")
Back to Journals » Clinical Ophthalmology » Volume 11
Uveal melanoma: epidemiology, etiology, and treatment of primary disease
Authors Krantz BA, Dave N, Komatsubara KM, Marr BP, Carvajal RD
Received 30 August 2016
Accepted for publication 22 November 2016
Published 31 January 2017 Volume 2017:11 Pages 279—289
DOI https://doi.org/10.2147/OPTH.S89591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Benjamin A Krantz,1 Nikita Dave,2 Kimberly M Komatsubara,2 Brian P Marr,3,4 Richard D Carvajal5
1Division of Hospital Medicine, 2Division of Hematology/Oncology, Columbia University Medical Center, 3Ophthalmic Oncology Service, Memorial Sloan Kettering Cancer Center, 4Department of Ophthalmology, Weill Cornell Medical College, 5Division of Hematology/Oncology, Columbia University Medical Center, New York, NY, USA
Abstract: Uveal melanoma (UM) is the most common intraocular malignancy and arises from melanocytes in the iris, ciliary body, or choroid. Early diagnosis and local treatment is crucial, as survival correlates with primary tumor size. However, approximately 50% of patients will develop metastatic disease with 6–12 months’ survival from metastatic diagnosis. Genomic analyses have led to the development of gene-expression profiles that effectively predict metastatic progression; unfortunately, no adjuvant therapy has been shown to prolong survival to date. New insights into the molecular biology of UM have found frequent activating mutations in genes encoding for the G-protein α-subunit, GNAQ and GNA11, and improved understanding of the downstream signaling pathways MAPK, PI3K/Akt, and Hippo have afforded an array of new targets for treatment of this disease. Studies are under way with rationally developed regimens targeting these pathways, and novel agents are under development. We review the diagnosis, management, and surveillance of primary UM and the adjuvant therapy trials under way.
Keywords: uveal melanoma, ocular melanoma, GNAQ, GNA11, MAP kinase, MEK
Introduction
Uveal melanoma (UM) is a rare malignancy that arises from melanocytes within the uveal tract of the eye. Although UM is often diagnosed at an early stage, local treatment modalities come with significant visual morbidity and metastatic progression is not uncommon, portending an extremely poor prognosis. Much has been learned about the pathophysiology of UM, but despite these advances improvements in overall survival have not been achieved. Only recently have novel therapeutics emerged that rationally target the known mechanisms of this disease, and a number of trials are under way attempting to alter the disease course. In this review, we focus on the diagnosis of UM, therapeutic options for local control, and the pursuit of effective adjuvant therapy.
Epidemiology, etiology, and prognosis
Epidemiology
UM is the most common primary intraocular malignancy in adults, representing 3%–5% of all melanomas.1,2 UM most commonly arises from choroidal melanocytes (85%–90%), but can also arise from the iris (3%–5%) and ciliary body (5%–8%).1,3 The median age of diagnosis is approximately 62 years; however, the peak range for diagnosis is between 70 and 79 years.1,4,5
UM incidence varies by sex, race, and country. Males have a 30% greater incidence than females.1 In the US, the incidence is approximately five per million individuals, with a significantly higher incidence in non-Hispanic whites (6.02 per million) when compared with blacks and Asians (0.31 and 0.39 per million, respectively). Incidence in Hispanics is in the middle, at 1.67 per million.1,6,7 In Europe, incidence increases with latitude, ranging from two per million in Spain and Italy, four to six per million in Central Europe, and greater than eight per million in Denmark and Norway.8 The incidence in South Korea is similar to Asians in the US, at 0.42 per million.9
Risk factors
A variety of risk factors have been identified, including the presence of light eyes, fair skin, an inability to tan, ocular melanocytosis, dysplastic nevus syndrome, and BAP1 mutations.10–13 BAP1 is a tumor-suppressor gene located on chromosome 3 that is mutated in 47% of UM.12 UM in BAP1 germ-line mutants is usually diagnosed between the ages of 30 and 59 years, and is driven by inactivating mutations in the lone functional BAP1 gene, analogous to the frequent loss of chromosome 3 observed in high-risk sporadic disease.14 Interestingly, patients with UM have an 11% higher risk of second malignancies, such as cutaneous melanoma and renal cell carcinoma, than the general population, which may be driven by the presence of germ-line BAP1 mutations.
The role of sun exposure as a risk factor for UM is unclear. Tucker et al found that time spent outdoors, tanning, and rare use of ocular sun protection were risk factors for the development of UM.15 A subsequent meta-analysis found that chronic ultraviolet-light exposure and geographic latitude were not associated with disease, but identified welding as a risk factor.16 Interestingly, a recent study found that posterior choroidal melanomas occurring in illuminated areas were associated with frequent adenine-to-cytosine mutations, whereas ciliochoroidal melanoma arising from unilluminated areas are associated with frequent adenine-to-thymine mutations and light eye color. This suggests both light eye color and sunlight may be independent risk factors associated with different anatomic and mutation profiles.17
Presentation and diagnosis
The most common presenting symptom in those with primary UM is blurred vision (37.8%); however, many patients are asymptomatic at the time of diagnosis (30.2%). Other common symptoms at presentation include photopsia (8.6%), floaters (7%), visual field loss (6.1%), visible tumor (3.1%), pain (2.4%), and metamorphopsia (2.2%).4
Differentiating UM from benign pigmented nevi can be challenging, and management is complicated by the low number of nevi that transform into UM (one in 8,000). Factors associated with an increased risk include thickness greater than 2 mm, subretinal fluid, symptoms, orange pigment, tumor margin less than 3 mm to disk, ultrasonographic hollowness, and lack of surrounding halo.18 Other diagnoses to be considered when assessing lesions concerning for UM are dependent upon location (Table 1). In a series of 200 patients referred for evaluation of iris lesions concerning for melanoma, 24% were confirmed to have UM: 38% were diagnosed with primary iris cysts, 31% with iris nevi, 5.7% with essential iris atrophy, 4.7% with foreign bodies, 2.5% with peripheral anterior synechia, and 2.5% with secondary metastases.19 Subsequently, in a series of 400 consecutive referrals for evaluation for posterior UM performed by the same group, 26.5% were diagnosed with choroidal nevi, 23.5% with disciform degeneration, 9.5% with retinal pigment-epithelium hypertrophy, and 8% with hemangiomas.20
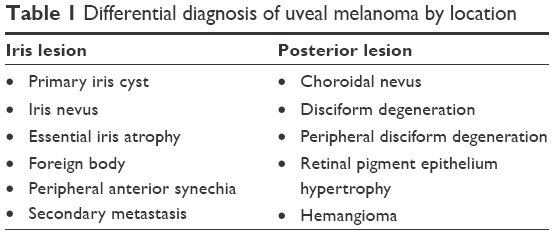 | Table 1 Differential diagnosis of uveal melanoma by location |
Despite the broad differentials, data from the practice-changing Collaborative Ocular Melanoma Study (COMS) found a clinical misdiagnosis rate of only 0.48%, suggesting that the majority of intraocular tumors can be diagnosed on the basis of clinical examination alone.21 COMS, however, had strict inclusion and exclusion criteria, and subsequent studies have estimated a need for diagnostic fine-needle aspiration biopsy ranging from 1% to 9% of cases.22,23 Specialized ocular imaging, including ultrasound and fluorescein angiography, are often performed for further characterization. Fine-needle aspiration biopsy is becoming used more routinely at the time of diagnosis, due to advancements in the use of cytogenetic analysis and gene-expression profiling for prognostication.24
