ng/L for the TT, TC and CC genotypes, respectively, and the individual AUC0–t from dosing to the last measurable time point (AUClast) was 3,360.57±346.58, 2,999.67±700.18 and 4,054.37±0.0 ng·h/L/mg, respectively.
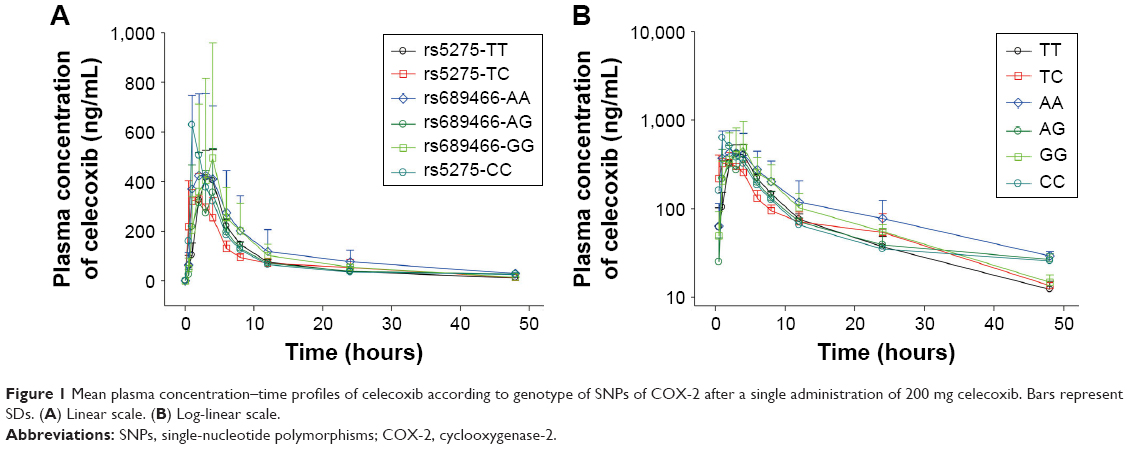 | Figure 1 Mean plasma concentration–time profiles of celecoxib according to genotype of SNPs of COX-2 after a single administration of 200 mg celecoxib. Bars represent SDs. (A) Linear scale. (B) Log-linear scale. |
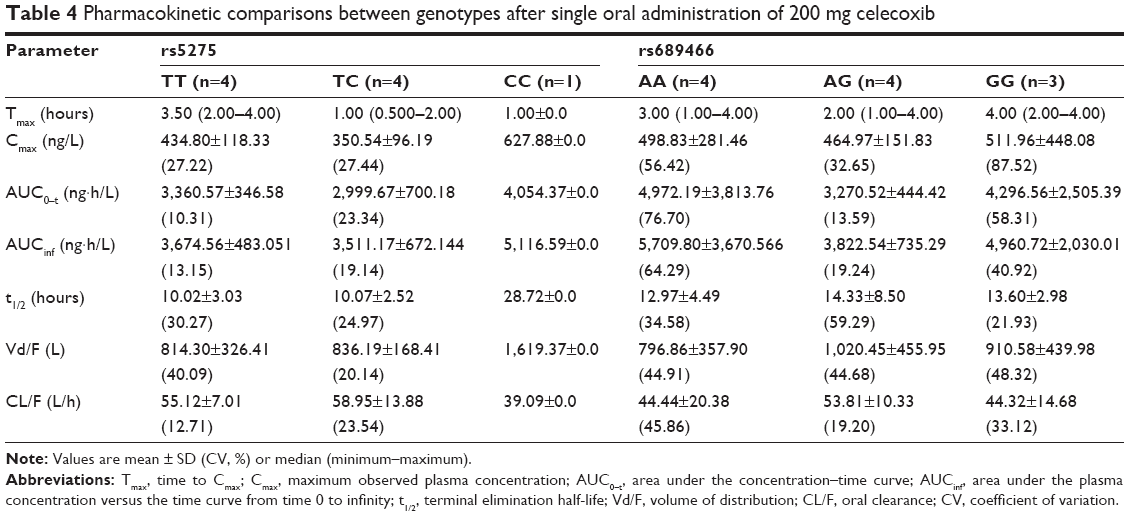 | Table 4 Pharmacokinetic comparisons between genotypes after single oral administration of 200 mg celecoxib |
For rs689466 (A>G), Cmax was 498.83±281.46, 464.97±151.83 and 511.96±448.08 ng/L for the AA, AG and GG genotypes, respectively, and AUClast was 4,972.19±3,813.76, 3,270.52±444.42 and 4,296.56±2,505.39 ng·h/L/mg, respectively. In the analysis of the pharmacokinetic parameters Cmax and AUClast, significant differences in drug exposure were not investigated for each SNP genotype (p>0.05).
