")
Back to Journals » The Application of Clinical Genetics » Volume 11
Utility of trio-based exome sequencing in the elucidation of the genetic basis of isolated syndromic intellectual disability: illustrative cases
Authors Carneiro TNR , Krepischi ACV , Costa SS , Tojal da Silva I, Vianna-Morgante AM , Valieris R, Ezquina SAM , Bertola DR, Otto PA, Rosenberg C
Received 17 February 2018
Accepted for publication 12 April 2018
Published 22 August 2018 Volume 2018:11 Pages 93—98
DOI https://doi.org/10.2147/TACG.S165799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Thaise NR Carneiro,1 Ana CV Krepischi,1 Silvia S Costa,1 Israel Tojal da Silva,2 Angela M Vianna-Morgante,1 Renan Valieris,2 Suzana AM Ezquina,1 Debora R Bertola,3 Paulo A Otto,1 Carla Rosenberg1
1Human Genome and Stem-Cell Research Center, Department of Genetics and Evolutionary Biology, Institute of Biosciences, University of São Paulo, São Paulo, Brazil; 2Laboratory of Computational Biology and Bioinformatics, International Research Center, A. C. Camargo Cancer Center, São Paulo, Brazil; 3Genetics Unit, Instituto da Criança, Hospital das Clínicas, Medical School, University of São Paulo, São Paulo, Brazil
Introduction: Exome sequencing is recognized as a powerful tool for identifying the genetic cause of intellectual disability (ID). It is uncertain, however, whether only the exome of the proband should be sequenced or if the sequencing of parental genomes is also required, and the resulting increase in diagnostic yield justifies the increase in costs.
Patients and methods: We sequenced the exomes of eight individuals with sporadic syndromic ID and their parents.
Results and discussion: Likely pathogenic variants were detected in eight candidate genes, namely homozygous or compound heterozygous variants in three autosomal genes (ADAMTSL2, NALCN, VPS13B), one in an X-linked gene (MID1), and de novo heterozygous variants in four autosomal genes (RYR2, GABBR2, CDK13, DDX3X). Two patients harbored rare variants in two or more candidate genes, while in three other patients no candidate was identified. In five probands (62%), the detected variants explained their clinical findings. The causative recessive variants would have led to diagnosis even without parental exome sequencing, but for the heterozygous dominant ones, the exome trio-based approach was fundamental in the identification of the de novo likely pathogenic variants.
Keywords: exome, intellectual disability, next-generation sequencing
Introduction
Intellectual disability (ID) is a complex and heterogeneous clinical condition that affects 1%–2% of the general population, and can result from genetic or environmental factors, or a combination of both. However, most severe forms of ID have a single genetic basis, ranging from chromosomal alterations to point mutations.1–3
About 700 genes have already been associated with ID;4 however, a clear genetic explanation for the phenotype of many patients remains unknown. The implementation of whole exome sequencing (WES) in the last decade increased the identification yield of new mutations and genes associated with various diseases, and led to the demonstration that de novo mutations are a frequent cause of ID.5 WES has also successfully identified autosomal recessive6,7 and X-linked8,9 causative mutations in ID cohorts.
In non-familial cases, the situation is complicated by the lack of information on the type of inheritance underlying the phenotype. To date, studies of sporadic cases that have been performed using WES to elucidate the causes of ID have led to the diagnosis of 15%–30% of the patients.10–12
Through the exome sequencing of probands and their unaffected parents (trio analysis), this work aimed at identifying variants, which could explain ID in sporadic cases, and evaluating the utility of trio-based exome sequencing in the identification of pathogenic variants.
Patients and methods
The patients were referred to the Genetic Counseling Service of the Department of Genetics and Evolutionary Biology, Institute of Biosciences, University of São Paulo. The study was approved by the Ethics Committee of the institution. Written informed consent was obtained from the parents of all patients.
The patients had ID ranging from moderate to severe and other associated clinical signs. Table 1 summarizes the main clinical findings of the patients in the cohort.