Prognosis
Despite excellent rates of local disease control with surgery or radiotherapy, up to 50% of patients will ultimately develop metastatic disease, with the most common initial sites being the liver (60.5%), the lung (24.4%), skin/soft tissue (10.9%), and bone (8.4%).25 The overall rate of survival from initial diagnosis is 69% at 5 years, 55% at 15 years, and 51% at 25 years; however, following the development of metastatic disease, median overall survival is approximately 13.4 months, with only 8% surviving 2 years.26–28
Various predictors of metastatic disease and survival have been studied, including clinical staging systems, gene-cytogenetic findings, and gene-expression panels. The American Joint Committee on Cancer (AJCC) UM T1–T4 staging system is based on tumor characteristics of base diameter, thickness, ciliary body involvement, and distance of extraocular extension. Survival decreases rapidly with increasing stage. Estimates of death at 5, 10, and 20 years are 4%, 8%, and 11% for T1, 8%, 13%, and 24% for T2, 19%, 27%, and 36% for T3, and 30%, 43%, and 51% for T4 lesions, respectively.29 When compared with AJCC stage I disease, the rate of metastasis/death is three times greater for stage II and nine to ten times greater for stage III disease.30
Recurrent cytogenetic alterations have provided an additional prognostic method. The most commonly studied abnormalities are the presence of 8q rearrangements and chromosome 3 loss, both of which are associated with a worse prognosis. Copy-number alterations in 8q are identified in up to 79% of UM. Those with a normal 8q copy number have 93% 5-year survival, while those with one copy gain have 67% 5-year survival and amplification of more than one copy have 29% 5-year survival.31 Monosomy 3 is present in 65% of UM and associated with 5-year survival of 37%, while those with no chromosome 3 alterations have 5-year survival of 90%. Monosomy 3 is often present concurrently with 8q rearrangements, with the combination associated with a particularly poor prognosis.31 Additional cytogenetic alterations associated with greater risk of development of distant recurrence include 8p loss, 6q loss, and 1p loss.32 Cytogenetic testing is commercially available from such companies as Impact Genetics.
Gene-expression profiling is emerging as a novel prognostic method that predicts metastatic risk with greater accuracy than tumor size or thickness.33 A commercially available gene-expression panel from Castle Biosciences utilizes a 15-gene assay to separate UM into two classes, with class 1 UM having low metastatic potential and class 2 UM having high metastatic potential. Onken et al initially validated this assay in primary posterior UM, where class 1 disease had a metastatic rate of 1% and class 2 25.9%.33 Recently, PRAME has been reported to be an independent biomarker for UM, and when combined with a 12-gene expression panel, PRAME messenger-RNA expression predicted a 5-year metastatic rate of 0 in class 1/PRAME−, 38% in class 1/PRAME+, and 71% in class 2.34 Further evaluation of the role of PRAME expression in UM is ongoing.
Pathophysiology
Unlike cutaneous melanoma, UM is genetically characterized by a small number of alterations; however, several of these alterations have been well characterized and been found to alter intracellular signaling, surface-receptor expression, and ligand production. While cutaneous melanomas are driven by MAPK activation through mutations in BRAF (~50% of cases), NRAS (10%–25% of cases), or loss of function in NF1 (14% of cases), UM rarely harbors such alterations and rather is characterized by point mutations in the G-protein α-subunit.35 GNAQ and GNA11 are genes that code for the α-subunit of G proteins that act in conjunction with G-protein-coupled receptors. G protein and G-protein-coupled receptor-signal transduction takes place through the conversion of GDP to GTP, which activates the G protein and signals via downstream effector proteins. In order for the G protein to return to its inactive state, GTP must be hydrolyzed to GDP. It has been demonstrated that glutamine at the 209 position is required for GTPase activity, and mutations that disrupt this activity cause a constitutively active GTP-bound state. This is similar in mechanism to the better-known RAS oncogenes that code for monomeric G proteins, which are commonly constitutively or inappropriately activated in many malignancies.36
Van Raamsdonk et al characterized a mutually exclusive mutation pattern affecting GNAQ/GNA11 in 83% of UM samples.37,38 Further analyses have shown GNAQ/GNA11-mutation rates as high as 96% in metastatic UM samples.38–40 Just as there are ethnic variations in UM incidence, there may also be ethnic differences in mutational landscape. Xu et al found a much lower GNAQ/GNA11-mutation burden of 38% in a small Chinese UM population sample.41 Recently, additional mutations that are mutually exclusive to GNAQ/GNA11 but work along similar pathways have been identified. PLCB4 is a downstream effector of GNAQ/GNA11 that has been found to be mutated in three of 28 UM samples without GNAQ/GNA11 mutations.42 Furthermore, recurrent activating mutations in the G-protein-coupled receptor CYSLTR2 have been found in UM without GNAQ/GNA11 or PCLB4 mutations.43 These less frequent mutations reaffirm the importance of the Gα-signaling pathway in this disease.
The effects of constitutively active Gα include activation of the MAPK, PI3K–Akt–mTOR, and Hippo pathways (Figure 1).44 The MAPK pathway contributes to the regulation of cell proliferation and apoptosis. Mutant GNAQ/GNA11 activates phospholipase C, which cleaves phosphatidylinositol bisphosphate into inositol trisphosphate and diacylglycerol that subsequently activates PKC.45,46 PKC begins a cascade of phosphorylation of Raf, MEK1/2, and ERK, which translocates to the nucleus to regulate cell proliferation and survival.47,48 The PI3K–Akt–mTOR pathway is a key pathway for growth and homeostasis, and may act as a parallel-growth pathway to MAPK in UM. The PI3–Akt–mTOR pathway is upregulated in UM, but does not appear to drive growth on its own.49 Inhibition of PI3K in isolation has little effect on cell proliferation; however, upregulation of this pathway has been demonstrated with MEK inhibition.50 Enhanced cell-growth inhibition and apoptosis have been observed when combined MEK and PI3–mTOR inhibition has been studied.51,52 The Hippo pathway acts through the proteins YAP and TAZ to activate the transcription factors TEAD and SMAD.53,54 Like the MAPK pathway, it is a regulator of cell proliferation and apoptosis. Traditionally, Hippo signals through the kinases MST1/2 and Lats1/2 to phosphorylate YAP.55 Alternatively, GNAQ/GNA11 mutations have been shown to increase YAP and TAZ activation through an alternative pathway utilizing Trio, a guanine nucleotide-exchange factor, and the downstream GTPases Rho and Rac.56 These multiple pathways provide an array of targets that are being studied for therapeutic intervention. The various parallel pathways suggest a role for combination therapy, with clinical studies under way for metastatic disease.