Pharmacodynamic analysis of celecoxib according to COX-2 SNPs
Compared with the pre-dose time point (0 hour), the maximum effect of COX-2 inhibition, based on changes in LPS-induced plasma PGE2 levels, was achieved at 2.0 hours (median Tmax) for all genotypes of COX-2 SNPs (rs5275, rs689466). Recovery from the inhibitory effects was observed at 4 hours after celecoxib administration, and the effects almost disappeared at 8 hours after administration in all genotype groups, except for the CC genotype of rs5275. Twelve hours after administration, the inhibitory effect of celecoxib nearly completely disappeared. The mean plasma PGE2 level–time profiles according to genotype are presented in Figure 2.
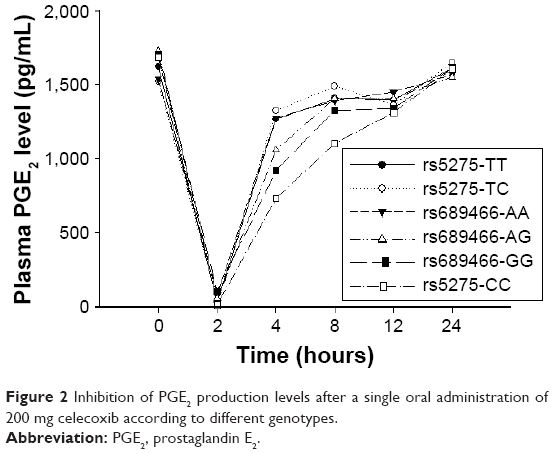 | Figure 2 Inhibition of PGE2 production levels after a single oral administration of 200 mg celecoxib according to different genotypes. |
The AUEC was calculated from the plasma PGE2 level–time profiles and compared with the genotypes of COX-2 SNPs; the results are described in Table 5. Considering the AUEC values between genotypes of rs5275 (T>C), genotype CC tended to be higher, but the statistical significance was not investigated. Among the genotypes of rs689466 (A>G), the AUEC value for GG genotype was significantly lower than those for the AA and GA genotypes (p=0.029), but the AUEC values of genotypes AA and GA did not differ. The differences in the AUECs between the genotypes of COX-2 SNPs reflect the time point of recovery from inhibited COX-2 activity, as the degree of the AUEC is compatible with the time point of recovery. For example, the CC genotype of rs5275 showed the latest time point of recovery among the evaluated genotypes (Figure 2).
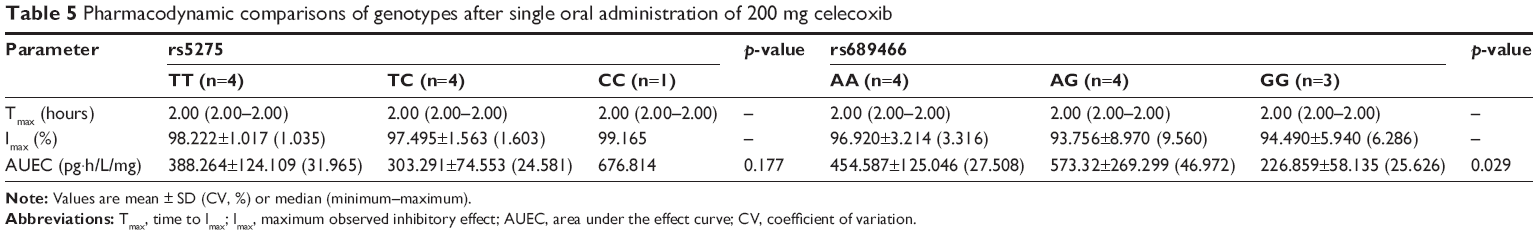 | Table 5 Pharmacodynamic comparisons of genotypes after single oral administration of 200 mg celecoxib |
Safety and tolerability in the clinical trial involving pharmacokinetic and pharmacodynamic analyses
During the course of the present study, no adverse events, serious adverse events or adverse drug responses were reported, and no clinically significant changes in vital signs, physical examination findings, ECG intervals or laboratory test parameters were observed compared with baseline findings.
