")
Back to Journals » Clinical Epidemiology » Volume 11
Validity of first-time diagnoses of congenital epidermolysis bullosa in the Danish National Patient Registry and the Danish Pathology Registry
Authors Kristensen MH , Schmidt SAJ , Kibsgaard L, Mogensen M, Sommerlund M , Koppelhus U
Received 21 August 2018
Accepted for publication 24 October 2018
Published 17 January 2019 Volume 2019:11 Pages 115—124
DOI https://doi.org/10.2147/CLEP.S184742
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Henrik Sørensen
Mattias Hedegaard Kristensen,1 Sigrún Alba Jóhannesdóttir Schmidt,2 Line Kibsgaard,1 Mette Mogensen,3 Mette Sommerlund,1 Uffe Koppelhus1
1Department of Dermatology, Aarhus University Hospital, Aarhus, Denmark; 2Department of Clinical Epidemiology, Aarhus University Hospital, Aarhus, Denmark; 3Department of Dermatology, Bispebjerg Hospital, University of Copenhagen, Copenhagen, Denmark
Purpose: Congenital epidermolysis bullosa (CEB) is a group of rare monogenic genodermatoses. Phenotypically, the diseases vary in both severity and dissemination, which complicates studies of their epidemiology. To investigate the potential of using the Danish National Patient Registry (DNPR) for epidemiological research on CEB, we examined the positive predictive value (PPV) of a first-time diagnosis of CEB.
Methods: We identified patients with a record of CEB in DNPR and the Danish Pathology Registry (DPR) during January 1, 1977, until December 31, 2015. We restricted diagnoses from two dermatological departments and one regional hospital. Diagnoses in the DNPR are coded by the eighth and tenth revisions of the ICD (ICD-8 and ICD-10) and in the DPR by the Systematized Nomenclature of Medicine (SNOMED). We used clinical description in medical records, family history, histological findings, and molecular genetic investigations to validate diagnoses and classified them as rejected and confirmed. We estimated PPVs for any diagnosis, according to coding systems used, and for additional subdivisions of ICD-10 codes.
Results: We identified 116 cases from the hospital departments investigated and evaluated 96 medical records for validity. The overall PPV for probable CEB was 62.5% (95% CI: 52.5–71.5). For ICD-8, ICD-10, and SNOMED codes, the PPVs were 30.8% (95% CI: 11.4–57.7), 76.7% (95% CI: 65.8–84.9), and 0.0% (95% CI: 0.0–21.7), respectively. For the ICD-10 codes, we found the highest PPVs for diagnoses arising from the dermatological departments. For subdivisions of ICD-10 codes, PPVs were high for epidermolysis bullosa simplex and dystrophica.
Conclusion: The PPVs for first-time diagnoses of CEB registered in the two Danish nationwide registries investigated, DNPR and DPR, ranged from low to average. We therefore recommend that these data be used with caution and restricted to ICD-10 diagnoses from specialized dermatological departments.
Keywords: Denmark, diagnosis, epidermolysis bullosa, health administrative data, registration, validity
Introduction
Congenital epidermolysis bullosa (CEB) is a group of rare genetic skin diseases with known monogenetic pathogenesis.1 CEB is characterized by blistering caused by fragile epithelial surfaces in the skin and mucosa.2 It is associated with variable levels of morbidity, some of which require highly specialized care.3 The wide range of clinical presentations and the limited knowledge on genotype–phenotype correlations make further investigations into the epidemiology, pathogenesis, and prognostic factors of epidermolysis bullosa (EB) both difficult and much needed.
The latest consensus report on the classification of CEB is from 2014 and was preceded by three earlier reports from 1991, 2000 and 2008.4–6 The relatively frequent modifications of the definitions illustrate uncertainties regarding the classification of this rare genetic disease.
A rare disease is defined by the European Union as a “life-threatening or chronically debilitating condition” with a prevalence below 1:2,000.7 The estimated prevalence of CEB ranges from 6:1,000,000 for EB simplex to less than 1:1,000,000 for junctional EB.8 These prevalence estimates are derived from single reports and may generalize poorly because of the heterogeneous nature of the diseases and the different diagnostic methodologies employed. Furthermore, the continuous changes in both the nomenclature and the subdivision of EB will impede the strength of epidemiological studies merely based on extraction and extrapolation of the few existing studies.
It is enticing to use population-based healthcare databases, such as those developed and broadly utilized in Denmark, as a cost-effective way to conduct epidemiological research on EB. With the whole population as a cohort and an uncensored inclusion process, the Danish registries provide the potential to obtain precise and unbiased epidemiological estimates.9 As the currently implemented diagnosis coding system (the tenth revision of the ICD) was developed in the late 1980s, epidemiological statements and surveillance based on the ICD-10 may lack precision compared to current diagnostic consensus. Thus, an assessment of validity of codes for these diseases in the Danish registries is essential. We therefore validated first-time diagnosis of CEB in two Danish patient registries – The Danish National Patient Registry (DNPR) and The Danish Pathology Registry (DPR) – using a review of medical records as reference standard.
Methods
Data sources
The Danish National Health Service provides tax-supported health care for all Danish residents. This facilitates free and unrestricted access to hospitals and treatment. Since 1968, each person living in Denmark has been given a unique 10-digit Civil Personal Registration (CPR) number by the Danish Civil Registration System.10 The CPR number allows accurate and individualized registrations of visits and diseases in various Danish registries. In this study, we used the DNPR and the DPR to identify all patients with a first-time diagnosis of CEB and validated the diagnoses based on information recorded in each patient’s individual medical record.
Danish National Patient Registry
The DNPR contains information on patient contacts to all Danish hospitals, including hospital admissions since 1977 and visits to all emergency rooms and outpatient hospital clinics since 1994.10 Each record includes, among other things, administrative measures, such as date and place of admission and discharge, along with coding of the primary reason for contact (primary diagnoses) and any conditions that contributed to the contact (secondary diagnoses). Diagnoses are recorded at discharge by a medical doctor, using the eighth revision (ICD-8) until the end of 1993 and the Danish version of the ICD-10 thereafter.
Danish Pathology Registry
Since 1997, all Danish Departments of Pathology have been legally obliged to report results from histopathological investigations to the DPR. The DPR was established to supplement registration of diseases in other registries (eg, DNPR) and contains information related to patient diagnosis and treatment from both hospitals and the primary sector. Registration is performed by the investigating pathologist and is based on the Danish version of the Systematized Nomenclature of Medicine (SNOMED).11
Medical records
We searched medical records and extracted relevant information on clinical descriptions, family histories, and results from histopathology and molecular genetic tests. We collected and managed data using the Research Electronic Data Capture (REDCap) tool hosted at Aarhus University. REDCap is a secure, web-based application designed to support data capture for research studies.12
Data collection and validation
We identified all patients registered with a first-time diagnosis of EB in the DNPR (ICD-8 code 757.23; ICD-10: Q81) or in the DPR (SNOMED-code S36300) during January 1, 1977, until December 31, 2015. The data extraction was specified to also include ongoing contacts.