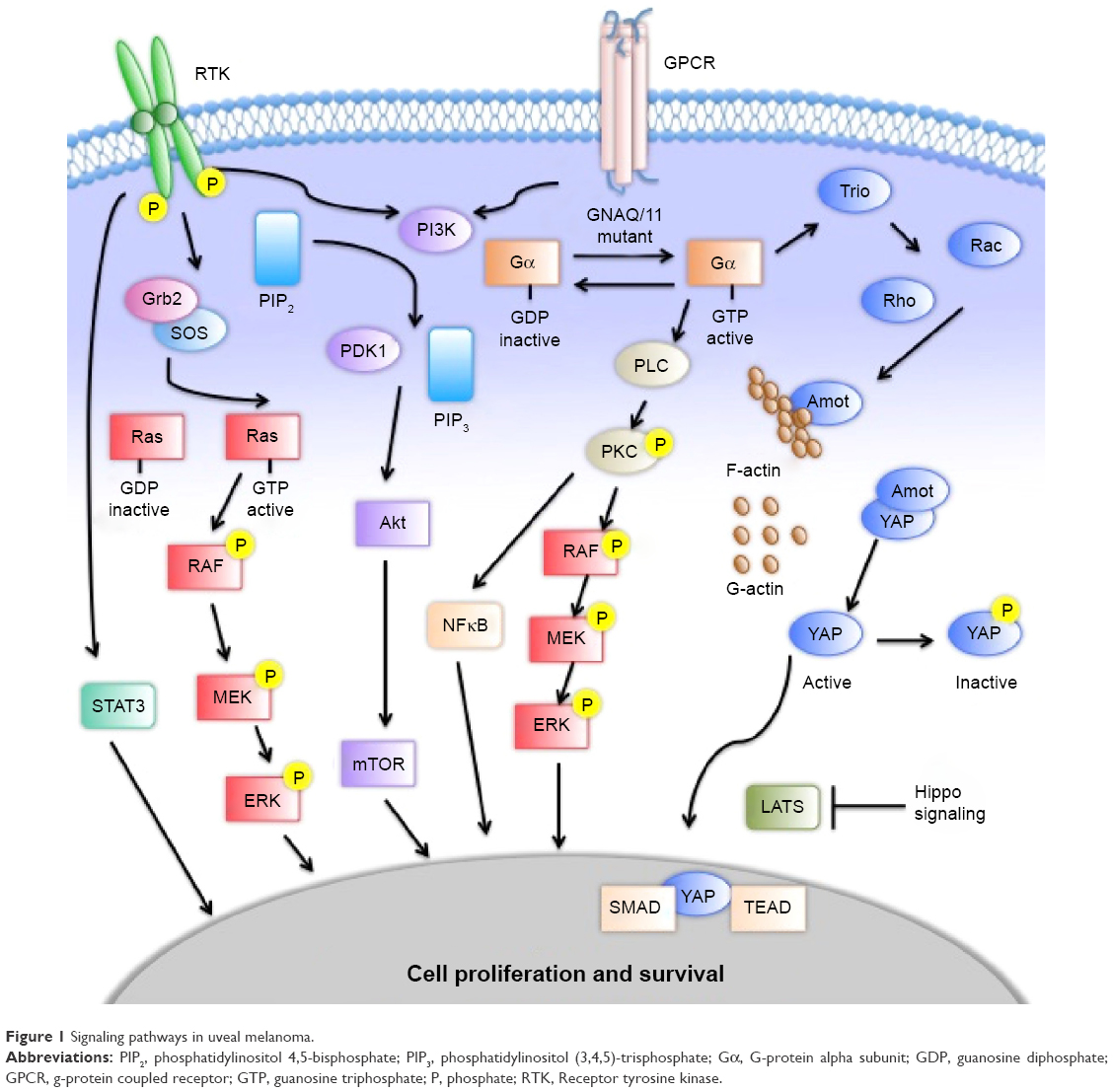 | Figure 1 Signaling pathways in uveal melanoma. |
Whereas the Gα pathway creates a cascade that leads to altered gene expression, several genes whose products are directly involved in transcription and translation are affected in UM. The best-described are BAP1, SF3B1, and EIF1AX. BAP1 is a nuclear deubiquitinase located on chromosome 3p. It functions as a tumor suppressor, and has an important role in transcription and the DNA-damage response. BAP1 has inactivating mutations in approximately 47% of primary UMs and 84% of metastatic UMs, consistent with the association of BAP1 mutations with inferior outcomes.12 SF3B1 encodes for splicing factor 3B subunit 1, which is involved in pre-messenger RNA splicing. SF3B1 alterations are associated with more favorable prognostic features, including younger age at diagnosis, fewer undifferentiated epithelioid cells, and disomy 3, as well as an inverse correlation with BAP1 mutations; however, SF3B1 mutations appear to be associated with the development of delayed metastasis, with a median of 8.2 years.57,58 EIF1AX encodes for eukaryotic translation initiation-factor 1A, X-linked, which stimulates transfer of Met RNA to the small ribosomal subunit. As with SF3B1, EIF1AX mutations are associated with disomy 3, but are less frequently associated with metastases.59 Of note, given the mutual exclusivity from BAP1 and SF3B1, it is not clear if the presence of EIF1AX mutations is protective or whether the association is due to a lack of negative effects of the other mutations.57
In addition to the genetic alterations described above, epigenetic factors, including DNA methylation, histone modification, and alterations in noncoding RNA, are increasingly felt to play a role in the development of UM. DNA methylation and histone deacetylation inactivate genes by inhibiting their transcription. Tumor suppressors, such as RASSF1A, and other molecules, such as the adhesion protein E-cadherin, are hypermethylated in 50%–70% of UM, correlate with metastatic disease, and may provide potential novel therapeutic strategies.60,61 Preclinical tumor models using the approved DNA methyltransferase inhibitor 5-aza-dC have restored E-cadherin transcription and reduced growth and invasiveness of UM cells.62,63 MicroRNAs (miRNAs) inhibit translation of complementary coding RNA and may act intracellularly, or when packaged in exosomes are capable of traveling in blood and modifying activity of distant cells.64 miR-124, miR-137, and miR-34b/c act as tumor suppressors by inhibiting oncogenes and are downregulated in UM, whereas many miRNAs, like miR-20a, -25b, -146a, and 199a, are overexpressed and may promote metastatic progression, in these examples likely through immunosuppression.65–68 Interestingly, the expression of miRNAs themselves may be epigenetically modified.
Management of primary uveal melanoma
The management of localized UM can be divided into globe-preserving therapy or enucleation. Globe-preserving therapies can broadly be classified into radiation, surgical, and laser therapy. The majority of primary UM lesions in the US are treated with plaque brachytherapy based upon results of the COMS trial, which randomized patients with medium-sized choroidal melanomas to primary therapy with 125I brachytherapy versus enucleation. No difference was observed in mortality between the two groups at up to 15 years of follow-up.69
Radiation therapy
Radiation therapy modalities include brachytherapy, photon-based external-beam radiation, and charged-particle radiation. These modalities have demonstrated excellent local control and globe preservation; however, long-term vision loss is common. Brachytherapy involves securing a radioactive plaque to the episclera to deliver a fixed dose of focal radiation to a tumor. The most common radioisotopes used are 125I and 106Ru. 125I emits γ-radiation, which penetrates more deeply into tissues than the β-emitting 106Ru (<6 mm tumor thickness).3,70 The American Brachytherapy Society recommends against brachytherapy in patients with tumors with extraocular extension, large basal diameters, blind painful eyes, and those with no light-perception vision.71 Local recurrence rates are 7%–10% for 125I, 14.7% for 106Ru, and 3.3% with 103Pd.69,70,72,73 Although associated with good local control, brachytherapy is associated with complications, including radiation-induced retinopathy (45%–67%), cataracts (44%), neovascular glaucoma (28.3%), and macular edema (24.5%).74,75 These complications lead to moderate vision loss in 58% of patients and poor visual acuity (best corrected worse than 5/200) in 28% within 2 years. Outcomes may be improved with the use of intravitreal bevacizumab, with 33% experiencing moderate vision loss and 15% developing poor visual acuity.76 Regular ophthalmologic exams for years are required to monitor complications.