Discussion
The major finding of the present study was that when pharmacokinetic characteristics are excluded, COX-2 SNPs affect the drug response to NSAIDs or selective COX-2 inhibitors (such as celecoxib) by modulating COX-2 activity.
Drug response variability – causative factors
Clinically, interindividual variability in drug responses is not unusual, and the causative mechanisms have also been well established in a previous study.14 Regarding pharmacokinetics, genetic polymorphisms in drug-metabolizing enzymes, such as hepatic cytochrome P450 isoenzymes, can lead to clinically significant variability in drug responses.15 Because the variability of pharmacokinetic parameters such as Cmax and AUC suggests that the exposure of the body to the administered drug is variable in each subject, prediction of the variability of drug response is not difficult. However, the pharmacokinetic parameters evaluated in the present study did not differ between the compared groups, which were classified based on the genotype of COX-2 SNPs. This finding indicates that the variability in COX-2 inhibition by celecoxib is not associated with the degree of exposure to celecoxib. When the effect of pharmacokinetic characteristics is excluded, the variability in COX-2 inhibition is only regulated by the corresponding genotype.
Drug response variability – pharmacodynamic analysis according to the genotype
In the present study, the identified genotype of the COX-2 SNP affecting the variability of the response to celecoxib was rs689466 (A>G). The AUEC for the GG genotype of rs689466 was significantly lower than for the AA or AG genotype, suggesting that a subject with the GG genotype of rs689466 would be more responsive to NSAIDs in terms of COX-2 inhibition.
However, it is difficult to exclude the effect of rs5275 (T>C) on the COX-2 inhibition by celecoxib. Although the significance of the results was not investigated, the inhibitory effects associated with the CC genotype of rs5275 tended to be strong. The lack of significance was most likely caused by the small sample size (only one subject with CC genotype of rs5275). Therefore, we expect that rs5275 (T>C) could be demonstrated to act as a factor that affects the variability of NSAIDs and their COX-2 inhibitory effect if further studies are conducted with larger sample size.
In contrast to the AUEC results, the maximum observed inhibitory effect (Imax) and time to Imax (Tmax) showed no significant differences in the inhibitory effect of celecoxib according to the COX-2 SNP genotype, and the median Tmax value was identical in all genotypes. It is thought that this finding reflects the sampling time of pharmacodynamic assessment; thus, it will be necessary to compensate for the frequency and time interval for pharmacodynamic assessment in future studies.
Inhibitory effect of celecoxib according to the time – pharmacodynamic evaluation
In the pharmacodynamic analysis, the time duration of the action of celecoxib was estimated. The maximum effect of COX-2 inhibition, based on changes in LPS-induced plasma PGE2 levels, was achieved at 2.0 hours for all genotypes of COX-2 SNPs after a single oral administration of 200 mg celecoxib. The inhibitory effect of celecoxib was reversed after Tmax and completely disappeared at 12 hours after dosing all genotype groups. However, the clinically recommended dosing interval for 200 mg celecoxib is 24 hours. Therefore, it is necessary to study the appropriateness of the current dosing interval for 200 mg celecoxib.