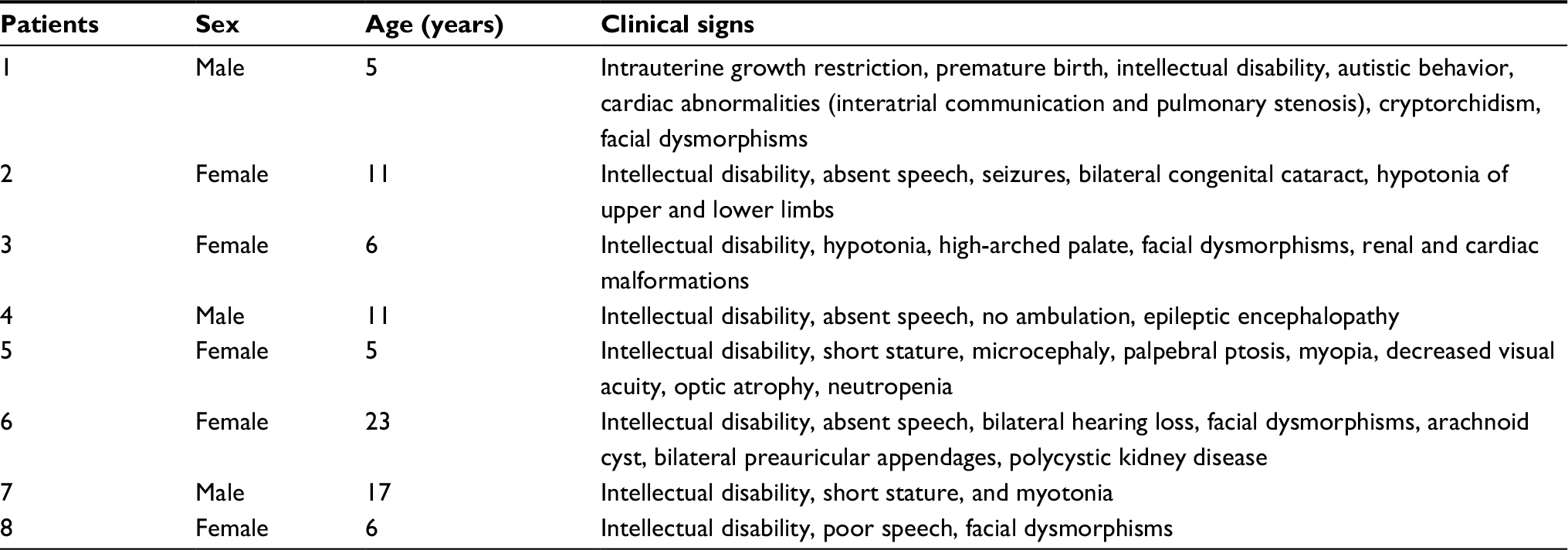 | Table 1 Main clinical findings of the patients in the cohort |
Genomic DNA from peripheral blood samples was extracted, according to standard procedures. Fragile-X syndrome (AmplideX® FMR1; Asuragen, Austin, TX, USA) and genomic imbalances (180K platform; Agilent Technologies, Santa Clara, CA, USA, or 850K platform; Illumina, San Diego, CA, USA) had previously been excluded in these families.
Genomic libraries were constructed using the SureSelect XT or SureSelect QXT kit V6 (Agilent SureSelect Whole Exome Enrichment kit), according to the manufacturer’s instructions, with 100× coverage and 90% of the targets covered at 20×; sequencing was performed on the Hiseq 2500 sequencer from Illumina. The quality of the sequencing was verified through the FastQC program (Babraham Institute). The raw reads were aligned to the reference genome (GRCh37/hg19), using the Burrows–Wheeler Aligner,13 and pre-processed according to GATK toolkit,14 which involves indel realignment, base quality score recalibration, base alignment quality scoring, and variant calling. Filtering and prioritization were conducted using VarSeq® software (Golden Helix, Bozeman, MT, USA) and variant effect predictor. After coding, non-synonymous variants fitting the models of dominant de novo or recessive homozygote/compound heterozygote/hemizygote were filtered per frequency (1%) against the databases: NHLBI ESP6500SI-V2 exomes variant frequencies, ClinVar,15 dbSNP138, 1000 Genome Project,16 ExAC Browser,17 and ABRAOM.18
After variant filtering, in silico prediction of pathogenicity of variants was performed using five prediction algorithms, namely SIFT,19 PolyPhen-2,20 Mutation Taster,21 Mutation Assessor, and FATHMM.22 The VarElect online tool was used to prioritize variants according to the phenotype. The OMIM database and scientific literature were used to compare the expected phenotypes with the clinical features of the patients.