Charged-particle radiotherapy may be used to treat medium-to-large tumors, including those that may not be good candidates for brachytherapy. A randomized controlled trial of helium-ion therapy versus brachytherapy for ciliochoroidal melanomas of thickness ≤11 mm and diameter ≤15 mm demonstrated improved local control, eye preservation, and disease-free survival for charged-particle therapy. Local control rates were 100% versus 84% at 5 years and 98% versus 79% at 12 years in favor of charged-particle therapy. Enucleation rates were 11% versus 22% at 5 years and 17% versus 37% at 12 years. Notably, this study was conducted at a single center, and may not be generalizable to the general population.77
Studies of proton-beam therapy have also shown optimistic results. A recent retrospective cohort study of patients with T3–T4 choroidal melanomas treated with proton-beam therapy demonstrated a 5-year local control rate of 94%, enucleation rate of 19.5%, and preservation of visual acuity ≥20/200 of 20%.78 A case series of tumors of all stages found primary proton-beam therapy to achieve a 96.4% local control rate and 95% eye-retention rate with median follow-up of 5 years.79
Surgery
Enucleation is the most common surgery performed for UM, and is appropriate for patients with vision loss, extensive extraocular growth, circumferential tumor invasion, and large tumor diameter.75 Preenucleation external-beam radiation therapy has been studied, without additional benefit observed, and thus is not recommended.80 Interestingly, the COMS quality-of-life report found that patients undergoing enucleation were less likely to have anxiety than patients treated with brachytherapy.81
Alternative surgical modalities include transretinal endoresection and transscleral resection. These procedures are site- and surgeon-dependent, with the majority of data coming from single-institution case series. Transscleral resection may be attempted in patients with large tumors who are not candidates for radiation therapy who seek eye-retaining treatment. The benefit of transscleral resection is improved vision preservation; however, this is a complex procedure with associated complications. Complications include retinal detachment (21%), ocular hypertension (21%), submacular hemorrhage (16%) and high rates of repeat vitreoretinal surgery (44%–70%). Hypotensive anesthesia may be used to minimize bleeding, but poses additional risks.74,75,82
Recurrence rates are higher with transscleral resection when compared to either enucleation or brachytherapy. In a retrospective review comparing transscleral resection to 125I brachytherapy for patients with tumor height >7.5 mm, 61.1% versus 5.6% maintained visual acuity >20/200, without significant difference in rates of metastasis. In this study, the majority of patients in the transscleral resection group received adjuvant 106Ru plaque therapy.75 A matched case-control study evaluating transscleral resection versus iodine brachytherapy found similar results, with improved vision preservation after transscleral resection but higher local recurrence in the transscleral group. Importantly, no difference in 8-year all-cause mortality, melanoma specific mortality, or quality of life was observed.83 The series of transscleral resections of large UM with longest follow-up comes from Innsbruck Medical University, where 5- and 10-year local tumor recurrence was 24% and 32%, respectively. Five- and 10-year metastatic rates of 28% and 44% were observed. In this study, lack of ruthenium adjuvant therapy carried a 4.4-fold greater risk of recurrence.84 The concern for local recurrence in transscleral resection was again seen when studying ciliochoroidal melanomas with large height, where the recurrence rate was 41% at 5 years with resection compared to 7% with brachytherapy.72
Neoadjuvant therapies have been utilized prior to transscleral resection in an effort to improve local control. Although mean follow-up was only 3.2 years, a case series of neoadjuvant proton-beam irradiation followed by resection demonstrated improved local recurrence rates, with no effect upon metastasis, when compared to historical controls.82 Of note, 70% underwent vitreoretinal surgery secondary to complications of tumor resection. Estimated risk of local recurrence was 4.2% and 10.4% at 3 and 5 years, respectively, with risk of metastasis 28.4% and 40.3%, respectively.
Laser therapy
Photodynamic laser photocoagulation and transpupillary thermal therapy (TTT) are modalities that direct focused energy to destroy tumor vascular supplies and reduce local recurrences by injecting and activating light-sensitive compounds and free radicals. TTT has shown some efficacy in treatment of residual choroidal melanomas and as adjuvant therapy after brachytherapy, when plaque tilt may have limited radiation delivery.85–87 TTT has been effective as primary therapy in up to 80% of cases of small or indeterminate lesions with few risk factors.88,89 A study of ruthenium brachytherapy with or without TTT demonstrated higher rates of tumor regression, globe preservation, and recurrence-free survival with adjuvant TTT.90 In contrast, Tarmann et al found that brachytherapy with TTT did not improve tumor control, but contributed to worse visual outcomes.70
Novel therapies for primary disease
Although current treatment modalities for primary disease achieve frequent local control, complications, including vision loss, are common, and novel approaches are much needed. Several novel approaches are currently in development.
ICON-1 is a structural variant of human factor VII being developed by Iconic Therapeutics. Factor VII is the natural ligand of tissue factor, which is commonly overexpressed and upregulated in UM and contributes to tumor growth, thrombosis, angiogenesis, and metastasis.91 ICON-1 binds to cells overexpressing tissue factor, initiating a signal cascade targeting immune cells to pathological tissue while leaving normal tissue intact. A Phase I study is ongoing to test safety and tolerability of intravitreal ICON-1 at three dosing regimens in patients with primary UM planning to undergo enucleation (NCT02771340).
Another tumor-targeted approach to treating UM is being developed by Aura Biosciences. Their lead drug, AU-011, consists of virus-like particles that selectively bind cancer cells, conjugated to infrared-activated molecules that destroy tumor membranes upon activation with an ophthalmic laser. Preclinical studies demonstrated human papillomavirus-modeled virus-like particles bind to heparin sulfate proteoglycans on disrupted epithelium, but do not bind to intact epithelium. Papillomavirus capsids bind various tumor-derived cell lines in vitro and in vivo in orthotopic models for ovarian and lung cancers, and complete tumor eradication was found histopathologically.92 AU-011 has been granted an “orphan” drug designation by the US Food and Drug Administration, and clinical trials are to begin in 2016.93
Adjuvant therapy
Despite aggressive management of localized disease, many ultimately develop metastasis. Indeed, circulating UM cells have been detected at diagnosis in patients with no clinically detectable lesions.94 While systemic adjuvant therapy has the potential to prevent metastases, no adjuvant therapy has been demonstrated to improve outcomes.
Adjuvant chemotherapy trials were initially investigated based on success in cutaneous melanoma. The first randomized controlled study of a systemic adjuvant therapy for UM evaluated dacarbazine, an intravenous alkylating agent, which despite its impact in melanoma demonstrated no survival advantage.95 Similarly, two nonrandomized studies of adjuvant interferon, which continues to be a recommended adjuvant in stage III cutaneous melanoma, found no impact on survival either.96–98 A study evaluating both agents in combination in the adjuvant setting has completed accrual, with final data anticipated in early 2017.