Importance of COX-2 genotype in various diseases
Currently, the COX-2 pathway or overexpression of COX-2 is recognized as important in various pathologies, including cancer development and cancer progression,16 based on extensive evidence.17 Although the causative mechanisms associating COX-2 with tumorigenesis are not predictable, the genetic variations in COX-2 are a remarkable mechanism. For example, the rs2143416 SNP is significantly associated with the risk of breast cancer,18 and rs5275 was shown to influence the risk of both lung cancer19 and breast cancer.20 The rs689466 SNP has also been associated with the risk of digestive system cancers.21 Additionally, an association between cardiovascular disease and a COX-2 SNP (rs20417) has also been reported.22 Based on these findings, we assumed that COX-2 SNPs may be related to the function of COX-2, which could contribute to the variability of the response to NSAIDs, including COX-2-specific inhibitors.
Objective measurements of the drug response – ex vivo model for clinical application
To evaluate the variability of drug response, the use of objective parameters for measuring the response to drugs is necessary because subjective parameters, such as the visual analog scale, exhibit poor reproducibility and show high interindividual variability. As the estimated body exposure to a drug must be considered when measuring the drug response, due to aforementioned factors, the method applied in the present study was consistent with that used in the clinical fields to predict or determine the drug response of patients using NSAIDs.23
In this clinical trial, we employed an LPS-derived COX-2 induction model in whole blood, which was easy to use and could objectively estimate the drug response.
Genotype differences – Koreans vs Caucasians
Another important finding of the present study that is important for the clinical use of NSAIDs, including COX-2-specific inhibitors, is that the allele frequencies of COX-2 SNPs in Koreans are different from those in patients with prostate cancer. According to Dossus et al,24 the minor allele frequencies of rs20417, rs689466 and rs5275 were 0.17, 0.20 and 0.34, respectively; however, the minor allele frequencies observed in the present study were 0.014, 0.467 and 0.180, respectively. Because the population assessed by Dossus et al primarily consisted of Caucasians from the US or Europe,24 the differences in the allele frequencies of the same COX-2 SNPs could reflect racial differences. Considering that rs689466 is a causative factor associated with the variability of the drug response to NSAIDs, this finding is of clinical interest. If genetic polymorphism is a factor affecting the variability of the response to drugs, then the causative SNP differs according to ethnic group in cases of unexpected drug response.
In this study, the obtained results described the relationship between the variability of the response to celecoxib and COX-2 SNPs. However, there were some limitations in the present work. For example, the clinical study was conducted in a small number of healthy, young, male volunteers, and the sample sizes of the genotype groups were not balanced; thus, the results could not fully reflect the clinical pathophysiology of COX-2 and the inhibitory effect of NSAIDs in patients with pain or inflammatory disease. Additionally, the variability of the response to COX-2 inhibitors was investigated in only two SNP types. Therefore, it is difficult to estimate the relationship of the causative SNP types of COX-2.
Conclusion
This study evaluated that the inhibitory effect of celecoxib in COX-2 induction was different according to the genotype of COX-2 SNPs. The pharmacodynamic analysis revealed that the maximum effect of COX-2 inhibition, based on changes in LPS-induced plasma PGE2 levels, was achieved at 2.0 hours for all genotypes of COX-2 SNPs after a single oral administration of 200 mg celecoxib. The inhibitory effect of celecoxib was reversed after Tmax and completely disappeared at 12 hours after dosing all genotype groups. The AUEC for the GG genotype of rs689466 was significantly lower than for the AA or AG genotype, suggesting that a subject with the GG genotype of rs689466 would be more responsive to NSAIDs in terms of COX-2 inhibition. Additionally, the allele frequencies of COX-2 SNPs (rs5275, rs689466) showed differences between Korean and Caucasian individuals, indicating ethnic differences.
Acknowledgment
SJL was supported by a Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF – 2014R1A1A3052625).