We validated the diagnoses against data collected from the medical records of patients identified from two dermatologic departments, the Dermatologic Departments at Aarhus University Hospital and at Bispebjerg Hospital, and one regional hospital, Regional Hospital West Jutland. We based first level of confirmation on clinical descriptions to exclude misdiagnoses and/or misclassifications. Histological evaluation of level of separation served as a relevant paraclinical finding. We defined positive family history as one or more similar or confirmed cases in the family. The required clinical findings for first level of confirmation of diagnosis were as follows: history of fragile skin with bullae; characteristic localizations; and age of onset before adulthood. Family history and relevant paraclinical findings provided confirmation of higher diagnostic specificity, with molecular genetic confirmation as the ultimate level. Consequently, we classified identified cases as rejected or probable, as specified in Table 1. As a subgroup of probable cases, we further identified those with the highest level of diagnostic certainty, including presence of histological, familial, or genetic evidence. We used other relevant clinical findings for subclassifying the conditions, including involvement of other epithelial surfaces than the skin; relative severity; involvement of teeth and/or gingiva; and other significant specific manifestations (Table S1).
 | Table 1 Criteria used for validating diagnoses of congenital epidermolysis bullosa |
One author, MHK, reviewed the medical records, extracted the relevant data, and classified diagnoses as described earlier. In cases where available material gave reason for doubt (ie, wording or other causes), MS or UK (both with broad expertise in diagnosis and treatment of patients with genodermatoses) were consulted.
Statistical analyses
We estimated the validity of first-time CEB diagnoses in the DNPR and the DPR using the positive predictive value (PPV). The PPV was defined as the proportion of patients with a probable or confirmed diagnosis of CEB of those identified as CEB through the aforementioned registries. We computed 95% CIs using the Wilson’s method for groups consisting of 40 patients or more and the Jeffrey’s method for groups consisting of less than 40 patients.13
We computed PPVs for the total sample and according to the diagnosis coding systems. Additionally, we stratified the PPV for ICD-10 diagnosis codes by the type of department (specialized or regional), calendar year (before 2001, 2001–2008, and 2009–2015), age at diagnosis (<1 year, 1–5 years, 6–15 years, 16–64 years, and >64 years), sex, and by type of diagnosis (primary or secondary) and contact (admitted or outpatient hospital clinic).
We performed statistical analyses using Stata software (version 14.2; StataCorp LP, College Station, TX, USA).
Permits
The study was approved by the Danish Data Protection Agency (record number: 2013-58-0026; case number: 1-16-02-668-15). The collection of data was approved by the National Board of Health (case number: 3-3013-1606/1). According to Danish legalization, written informed consent was not required. All investigations were carried out in accordance with the principles of the Declaration of Helsinki.
Results
We identified 618 patients diagnosed with EB in the DNPR (n=512) and the DPR (n=106) during the study period. Among them, 116 patients (18.8%) were registered at one of three departments selected for validation (Figure 1). Medical records were available for 96 patients (82.8%): 90.9% at Aarhus Dermatological Department, 69.0% at Bispebjerg Dermatological Department, and 100% at Regional Hospital West Jutland. We were able to retrieve as much as 100% of the ICD-10 identified records for the period of 2009–2015 (Table S2). For all patients with medical records available for review, the median age at first-time diagnosis was 23 years (IQR 5–40 years) and 42.7% were female (Table 2). The total study population had higher median age at diagnosis and a higher proportion of females (Table 2). For the ICD-10-coded validation sample, we found similar distribution of characteristics for those with nonmissing and missing medical records (Table S3).