Fotemustine, an alkylating agent with high hepatic uptake and a short half-life, was initially studied by intra-arterial delivery for hepatic lesions, with good response.99 It has since been studied in UM patients with high risk of metastases by intra-arterial hepatic delivery as adjuvant therapy, with a suggestion of survival benefit but not statistical significance.100 Subsequently, intravenous fotemustine versus intra-arterial hepatic delivery for isolated liver metastases showed no difference in overall survival, but intra-arterial delivery increased progression-free survival and response rate.101 These studies led to the development of a randomized Phase III trial of adjuvant intravenous fotemustine versus observation in high-risk patients.
Various novel therapies are being investigated in UM. Both c-Met and c-Kit are tyrosine-kinase receptors that are highly expressed in UM and activate the MAPK and PI3–Akt pathways.102–104 Crizotinib is an inhibitor of c-Met, ALK, and ROS1, and has been shown to inhibit phosphorylation of c-Met but not ALK or ROS1 in UM. Using a mouse model of UM, Surriga et al demonstrated inhibition of the establishment of metastases with crizotinib administration.105 Crizotinib is currently being studied for UM in the adjuvant setting (NCT02223819).
In a retrospective study of sunitinib, a multitargeted tyrosine-kinase inhibitor that inhibits c-Kit, VEGF receptors, and other targets, patients deemed at high risk of metastasis based on cytogenetics and tumor size received 6 months of therapy. The estimated 5-year survival in the sunitinib group was 91% vs 61% in matched controls.106 Sunitinib is now being investigated as adjuvant therapy for high-risk tumors in a Phase II trial versus the histone deacetylase inhibitor valproic acid (NCT02068586). The rationale for histone deacetylase inhibition stems from histone-acetylation regulation of DNA expression. Histone deacetylase inhibitors, including valproic acid, promote cell differentiation and induce transition from a class 2 to class 1 gene-expression profile in cultured UM cells.107 Table 2 lists these and other trials evaluating various systemic therapies in the adjuvant setting.
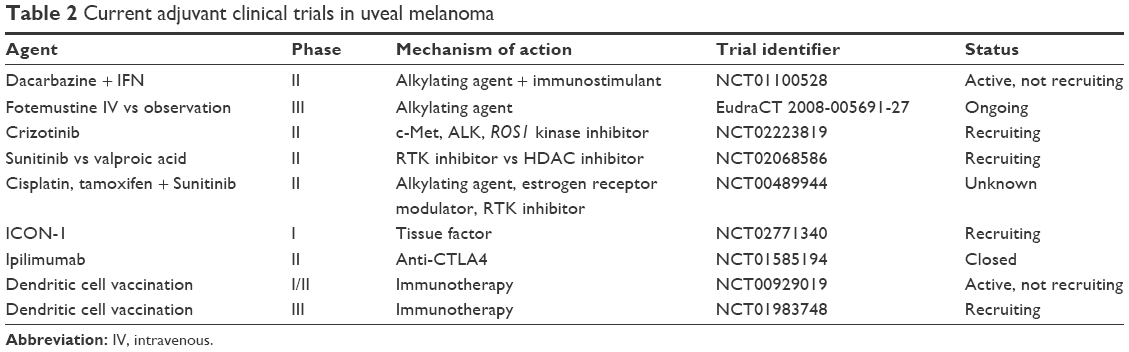 | Table 2 Current adjuvant clinical trials in uveal melanoma |
Posttreatment follow-up
There is no consensus as to what radiological or serum surveillance tests, if any, are appropriate after management of local disease.108,109 Various surveillance imaging modalities have been used, including chest X-ray, abdominal ultrasound, magnetic resonance imaging, computed tomography, and fludeoxyglucose positron-emission tomography imaging.110,111 Compared to liver function tests alone, biannual magnetic resonance imaging has greater predictive value for metastasis detection and may be a more effective, radiation-free alternative to other imaging modalities, as it also detects small lesions that may be missed on ultrasonography, computed tomography, and positron-emission tomography imaging.112–115 In the absence of data, consideration of routine imaging, including imaging of the liver, and examination at intervals between 3 and 12 months based upon predicted risk of recurrence is reasonable in an effort to identify disease when resection of metastases or institution of therapy for metastatic disease may be possible.
Metastatic uveal melanoma therapies
Unfortunately, up to 50% of patients with UM will develop metastatic disease despite primary therapy and active surveillance. A detailed discussion of treatments for metastatic UM is beyond the scope of this review, and has recently been reviewed by our group; however, it is important to note that there is no standard of care for the treatment of metastatic disease nor has any therapy been shown to improve overall survival.116 Therefore, clinical trial participation is encouraged for those who do develop metastatic disease. Some areas of ongoing investigation include therapies targeting the MAPK and/or PI3K pathway, and epigenetic modification with an HDAC inhibitor. While immunotherapy has shown durable responses in cutaneous melanoma, its clinical activity is limited in UM.117 Ongoing trials of other immune-based therapies include investigation of tumor-infiltrating lymphocyte therapy, as well as IMCgp100, a novel bispecific immunotherapeutic targeting gp100 and the CD3 protein complex on T cells. IMCgp100 increases the affinity and activation of CD8+ cytotoxic T lymphocytes against tumors expressing gp100 peptide.118,119 Results from a Phase I trial in metastatic melanoma were presented at the 2016 American Society of Clinical Oncology meeting, demonstrating a favorable safety profile and tumor shrinkage in patients with UM.120 A Phase I study of IMCgp100 in metastatic UM is currently recruiting (NCT02570308).
Conclusion and future directions
UM is the most common intraocular malignancy, and despite excellent local control with available therapies, little progress has been made to alter the disease course. Radiation therapy and enucleation have been the mainstays of therapy for decades, with little to no efficacy demonstrated from traditional chemotherapy in the primary, adjuvant, or metastatic setting. The future of UM treatment is evolving rapidly. Novel therapeutic strategies are emerging for the management of primary UM, such as the targeting of tissue factor and heparin sulfate proteoglycans, which have led to the initiation of early phase trials. In the adjuvant and metastatic settings, an improved understanding of UM pathophysiology has led to the study of new therapies that target the dysregulated pathways, as well as the immunological response to this disease, and will hopefully lead to improved outcomes; however, further work is needed. As our understanding of oncogene addiction evolves, combination regimens that prevent feedback escape will be further investigated and new therapeutics that target driving genetic events developed. Agents that modify gene expression and epigenetics, like HDAC and DNA methyltransferase inhibitors, will be further studied and next-generation immunotherapies that not only release inhibition of existing immune responses but actively direct our immune system to malignant cells, like IMCgp100, will move through development. In order for this to happen, extensive study must take place, and continued funding of clinical trials and development of novel therapies will be critical to improving outcomes for patients with this challenging disease.
Disclosure
RDC serves a consultant and/or advisory board member for AstraZeneca, Aura Biosciences, Iconic Therapeutics, Janssen, Merck, Novartis, Rgenix, and Thomson Reuters. BPM serves as a consultant for Aura Biosciences. The other authors report no conflicts of interest in this work.