Potentially pathogenic variants in the proband were validated by Sanger sequencing also performed to analyze the presence or absence of the same variants in their parents. Variants were classified according to the ACMG guideline.23
Results and discussion
We sequenced the exomes (WES) of eight patients with idiopathic syndromic ID and their parents (trios). Rare variants in eight genes were detected in five patients: a homozygous variant in ADAMTSL2; compound heterozygous variants in NALCN and VPS13B; a variant in the X-linked gene MID1; and four heterozygous de novo variants in the autosomal genes RYR2, GABBR2, DDX3X, and CDK13. Table 2 summarizes the WES findings; the variants in bold were considered as causative of the clinical phenotypes. The likely pathogenic variants found in this study were missense, except for those in VPS13B (stop gain), and the clinical impact could only be estimated, even with the help of prediction algorithms. Figure 1 illustrates the results, showing the de novo variant detected in GABBR2 and the maternally inherited variant in MID1.
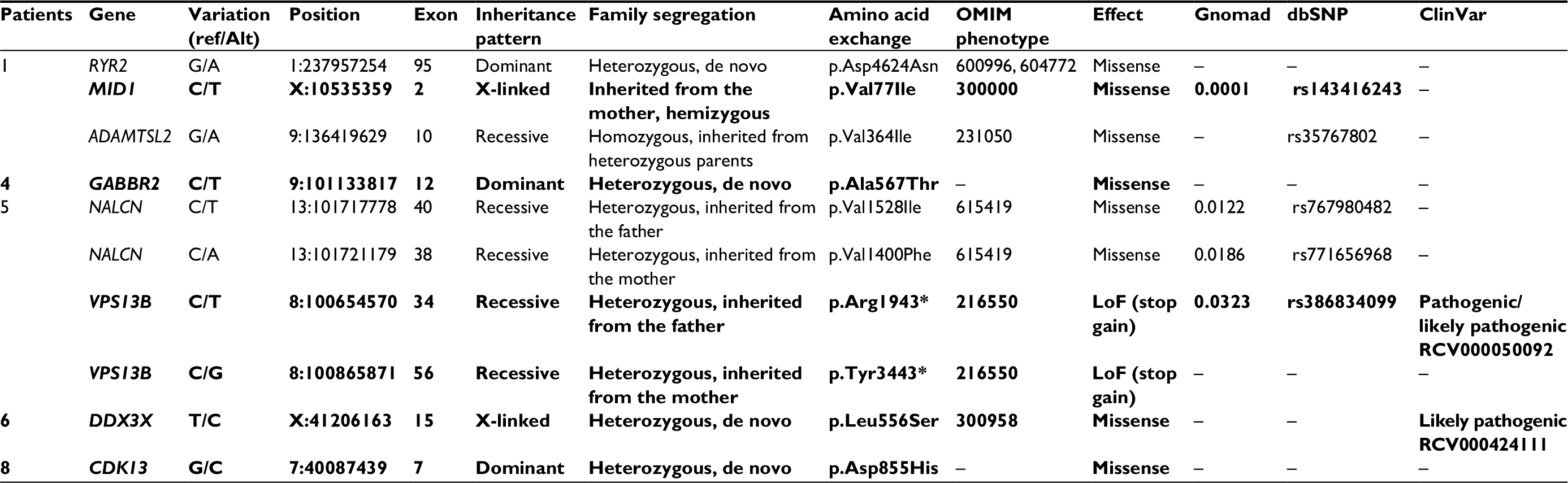 | Table 2 The candidate mutations identified in five of the patients of the cohort Abbreviations: Alt, alternate nucleotide; LoF, loss of function; OMIM, Online Mendelian Inheritance in Man; Ref, reference nucleotide. Note: Variants in bold are considered to be causative of the phenotypes. |