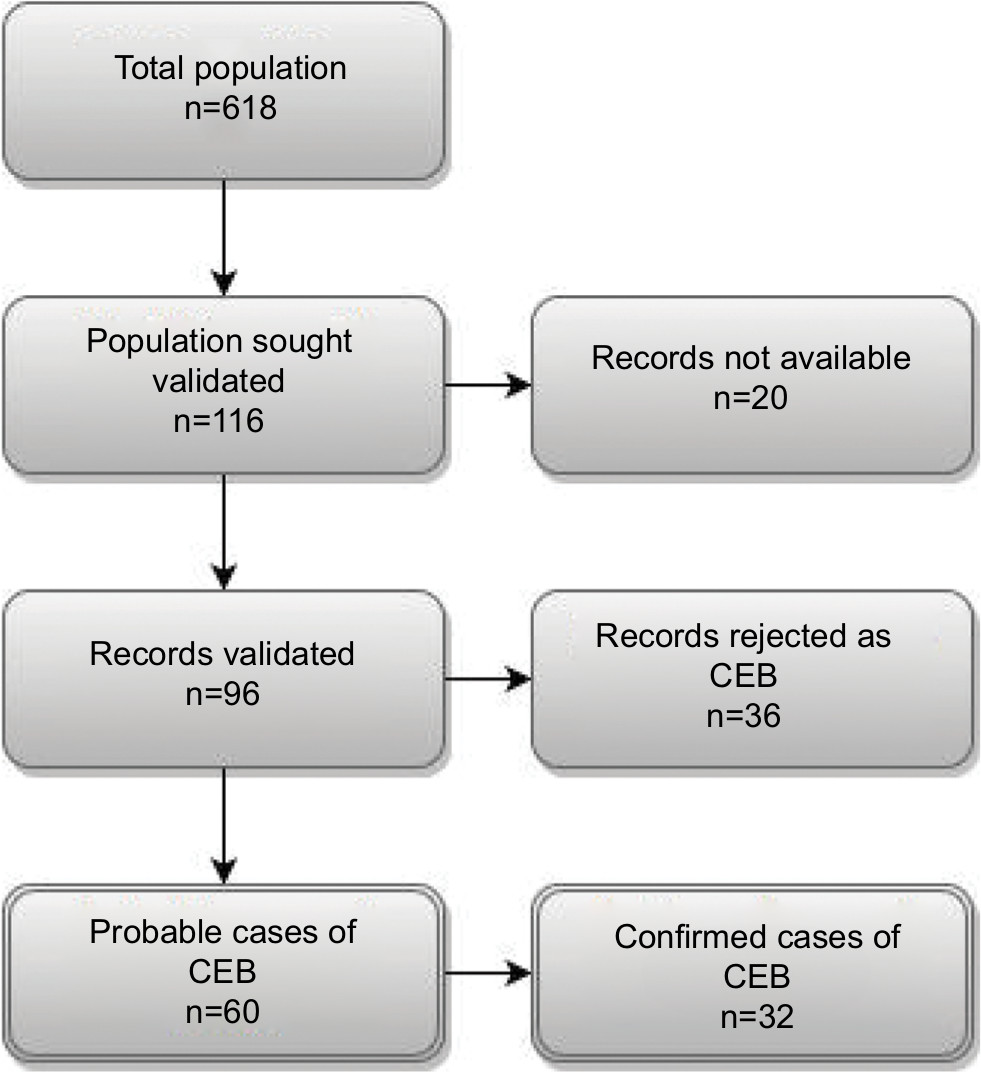 | Figure 1 Flowchart illustrating the validation process of the CEB diagnoses. Abbreviation: CEB, congenital epidermolysis bullosa. |
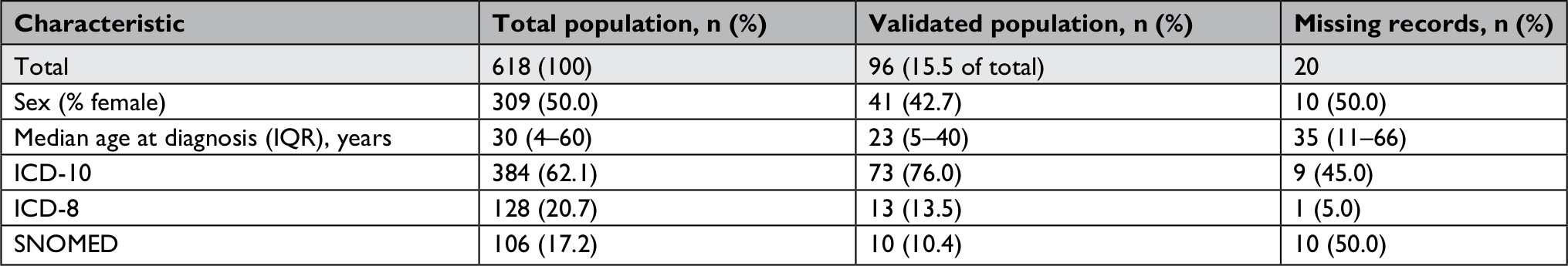 | Table 2 Demographics and distribution of the total study population, the validation sample, and missing records Abbreviation: SNOMED, Systematized Nomenclature of Medicine. |
In the validation sample, we classified 36 diagnoses (38%) as rejected: 22 (23%) represented other diagnoses than EB, 13 (14%) had insufficient evidence to satisfy validation, and in one case (1%), available information was too scarce to suggest any diagnosis. Of those classified as other diagnoses, 16 were acquired EB, one was a blister in a factitious disorder, one was toxic epidermal necrolysis, one was x-linked ichthyosis, one was neurofibromatosis, one was localized epidermolytic rash, and one was a traumatic blister. Among records where available data were insufficient to suggest other diagnoses or to satisfy validation, 12 had evidence suggesting CEB but record information did not fulfill the listed criteria for probable CEB (Table 1) and one had clinical evidence suggesting cutaneous vasculitis.
This medical record review yielded an overall PPV for CEB of 62.5% (95% CI: 52.5–71.5), including probable and confirmed diagnoses (Table 3). The PPV decreased to 33.3% (95% CI: 24.7–43.2) when limiting to confirmed diagnoses, exclusively. When comparing the three coding systems, the PPVs for probable diagnoses were 30.8% (95% CI: 11.4–57.7) for ICD-8, 76.7% (95% CI: 65.8–84.9) for ICD-10, and 0.0% (95% CI: 0.0–21.7) for SNOMED. For confirmed diagnoses, the corresponding values were 15.4% (95% CI: 3.3–40.9), 41.1% (95% CI: 30.5–52.6), and 0.0% (95% CI: 0.0–21.7).
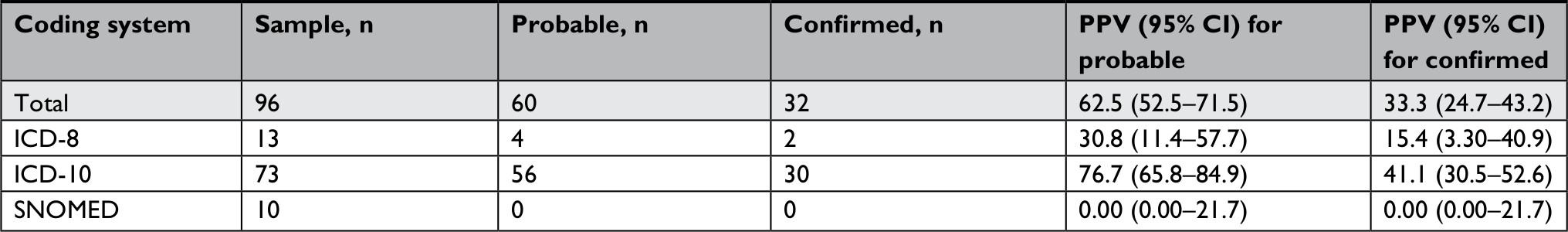 | Table 3 PPV for the coding of epidermolysis bullosa in the DNPR and the DPR Abbreviations: DNPR, Danish national Patient Registry; DPR, Danish Pathology registry; PPV, positive predictive value; SNOMED, Systematized Nomenclature of Medicine. |
Results from analyses restricted to ICD-10 codes are shown in Table 3. For the ICD-10 codes, the PPVs for the probable diagnoses at specialized dermatological departments and the regional department were 78.3% (95% CI: 67.2–86.4) and 50.0% (95% CI: 12.3–87.7), respectively. For the confirmed diagnoses, the corresponding PPVs were 40.6% (95% CI: 29.8–52.4) and 50.0% (95% CI: 12.3–87.7). We found no clear variation in PPVs according to the calendar period of diagnosis. We observed comparable PPVs in subgroups of sex and age. The lowest estimates were found at the extremes of the age categories. The PPVs including probable diagnosis was 81.3% (95% CI: 70.0–88.9) for primary diagnoses and 44.4% (95% CI: 17.3–74.6) for secondary diagnoses. This difference was less pronounced for confirmed diagnoses: 42.2% (95% CI: 30.9–54.4) for primary diagnoses and 33.3% (95% CI: 10.4–65.2) for secondary diagnoses. Similarly, the PPVs for probable diagnoses were 78.6% (95% CI: 67.6–86.6) for outpatients and 33.3% (95% CI: 3.9–82.3) for inpatients. Corresponding PPVs for confirmed diagnoses were 41.4% (95% CI: 30.6–53.1) and 33.3% (95% CI: 3.9–82.3). A more detailed comparison of data from the two dermatological departments is provided in Tables S4–S6. In short, the distribution of diagnosis coding systems differed slightly at the respective departments, but the characteristics were similar overall (Table S4) and when restricting to those recorded with the ICD-10 system (Table S5). We also found comparable PPVs in subgroup analyses for the two dermatological departments (Table S6).
At the four-digit ICD-10 level, we found PPVs of 83.3% (95% CI: 67.3–93.3) for EB simplex and 100% (95% CI: 85.7–100) for dystrophic EB (Table S7). For the remaining ICD diagnoses (junctional and others), we observed PPVs of 0, as records found yielded no findings of probable during this validation.