References
McLaughlin CC, Wu XC, Jemal A, Martin HJ, Roche LM, Chen VW. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005;103(5):1000–1007. | ||
Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110(5):956–961. | ||
Damato B. Progress in the management of patients with uveal melanoma: the 2012 Ashton Lecture. Eye (Lond). 2012;26(9):1157–1172. | ||
Damato EM, Damato BE. Detection and time to treatment of uveal melanoma in the United Kingdom: an evaluation of 2,384 patients. Ophthalmology. 2012;119(8):1582–1589. | ||
Andreoli MT, Mieler WF, Leiderman YI. Epidemiological trends in uveal melanoma. Br J Ophthalmol. 2015;99(11):1550–1553. | ||
Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118(9):1881–1885. | ||
Hu DN, Yu GP, McCormick SA, Schneider S, Finger PT. Population-based incidence of uveal melanoma in various races and ethnic groups. Am J Ophthalmol. 2005;140(4):612–617. | ||
Virgili G, Gatta G, Ciccolallo L, et al. Incidence of uveal melanoma in Europe. Ophthalmology. 2007;114(12):2309–2315. | ||
Park SJ, Oh CM, Kim BW, Woo SJ, Cho H, Park KH. Nationwide incidence of ocular melanoma in South Korea by using the National Cancer Registry Database (1999-2011). Invest Ophthalmol Vis Sci. 2015;56(8):4719–4724. | ||
Shields CL, Kaliki S, Livesey M, et al. Association of ocular and oculodermal melanocytosis with the rate of uveal melanoma metastasis: analysis of 7872 consecutive eyes. JAMA Ophthalmol. 2013;131(8):993–1003. | ||
Weis E, Shah CP, Lajous M, Shields JA, Shields CL. The association of cutaneous and iris nevi with uveal melanoma: a meta-analysis. Ophthalmology. 2009;116(3):536–543.e2. | ||
Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–1413. | ||
Gallagher RP, Elwood JM, Rootman J, et al. Risk factors for ocular melanoma: Western Canada Melanoma Study. J Natl Cancer Inst. 1985;74(4):775–778. | ||
Lains I, Bartosch C, Mondim V, et al. Second primary neoplasms in patients with uveal melanoma: a SEER database analysis. Am J Ophthalmol. 2016;165:54–64. | ||
Tucker MA, Shields JA, Hartge P, Augsburger J, Hoover RN, Fraumeni JF Jr. Sunlight exposure as risk factor for intraocular malignant melanoma. N Engl J Med. 1985;313(13):789–792. | ||
Shah CP, Weis E, Lajous M, Shields JA, Shields CL. Intermittent and chronic ultraviolet light exposure and uveal melanoma: a meta-analysis. Ophthalmology. 2005;112(9):1599–1607. | ||
de Lange MJ, Razzaq L, Versluis M, et al. Distribution of GNAQ and GNA11 mutation signatures in uveal melanoma points to a light dependent mutation mechanism. PLoS One. 2015;10(9):e0138002. | ||
Shields CL, Furuta M, Berman EL, et al. Choroidal nevus transformation into melanoma: analysis of 2514 consecutive cases. Arch Ophthalmol. 2009;127(8):981–987. | ||
Shields JA, Sanborn GE, Augsburger JJ. The differential diagnosis of malignant melanoma of the iris: a clinical study of 200 patients. Ophthalmology. 1983;90(6):716–720. | ||
Shields JA, Augsburger JJ, Brown GC, Stephens RF. The differential diagnosis of posterior uveal melanoma. Ophthalmology. 1980;87(6):518–522. | ||
[No authors listed]. Accuracy of diagnosis of choroidal melanomas in the Collaborative Ocular Melanoma Study: COMS report no. 1. Arch Ophthalmol. 1990;108(9):1268–1273. | ||
Shields JA, Shields CL, Ehya H, Eagle RC Jr, De Potter P. Fine-needle aspiration biopsy of suspected intraocular tumors. Int Ophthalmol Clin. 1993;33(3):77–82. | ||
Char DH, Miller T. Accuracy of presumed uveal melanoma diagnosis before alternative therapy. Br J Ophthalmol. 1995;79(7):692–696. | ||
McCannel TA. Fine-needle aspiration biopsy in the management of choroidal melanoma. Curr Opin Ophthalmol. 2013;24(3):262–266. | ||
Rietschel P, Panageas KS, Hanlon C, Patel A, Abramson DH, Chapman PB. Variates of survival in metastatic uveal melanoma. J Clin Oncol. 2005;23(31):8076–8080. | ||
Diener-West M, Reynolds SM, Agugliaro DJ, et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group report no. 26. Arch Ophthalmol. 2005;123(12):1639–1643. | ||
Kujala E, Makitie T, Kivela T. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2003;44(11):4651–4659. | ||
Kuk D, Shoushtari AN, Barker CA, et al. Prognosis of mucosal, uveal, acral, nonacral cutaneous, and unknown primary melanoma from the time of first metastasis. Oncologist. 2016;21(7):848–854. | ||
Shields CL, Kaliki S, Furuta M, Fulco E, Alarcon C, Shields JA. American Joint Committee on Cancer classification of posterior uveal melanoma (tumor size category) predicts prognosis in 7,731 patients. Ophthalmology. 2013;120(10):2066–2071. | ||
Shields CL, Kaliki S, Furuta M, Fulco E, Alarcon C, Shields JA. American Joint Committee on Cancer classification of uveal melanoma (anatomic stage) predicts prognosis in 7,731 patients: the 2013 Zimmerman Lecture. Ophthalmology. 2015;122(6):1180–1186. | ||
Versluis M, de Lange MJ, van Pelt SI, et al. Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PLoS One. 2015;10(3):e0116371. | ||
Ewens KG, Kanetsky PA, Richards-Yutz J, et al. Genomic profile of 320 uveal melanoma cases: chromosome 8p-loss and metastatic outcome. Invest Ophthalmol Vis Sci. 2013;54(8):5721–5729. | ||
Onken MD, Worley LA, Char DH, et al. Collaborative Ocular Oncology Group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology. 2012;119(8):1596–1603. | ||
Field MG, Decatur CL, Kurtenbach S, et al. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin Cancer Res. 2016;22(5):1234–1242. | ||
Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–1696. | ||
Kalinec G, Nazarali AJ, Hermouet S, Xu N, Gutkind JS. Mutated α subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol Cell Biol. 1992;12(10):4687–4693. | ||
Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. | ||
Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191–2199. | ||
Piperno-Neumann S, Kapiteijn E, Larkin JM, et al. Landscape of genetic alterations in patients with metastatic uveal melanoma. J Clin Oncol. 2014;32(5 Suppl):9043. | ||
Griewank KG, van de Nes J, Schilling B, et al. Genetic and clinico-pathologic analysis of metastatic uveal melanoma. Mod Pathol. 2014;27(2):175–183. | ||
Xu X, Wei WB, Li B, Gao F, Zhang Z, Jonas JB. Oncogenic GNAQ and GNA11 mutations in uveal melanoma in Chinese. PLoS One. 2014;9(10):e109699. | ||
Johansson P, Aoude LG, Wadt K, et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7(4):4624–4631. | ||
Moore AR, Ceraudo E, Sher JJ, et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48(6):675–680. | ||
Shoushtari AN, Carvajal RD. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014;24(6):525–534. | ||