Author contributions
SJL and MKP contributed to statistical oversight and interpretation. SJL and MHC developed the protocol. MKP and DSS conducted the study. SJL and MKP involved in medical monitoring. MKP and DSS analyzed and interpreted the data. SJL, MKP, DSS and MHC developed and reviewed the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat Struct Biol. 1996;3(11):927–933. | ||
Lu G, Tsai AL, Van Wart HE, Kulmacz RJ. Comparison of the peroxidase reaction kinetics of prostaglandin H synthase-1 and -2. J Biol Chem. 1999;274(23):16162–16167. | ||
Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes. J Clin Invest. 2001;107(12):1491–1495. | ||
Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14(10):461–469. | ||
Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50 Suppl:S423–S428. | ||
Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56(3):387–437. | ||
Li-Wan-Po A, Girard T, Farndon P, Cooley C, Lithgow J. Pharmacogenetics of CYP2C19: functional and clinical implications of a new variant CYP2C19*17. Br J Clin Pharmacol. 2010;69(3):222–230. | ||
Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007;116(3):496–526. | ||
Dai ZJ, Shao YP, Ma XB, et al. Association of the three common SNPs of cyclooxygenase-2 gene (rs20417, rs689466, and rs5275) with the susceptibility of breast cancer: an updated meta-analysis involving 34,590 subjects. Dis Markers. 2014;2014:484729. | ||
Fritsche E, Baek SJ, King LM, Zeldin DC, Eling TE, Bell DA. Functional characterization of cyclooxygenase-2 polymorphisms. J Pharmacol Exp Ther. 2001;299(2):468–476. | ||
Szczeklik W, Sanak M, Szczeklik A. Functional effects and gender association of COX-2 gene polymorphism G-765C in bronchial asthma. J Allergy Clin Immunol. 2004;114(2):248–253. | ||
Shi J, Misso NL, Duffy DL, Thompson PJ, Kedda MA. A functional polymorphism in the promoter region of the cyclooxygenase-2 gene is not associated with asthma and atopy in an Australian population. Clin Exp Allergy. 2004;34(11):1714–1718. | ||
Kosaka T, Miyata A, Ihara H, et al. Characterization of the human gene (PTGS2) encoding prostaglandin-endoperoxide synthase 2. Eur J Biochem. 1994;221(3):889–897. | ||
Wood AJ. Racial differences in the response to drugs – pointers to genetic differences. N Engl J Med. 2001;344(18):1394–1396. | ||
Bae JW, Choi CI, Jang CG, Lee SY. Effects of CYP2C9*1/*13 on the pharmacokinetics and pharmacodynamics of meloxicam. Br J Clin Pharmacol. 2011;71(4):550–555. | ||
Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4(6):431–436. | ||
Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17(2):55–67. | ||
Gallicchio L, McSorley MA, Newschaffer CJ, et al. Nonsteroidal antiinflammatory drugs, cyclooxygenase polymorphisms, and the risk of developing breast carcinoma among women with benign breast disease. Cancer. 2006;106(7):1443–1452. | ||
Vogel U, Christensen J, Wallin H, et al. Polymorphisms in genes involved in the inflammatory response and interaction with NSAID use or smoking in relation to lung cancer risk in a prospective study. Mutat Res. 2008;639(1–2):89–100. | ||
Cox DG, Buring J, Hankinson SE, Hunter DJ. A polymorphism in the 3′ untranslated region of the gene encoding prostaglandin endoperoxide synthase 2 is not associated with an increase in breast cancer risk: a nested case-control study. Breast Cancer Res. 2007;9(1):R3. | ||
Dong J, Dai J, Zhang M, Hu Z, Shen H. Potentially functional COX-2-1195G>A polymorphism increases the risk of digestive system cancers: a meta-analysis. J Gastroenterol Hepatol. 2010;25(6):1042–1050. | ||
Zhang MM, Xie X, Ma YT, et al. Association of COX-2-765G>C genetic polymorphism with coronary artery disease: a meta-analysis. Int J Clin Exp Med. 2015;8(5):7412–7418. | ||
Shin D, Lee SJ, Ha YM, et al. Pharmacokinetic and pharmacodynamic evaluation according to absorption differences in three formulations of ibuprofen. Drug Des Devel Ther. 2017;11:135–141. | ||
Dossus L, Kaaks R, Canzian F, et al. PTGS2 and IL6 genetic variation and risk of breast and prostate cancer: results from the Breast and Prostate Cancer Cohort Consortium (BPC3). Carcinogenesis. 2010;31(3):455–461. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.