Discussion
In this study, we investigated the validity of the coding of CEB in the DNPR and the DPR. This included the following three diagnosis coding systems: ICD-8, ICD-10, and SNOMED. We estimated the coding systems’ validity for probable diagnoses, where we required only clinical findings, and for confirmed diagnoses, requiring positive family history and/or conclusive paraclinical findings. In the daily clinical setting, diagnoses of most cases of EB are based primarily on clinical findings. Consequently, the confirmed group provides insights into how often further investigations are deemed necessary to be certain of the diagnosis, or when it judged to affect the prognosis and/or the treatment. The probable group on the other hand is the most relevant measure of the precision of the clinical diagnostic process and therefore we focus mainly on this subset subsequently.
The PPVs for ICD-10 diagnoses were higher than for ICD-8 and SNOMED and were particularly high for the specialized dermatological departments. In fact, only a small proportion of CEB patients were identified through the DPR, and we were not able to identify any as probable. This finding may be explained by avoidance in using histopathology in diagnosis of this population consisting of mainly young children and infants with a positive family history. Regardless, identifying CEB cases through the DPR is a poor choice of method. Similarly, we note that the number of patients identified in regional departments was low (n=4), limiting precision. When stratifying by age at diagnosis, PPV improved as age decreased; however, only at age above 1 year. The high PPV at low age may indicate that the more severe phenotypes, that are easier to diagnose, are seen earlier in life with few differential diagnoses. The reason for PPV dropping at age below 1 year is unknown, but may be attributed to poor recording of relevant manifestations in the medical records at the low patient age. When stratifying by calendar year, we observed subtle differences between the time periods (Table S4). At both Aarhus and Bispebjerg Dermatological Departments, we found the lowest PPV in the period from 2001 to 2008. However, while there was a trend toward higher PPVs, and thus higher diagnostic precision in the more recent time periods in Aarhus, this was not clearly the case at Bispebjerg. The reason for this is unknown. There is a noteworthy difference between the PPVs of primary and secondary diagnoses. The PPV for primary diagnoses is approximately twice as good as the secondary diagnoses (Table 4). This difference is expected as secondary diagnoses may not be confirmed or focused on during treatment of a patient. Moreover, we note that EB rarely is a secondary finding, as it was the primary reason for patient contact for 64 of the 73 validated cases. EB patients were diagnosed at an outpatient hospital clinic assuming that EB was the primary cause for the consultation.
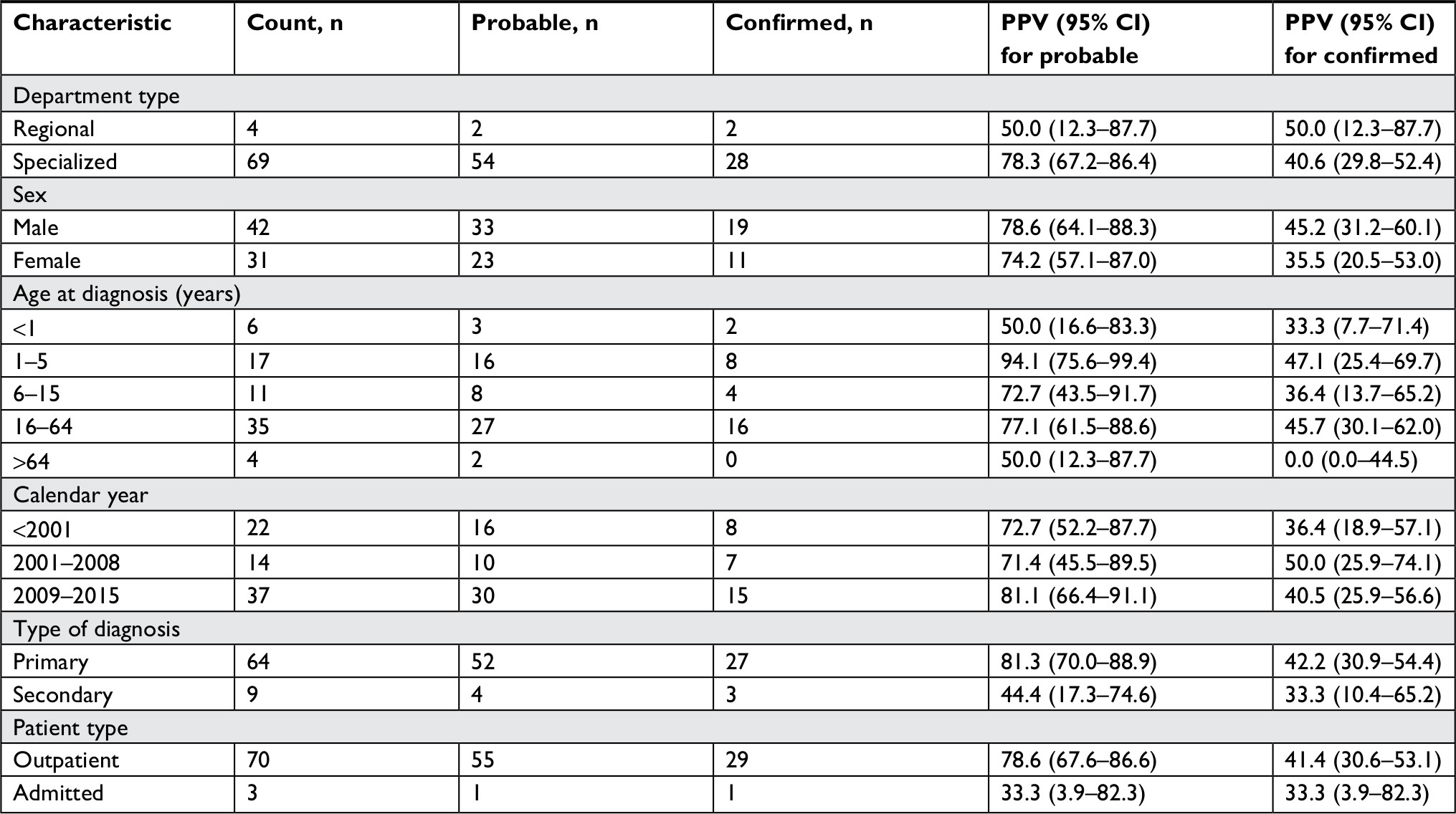 | Table 4 Results for PPV for probable and confirmed ICD-10 epidermolysis bullosa diagnoses stratified by department type, sex, age at diagnosis, calendar year, diagnosis type, and patient type Abbreviation: PPV, positive predictive value. |
A recent systematic review validating codes from the DNPR have found that PPVs herein ranges from 15% to 100%, emphasizing the need for validation of diagnoses before using data for research purposes.9 To our knowledge, the validity of EB diagnoses in the Danish registries has not been investigated, and the results from the study presented here show PPVs in the range low to medium in comparison.