Lee CH, Park D, Wu D, Rhee SG, Simon MI. Members of the Gq α subunit gene family activate phospholipase C β isozymes. J Biol Chem. 1992;267(23):16044–16047. | ||
Wu DQ, Lee CH, Rhee SG, Simon MI. Activation of phospholipase C by the α subunits of the Gq and G11 proteins in transfected Cos-7 cells. J Biol Chem. 1992;267(3):1811–1817. | ||
Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213(3):589–602. | ||
Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270(25):14843–14846. | ||
Populo H, Soares P, Rocha AS, Silva P, Lopes JM. Evaluation of the mTOR pathway in ocular (uvea and conjunctiva) melanoma. Melanoma Res. 2010;20(2):107–117. | ||
Babchia N, Calipel A, Mouriaux F, Faussat AM, Mascarelli F. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: interaction with B-Raf/ERK. Invest Ophthalmol Vis Sci. 2010;51(1):421–429. | ||
Khalili JS, Yu X, Wang J, et al. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012;18(16):4345–4355. | ||
Ho AL, Musi E, Ambrosini G, et al. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS One. 2012;7(7):e40439. | ||
Overholtzer M, Zhang J, Smolen GA, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103(33):12405–12410. | ||
Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. | ||
Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008;283(9):5496–5509. | ||
Feng X, Degese MS, Iglesias-Bartolome R, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated Rho GTPase signaling circuitry. Cancer Cell. 2014;25(6):831–845. | ||
Yavuzyigitoglu S, Koopmans AE, Verdijk RM, et al. Uveal melanomas with SF3B1 mutations: a distinct subclass associated with late-onset metastases. Ophthalmology. 2016;123(5):1118–1128. | ||
Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45(2):133–135. | ||
Martin M, Masshöfer L, Temming P, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45(8):933–936. | ||
Maat W, van der Velden PA, Out-Luiting C, et al. Epigenetic inactivation of RASSF1a in uveal melanoma. Invest Ophthalmol Vis Sci. 2007;48(2):486–490. | ||
Merhavi E, Cohen Y, Avraham BC, et al. Promoter methylation status of multiple genes in uveal melanoma. Invest Ophthalmol Vis Sci. 2007;48(10):4403–4406. | ||
Rajaii F, Asnaghi L, Enke R, Merbs SL, Handa JT, Eberhart CG. The demethylating agent 5-Aza reduces the growth, invasiveness, and clonogenicity of uveal and cutaneous melanoma. Invest Ophthalmol Vis Sci. 2014;55(10):6178–6186. | ||
Venza M, Visalli M, Catalano T, et al. DNA methylation-induced E-cadherin silencing is correlated with the clinicopathological features of melanoma. Oncol Rep. 2016;35(4):2451–2460. | ||
Eldh M, Bagge RO, Lasser C, et al. MicroRNA in exosomes isolated directly from the liver circulation in patients with metastatic uveal melanoma. BMC Cancer. 2014;14:962. | ||
Chen X, He D, Da Dong X, et al. MicroRNA-124a is epigenetically regulated and acts as a tumor suppressor by controlling multiple targets in uveal melanoma miR-124a is a tumor suppressor in uveal melanoma. Invest Ophthalmol Vis Sci. 2013;54(3):2248–2256. | ||
Chen X, Wang J, Shen H, et al. Epigenetics, microRNAs, and carcinogenesis: functional role of microRNA-137 in uveal melanoma. Invest Ophthalmol Vis Sci. 2011;52(3):1193–1199. | ||
Dong F, Lou D. MicroRNA-34b/c suppresses uveal melanoma cell proliferation and migration through multiple targets. Mol Vis. 2012;18:537–546. | ||
Wen X, Lu L, He Z, Fan X. Orchestrating epigenetic roles targeting ocular tumors. Onco Targets Ther. 2016;9:1001–1009. | ||
Collaborative Ocular Melanoma Study Group. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report no. 28. Arch Ophthalmol. 2006;124(12):1684–1693. | ||
Tarmann L, Wackernagel W, Avian A, et al. Ruthenium-106 plaque brachytherapy for uveal melanoma. Br J Ophthalmol. 2015;99(12):1644–1649. | ||
American Brachytherapy Society – Ophthalmic Oncology Task Force. The American Brachytherapy Society consensus guidelines for plaque brachytherapy of uveal melanoma and retinoblastoma. Brachytherapy. 2014;13(1):1–14. | ||
Puusaari I, Damato B, Kivelä T. Transscleral local resection versus iodine brachytherapy for uveal melanomas that are large because of tumour height. Graefes Arch Clin Exp Ophthalmol. 2007;245(4):522–533. | ||
Finger PT, Chin KJ, Duvall G. Palladium-103 ophthalmic plaque radiation therapy for choroidal melanoma: 400 treated patients. Ophthalmology. 2009;116(4):790–796.e1. | ||
Caminal J, Padrón-Pérez N, Arias L, et al. Transscleral resection without hypotensive anaesthesia vs iodine-125 plaque brachytherapy in the treatment of choroidal melanoma. Eye (Lond). 2016;30(6):833–842. | ||
Bechrakis NE, Bornfeld N, Zöller I, Foerster MH. Iodine 125 plaque brachytherapy versus transscleral tumor resection in the treatment of large uveal melanomas. Ophthalmology. 2002;109(10):1855–1861. | ||
Shah SU, Shields CL, Bianciotto CG, et al. Intravitreal bevacizumab at 4-month intervals for prevention of macular edema after plaque radiotherapy of uveal melanoma. Ophthalmology. 2014;121(1):269–275. | ||
Mishra KK, Quivey JM, Daftari IK, et al. Long-term results of the UCSF-LBNL randomized trial: charged particle with helium ion versus iodine-125 plaque therapy for choroidal and ciliary body melanoma. Int J Radiat Oncol Biol Phys. 2015;92(2):376–383. | ||
Bensoussan E, Thariat J, Maschi C, et al. Outcomes after proton beam therapy for large choroidal melanomas in 492 patients. Am J Ophthalmol. 2016;165:78–87. | ||
Seibel I, Cordini D, Rehak M, et al. Local recurrence after primary proton beam therapy in uveal melanoma: risk factors, retreatment approaches, and outcome. Am J Ophthalmol. 2015;160(4):628–636. | ||
Hawkins BS. The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma: IV. Ten-year mortality findings and prognostic factors. COMS report number 24. Am J Ophthalmol. 2004;138(6):936–951. | ||
Melia M, Moy CS, Reynolds SM, et al. Quality of life after iodine 125 brachytherapy vs enucleation for choroidal melanoma: 5-year results from the Collaborative Ocular Melanoma Study: COMS QOLS report no. 3. Arch Ophthalmol. 2006;124(2):226–238. | ||
Willerding GD, Cordini D, Moser L, Krause L, Foerster MH, Bechrakis NE. Neoadjuvant proton beam irradiation followed by transscleral resection of uveal melanoma in 106 cases. Br J Ophthalmol. 2016;100(4):463–467. | ||
Kivela T, Puusaari I, Damato B. Transscleral resection versus iodine brachytherapy for choroidal malignant melanomas 6 millimeters or more in thickness: a matched case-control study. Ophthalmology. 2003;110(11):2235–2244. | ||