There are some methodological limitations of this study that should be mentioned. First, a small proportion of medical records could not be retrieved. However, it is unlikely that the absence of records depends on factors affecting the PPV, as we observed similar characteristics for validated and missing records. When looking at the comparability between the validated population and the total population, the demographics and diagnosis system distribution differed to some extent. The validated population consisted of a larger proportion of ICD-10 diagnoses, a lower proportion of females, and a lower median age at diagnosis. This may be explained by the predominance of dermatological departments, where more severe cases are seen, in the validation sample. In general, it is questionable what can be inferred about diagnoses from regional departments, as the number of cases validated here was quite low. Our overall findings are most likely not generalizable to non-specialized departments. Second, we included records that had insufficient evidence to satisfy validation or where available material was too scarce to suggest another diagnosis, which probably results in an underestimation of PPVs. Conversely, as the clinical manifestations differ widely both within and between diagnoses, our criteria for “probable” diagnoses needed to be quite inclusive, which would result in an overestimation of PPVs. We sought to minimize this bias by identifying the most common misdiagnosis (acquired forms) and, as aforementioned, not excluding records with unsatisfactory data. Third, records were evaluated by only one person. Fourth, the study sample reflects the rarity of CEB and limits the study conclusions that can be drawn from in subgroup analyses. In particular, the specific diagnoses are so rare that interpretation and generalizability must be done with caution. Finally, we were not able to estimate completeness of diagnoses. It is possible that cases with milder disease manifestations, that are inherited dominantly, are never seen at a hospital because the condition and its treatment are known in the family and is manageable by the general practitioner. Validity of results of four-digit ICD-10 level diagnoses is limited to the more common forms identified, which are EB simplex and dystrophic EB, leaving junctional EB and the nonspecified group with results of low validity.
The strength of this study relies on the ability to identify patients nationwide from a population-based cohort using the Danish CPR number, thus avoiding selection bias of the primary data. Also, we were able to retrieve most medical records of the identified patients, yielding a validation sample of 16% of the total population. We focused on validating diagnoses from dermatological departments, because insights into the validity of diagnoses from dermatological departments are particularly important, eg, when planning future observational studies or clinical trials, which will typically be based in a setting where EB patients are diagnosed and followed. The registration of diagnosis at Danish hospitals is performed manually and often involves only one or a few doctors, which makes the systems susceptible to miscoding. This point may explain why many registrations were either acquired forms or completely unrelated diagnoses having ICD-10 codes that were numerically related to the ones investigated. A method for rooting out misclassifications is to require two or more independent registrations of the diagnosis of interest. Such an approach was used in a study investigating acromegaly, which showed an increase in PPV although this came at the expense of the total number of confirmed cases.14
The DNPR and the DPR are broad administrative and research registries comprising any condition leading to hospital contact or pathological examination, respectively. They are invaluable data sources for research, and high validity has been reported for many common diseases.9 However, as suggested by this study, the registries may be suboptimal for studying conditions, such as CEB, which is rare and diagnostically challenging for physicians without experience in recognizing and treating genodermatoses. Furthermore, relevant clinical and paraclinical details, eg, manifestations, severity, family history, and identified mutations, are not recorded in the general registries. These limitations underscore the need for a national disease-specific registry for CEB, such as The International Registry of Dystrophic Epidermolysis Bullosa Patients and Associated COL7A1 Mutations15and The National Epidermolysis Bullosa Registry,16 with validated diagnoses from specialized departments and requirements of details. This work has already been initiated with the recent establishment of The Danish Database for Genodermatoses,17 which has been driven in part by this work.
Conclusion
This study found that PPVs for diagnoses of CEB in the DNPR and DPR ranged from very low for ICD-8 and SNOMED codes to average for ICD-10 codes. Furthermore, when restricting to ICD-10 codes, the validity of the diagnoses was much higher for diagnoses from dermatological departments compared with a regional department. Consequently, CEB diagnoses identified through the Danish national health registries should be used with caution and if used should be restricted to ICD-10 codes from specialized dermatological departments.
Acknowledgments