Bechrakis NE, Petousis V, Willerding G, et al. Ten-year results of transscleral resection of large uveal melanomas: local tumour control and metastatic rate. Br J Ophthalmol. 2010;94(4):460–466. | ||
Badiyan SN, Rao RC, Apicelli AJ, et al. Outcomes of iodine-125 plaque brachytherapy for uveal melanoma with intraoperative ultrasonography and supplemental transpupillary thermotherapy. Int J Radiat Oncol Biol Phys. 2014;88(4):801–805. | ||
Saakian SV, Val’skii VV, Semenova EA, Amirian AG. [Transpupillary thermotherapy in the treatment of recurrent and residual choroidal melanomas: preliminary results]. Vestn Oftalmol. 2009;125(6):11–15. Russian. | ||
Harbour JW, Meredith TA, Thompson PA, Gordon ME. Transpupillary thermotherapy versus plaque radiotherapy for suspected choroidal melanomas. Ophthalmology. 2003;110(11):2207–2215. | ||
Turcotte S, Bergeron D, Rousseau AP, Mouriaux F. Primary transpupillary thermotherapy for choroidal indeterminate melanocytic lesions. Can J Ophthalmol. 2014;49(5):464–467. | ||
Mashayekhi A, Shields CL, Rishi P, et al. Primary transpupillary thermotherapy for choroidal melanoma in 391 cases: importance of risk factors in tumor control. Ophthalmology. 2015;122(3):600–609. | ||
Yarovoy AA, Magaramov DA, Bulgakova ES. The comparison of ruthenium brachytherapy and simultaneous transpupillary thermotherapy of choroidal melanoma with brachytherapy alone. Brachytherapy. 2012;11(3):224–229. | ||
Walker TM, Van Ginkel PR, Gee RL, et al. Expression of angiogenic factors Cyr61 and tissue factor in uveal melanoma. Arch Ophthalmol. 2002;120(12):1719–1725. | ||
Kines RC, Cerio RJ, Roberts JN, et al. Human papillomavirus capsids preferentially bind and infect tumor cells. Int J Cancer. 2016;138(4):901–911. | ||
Business Wire. FDA grants orphan drug designation to Aura Biosciences’ novel treatment for uveal melanoma. 2015. Available from: http://www.businesswire.com/news/home/20150521005822/en/FDA-Grants-Orphan-Drug-Designation-Aura-Biosciences%E2%80%99. Accessed November 26, 2016. | ||
Torres V, Triozzi P, Eng C, et al. Circulating tumor cells in uveal melanoma. Future Oncol. 2011;7(1):101–109. | ||
Desjardins L, Dorval T, Levy C, et al. Etude randomisée de chimiothérapie adjuvante par le deticène dans le mélanome choroïdien [Randomized study of adjuvant chemotherapy with dacarbazine in choroidal melanoma]. Ophtalmologie. 1998;12(3):168–173. French. | ||
Lane AM, Egan KM, Harmon D, Holbrook A, Munzenrider JE, Gragoudas ES. Adjuvant interferon therapy for patients with uveal melanoma at high risk of metastasis. Ophthalmology. 2009;116(11):2206–2212. | ||
Richtig E, Langmann G, Schlemmer G, et al. [Safety and efficacy of interferon alfaα-2b in the adjuvant treatment of uveal melanoma]. Ophthalmologe. 2006;103(6):506–511. German. | ||
National Comprehensive Cancer Network. NCCN Guidelines: Melanoma Version 3.2016. Fort Washington (PA): NCCN; 2016. | ||
Leyvraz S, Spataro V, Bauer J, et al. Treatment of ocular melanoma metastatic to the liver by hepatic arterial chemotherapy. J Clin Oncol. 1997;15(7):2589–2595. | ||
Voelter V, Schalenbourg A, Pampallona S, et al. Adjuvant intra-arterial hepatic fotemustine for high-risk uveal melanoma patients. Melanoma Res. 2008;18(3):220–224. | ||
Leyvraz S, Piperno-Neumann S, Suciu S, et al. Hepatic intra-arterial versus intravenous fotemustine in patients with liver metastases from uveal melanoma (EORTC 18021): a multicentric randomized trial. Ann Oncol. 2014;25(3):742–746. | ||
Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15(7):2207–2214. | ||
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The c-Kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9(5):435–443. | ||
Sato T, Han F, Yamamoto A. The biology and management of uveal melanoma. Curr Oncol Rep. 2008;10(5):431–438. | ||
Surriga O, Rajasekhar VK, Ambrosini G, Dogan Y, Huang R, Schwartz GK. Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol Cancer Ther. 2013;12(12):2817–2826. | ||
Valsecchi ME, Orloff MM, Chervoneva I, et al. Potential survival benefit of adjuvant sunitinib in high risk uveal melanoma. J Clin Oncol. 2015;33 Suppl:e20046. | ||
Landreville S, Agapova OA, Matatall KA, et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2012;18(2):408–416. | ||
Kaiserman I, Amer R, Pe’er J. Liver function tests in metastatic uveal melanoma. Am J Ophthalmol. 2004;137(2):236–243. | ||
Koutsandrea C, Moschos MM, Dimissianos M, Georgopoulos G, Ladas I, Apostolopoulos M. Metastasis rates and sites after treatment for choroidal melanoma by proton beam irradiation or by enucleation. Clin Ophthalmol. 2008;2(4):989–995. | ||
Francis JH, Patel SP, Gombos DS, Carvajal RD. Surveillance options for patients with uveal melanoma following definitive management. Am Soc Clin Oncol Educ Book. 2013:382–387. | ||
Francken AB, Fulham MJ, Millward MJ, Thompson JF. Detection of metastatic disease in patients with uveal melanoma using positron emission tomography. Eur J Surg Oncol. 2006;32(7):780–784. | ||
Chattopadhyay C, Kim DW, Gombos DS, et al. Uveal melanoma: from diagnosis to treatment and the science in between. Cancer. 2016;122(15):2299–2312. | ||
Piperno-Neumann S, Servois V, Mariani P, et al. Prospective study of surveillance testing for metastasis in 100 high-risk uveal melanoma patients. J Fr Ophtalmol. 2015;38(6):526–534. | ||
Marshall E, Romaniuk C, Ghaneh P, et al. MRI in the detection of hepatic metastases from high-risk uveal melanoma: a prospective study in 188 patients. Br J Ophthalmol. 2013;97(2):159–163. | ||
Wen JC, Sai V, Straatsma BR, McCannel TA. Radiation-related cancer risk associated with surveillance imaging for metastasis from choroidal melanoma. JAMA Ophthalmol. 2013;131(1):56–61. | ||
Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. Epub 2016 Aug 29. | ||
Guan X, Wang H, Ma F, Qian H, Yi Z, Xu B. The efficacy and safety of programmed cell death 1 and programmed cell death 1 ligand inhibitors for advanced melanoma: a meta-analysis of clinical trials following the PRISMA guidelines. Medicine (Baltimore). 2016;95(11):e3134. | ||
Bossi G, Buisson S, Oates J, Jakobsen BK, Hassan NJ. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol Immunother. 2014;63(5):437–448. | ||
Marquez-Rodas I, Cerezuela P, Soria A, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3(18):267. | ||
Middleton MR, Steven NM, Evans TJ, et al. Safety, pharmacokinetics and efficacy of IMCgp100, a first-in-class soluble TCR-antiCD3 bispecific T cell redirector with solid tumour activity: results from the FIH study in melanoma. J Clin Oncol. 2016;34 Suppl:3016. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.