Doctors and secretaries at the departments participating in the validation study are thanked for their cooperation and assistance in retrieving the medical records. This work was supported by research grants from Aarhus University’s Faculty of Medicine, Aage Bangs Foundation, and the Danish Society of Dermatology. The financial support given by these institutions is greatly appreciated.
Disclosure
The authors report no conflicts of interest in this work.
References
Leech SN, Moss C. A current and online genodermatosis database. Br J Dermatol. 2007;156(6):1115–1148. | ||
Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12. | ||
Hsu CK, Wang SP, Lee JY, Mcgrath JA. Treatment of hereditary epidermolysis bullosa: updates and future prospects. Am J Clin Dermatol. 2014;15(1):1–6. | ||
Fine JD, Bauer EA, Briggaman RA, et al. Revised clinical and laboratory criteria for subtypes of inherited epidermolysis bullosa. A consensus report by the Subcommittee on Diagnosis and Classification of the National Epidermolysis Bullosa Registry. J Am Acad Dermatol. 1991;24(1):119–135. | ||
Fine JD, Eady RA, Bauer EA, et al. Revised classification system for inherited epidermolysis bullosa: report of the second international consensus meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42(6):1051–1066. | ||
Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the third international consensus meeting on diagnosis and classification of EB. J Am Acad Dermatol. 2008;58(6):931–950. | ||
Baldovino S, Moliner AM, Taruscio D, Daina E, Roccatello D. Rare diseases in Europe: from a wide to a local perspective. Isr Med Assoc J. 2016;18(6):359–363. | ||
Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016;152(11):1231. | ||
Schmidt M, Schmidt SA, Sandegaard JL, Ehrenstein V, Pedersen L, Sørensen HT. The Danish National Patient Registry: a review of content, data quality, and research potential. Clin Epidemiol. 2015;7:449–490. | ||
Pedersen CB, Gøtzsche H, Møller JO, Mortensen PB. The Danish civil registration system. a cohort of eight million persons. Dan Med Bull. 2006;53(4):441–449. | ||
Bjerregaard B, Larsen OB. The Danish pathology register. Scand J Public Health. 2011;39(7):72–74. | ||
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap): a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. | ||
Brown LD, Cai TT, Dasgupta A. Interval estimation for a binomial proportion. Statistical Science. 2001;16(2):101–117. | ||
Dal J, Skou N, Nielsen EH, Jørgensen JO, Pedersen L. Acromegaly according to the Danish National Registry of Patients: how valid are ICD diagnoses and how do patterns of registration affect the accuracy of registry data? Clin Epidemiol. 2014;6:295–299. | ||
van den Akker PC, Jonkman MF, Rengaw T, et al. The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum Mutat. 2011;32(10):1100–1107. | ||
Fine JD, Johnson LB, Suchindran CM. The National Epidermolysis Bullosa Registry. J Invest Dermatol. 1994;102(6):54S–56S. | ||
Dorf IL, Sommerlund M, Skytte AB, Koppelhus U. Dyskeratosis follicularis. Ugeskr Laeger. 2018;180(19). |
Supplementary materials
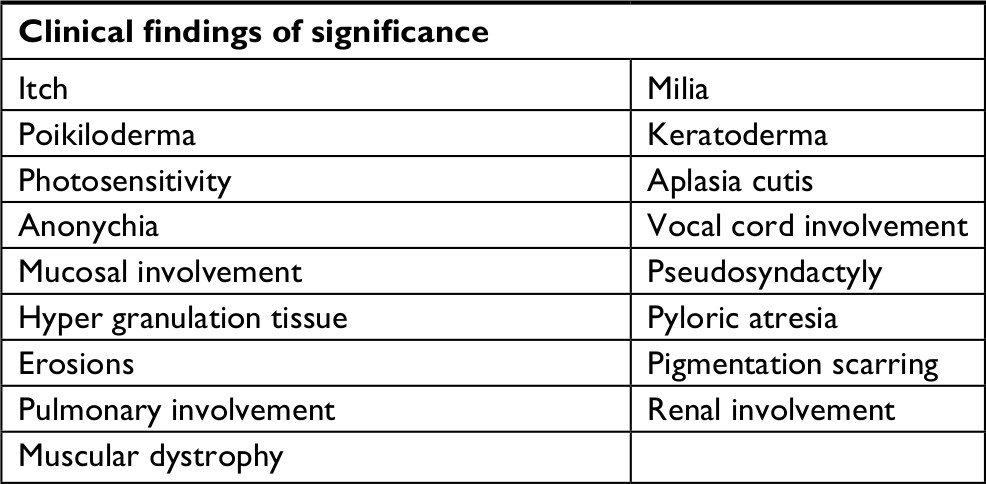 | Table S1 List of specific clinical symptoms and findings related to and indicating specific subtype of congenital epidermolysis bullosa Notes: The list was chosen as representative presentation for the wide range of specific subtypes of congenital epidermolysis bullosa by review of the latest consensus report. No single item on the list is pathognomonic but must be seen in correlation to other clinical and paraclinical findings.1 All patients were required to have “bullae” mentioned in the medical record to be considered probable or confirmed. Data from Fine JD, Bruckner-Tuderman L, Eady RA, et al.1 |
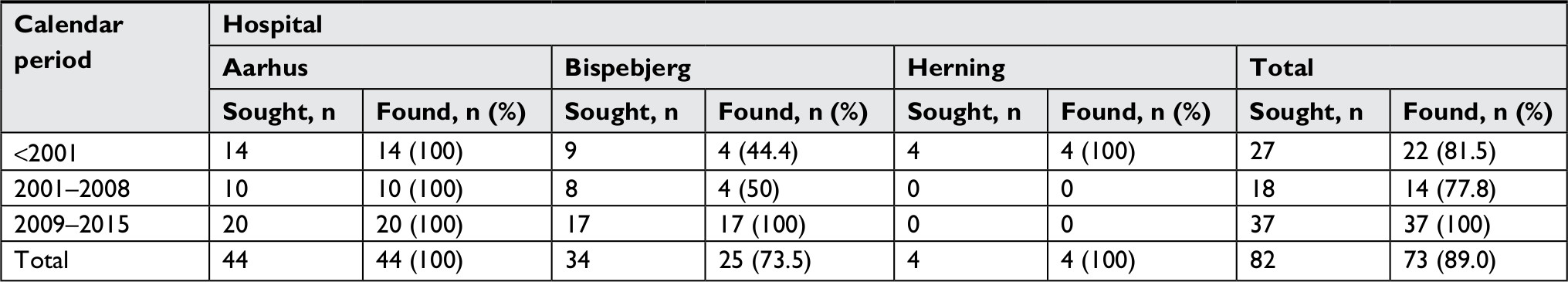 | Table S2 Proportion of patients identified by ICD-10 codes who had medical records available for validation, overall, and by hospital and calendar period of diagnosis |
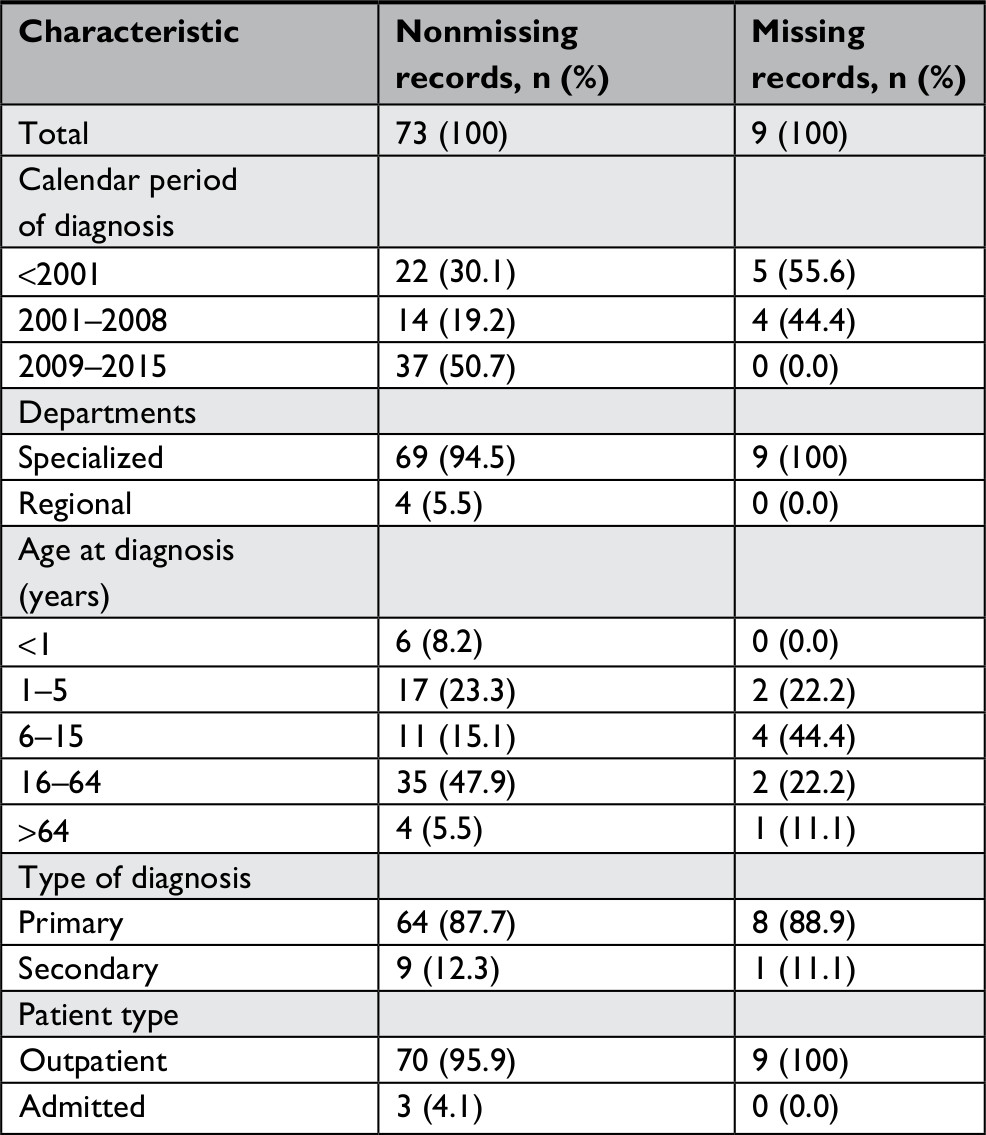 | Table S3 Comparison of characteristics of nonmissing records and missing records for those from the validations sample, ICD-10 diagnoses only |
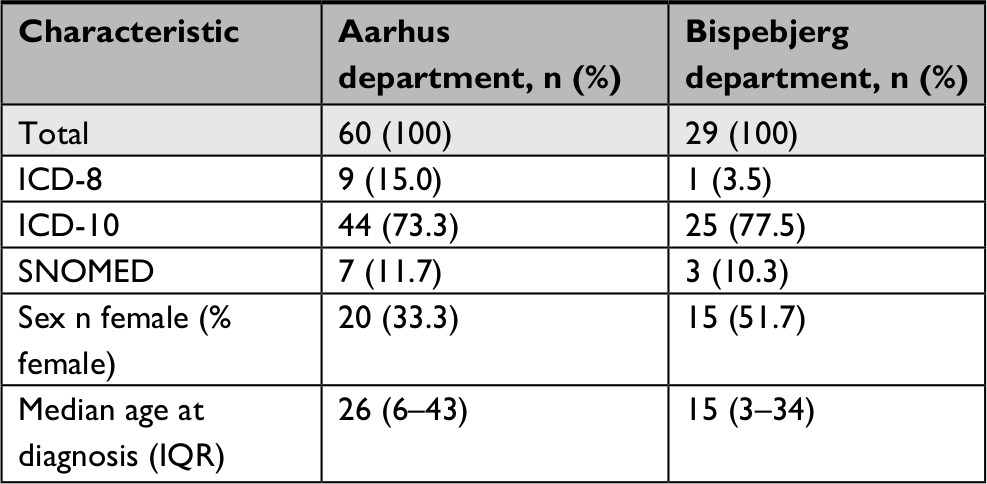 | Table S4 Demographic characteristics between validated population from Aarhus and Bispebjerg Dermatological Departments and the distribution of the diagnosis code systems (ICD-8, ICD-10, and SNOMED), sex, and median age at diagnosis Abbreviation: SNOMED, Systematized Nomenclature of Medicine. |
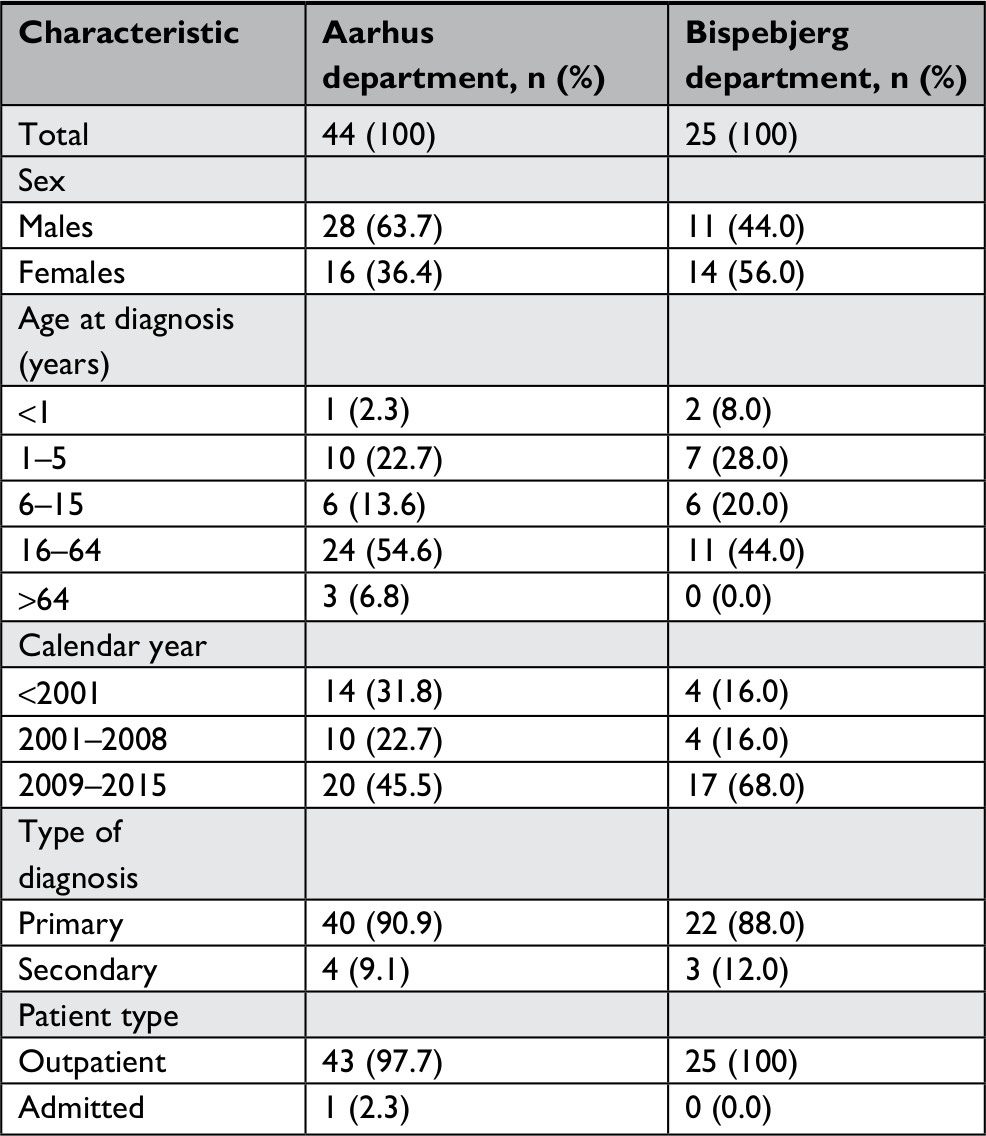 | Table S5 Demographic and descriptive parameters of the ICD-10 coded populations of the two specialized dermatological departments |
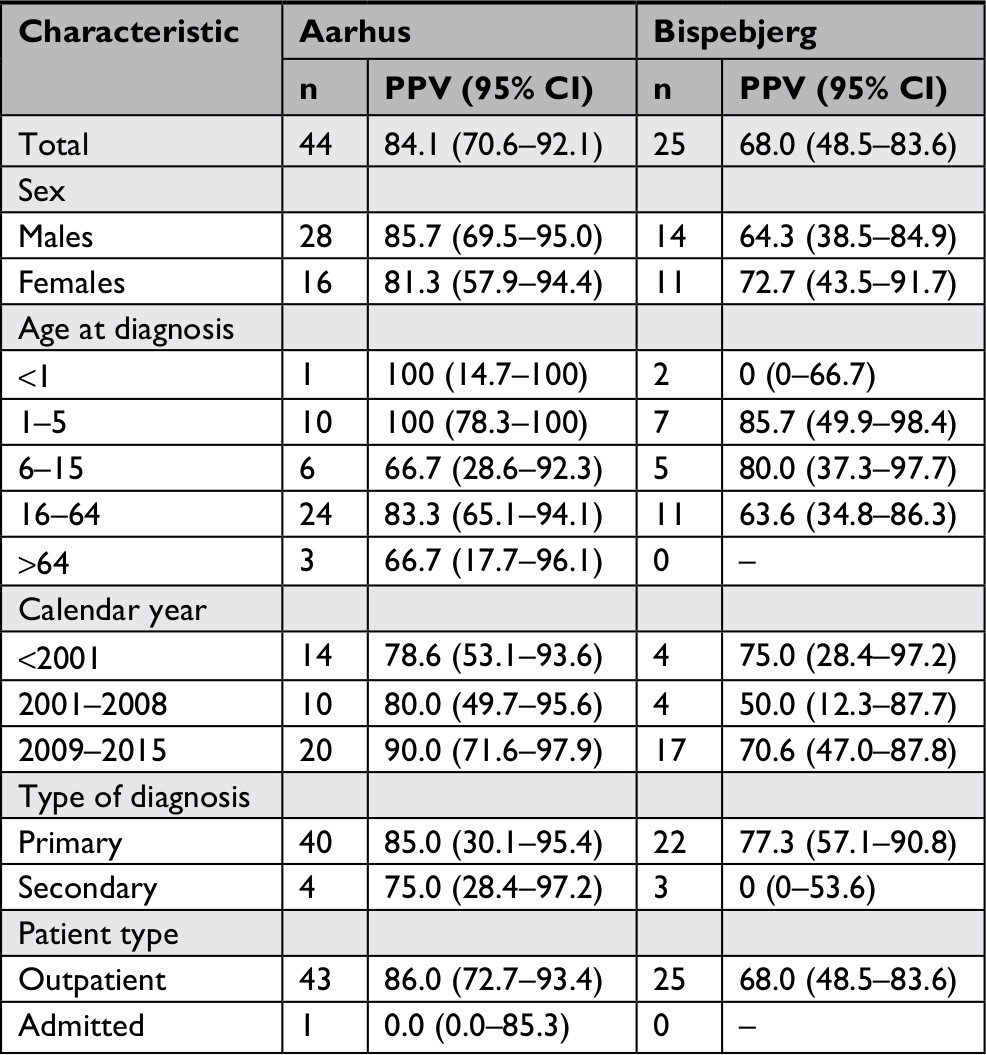 | Table S6 Comparing the PPV of the two dermatological departments in the presented subgroups Abbreviation: PPV, positive predictive value. |
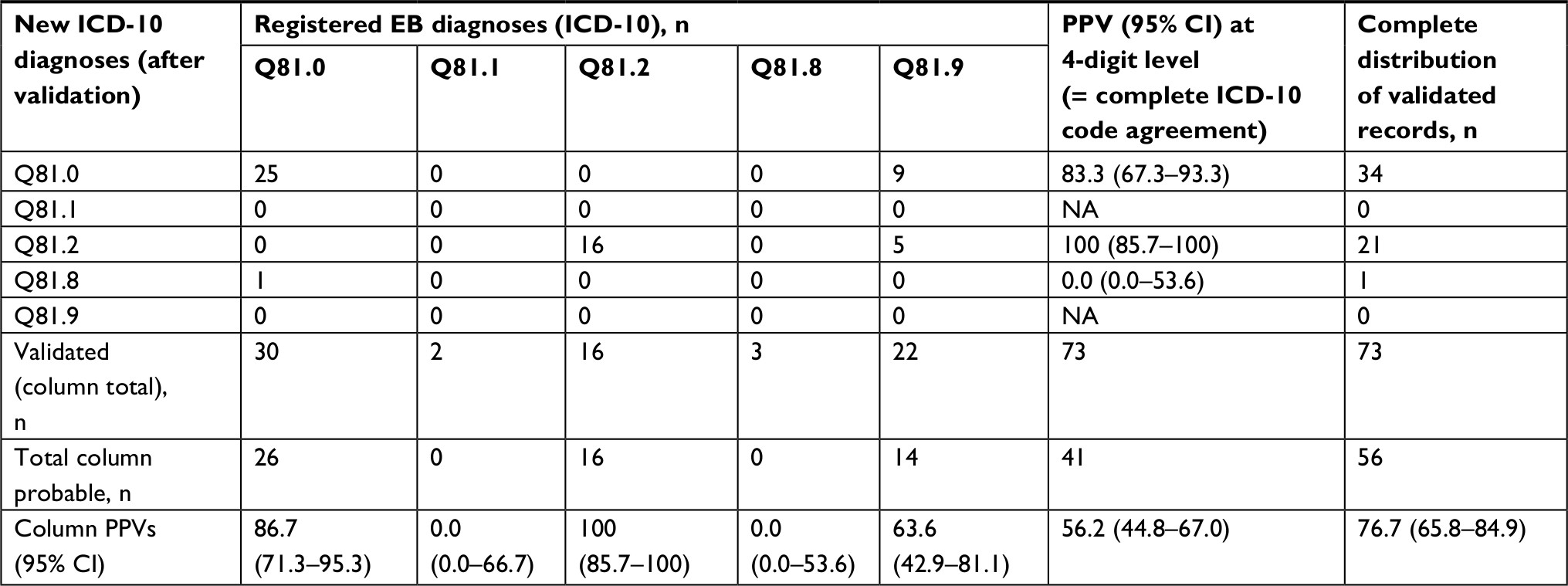 | Table S7 Distribution of specific ICD-10 EB diagnoses classified as probable against the new ICD-10 diagnosis based on validation including the PPV for each Abbreviations: EB, epidermolysis bullosa; PPV, positive predictive value; Q81.0, epidermolysis bullosa simplex; Q81.1, epidermolysis bullosa letalis; Q81.2, epidermolysis bullosa dystrophica; Q81.8, other epidermolysis bullosa; Q81.9, epidermolysis bullosa unspecified. |
Reference
Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–1126. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.