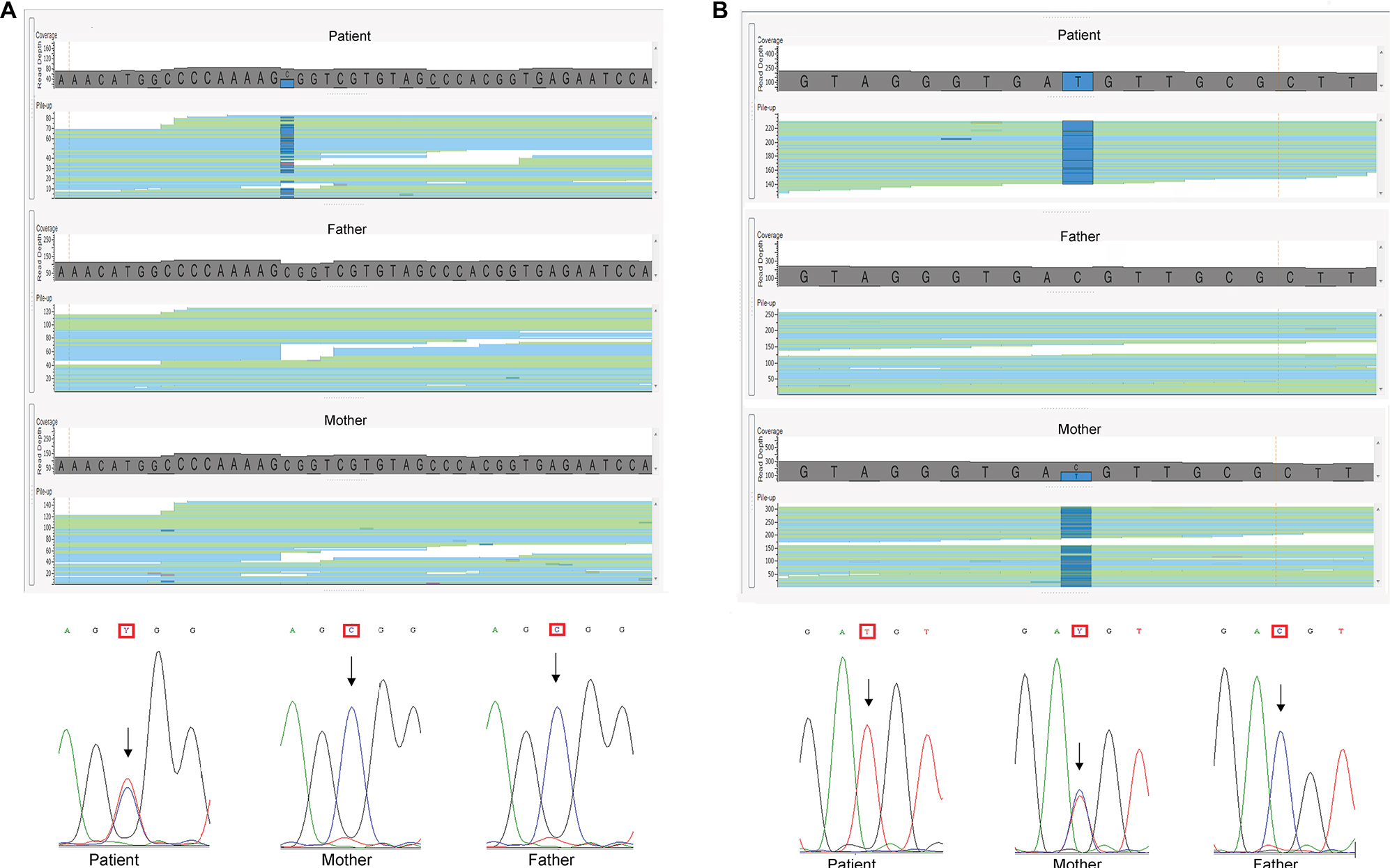 | Figure 1 Example of pathogenic mutations identified in the cohort. Notes: (A) Image of the binary alignment map files showing the forward and reverse reads of a segment of the GABBR2 gene; the mutation from C to T in heterozygosity in the proband, not present in his parents, can be seen in dark blue. Underneath, Sanger sequencing validation of the C/T substitution, (B) Image of the BAM files showing the forward and reverse reads of a segment of the MID1 gene; the mutated allele, in heterozygosity in the mother and hemizygosity in the proband, is seen in dark blue. Underneath, Sanger sequencing validation of the C/T substitution. |
As shown in Table 2, Patient 1 had rare variants of uncertain significance (VUS) in three different genes:a missense variant in RYR2, a gene associated with dominant arrhythmogenic right ventricular dysplasia or ventricular tachycardia, neither of the conditions documented in the patient; a homozygous missense variant in ADAMTSL2, whose mutations are known to cause recessive Ehlers–Danlos syndrome, not compatible with the patient’s phenotype; and a maternally inherited missense variant in the MID1 gene, associated with Opitz G/BBB syndrome, which could explain the cognitive impairment, cardiac defects, and cryptorchidism exhibited by the patient. The variant in MID1 was predicted as damaging (SIFT – http://sift.jcvi.org/) or probably damaging (PolyPhen-2 – http://genetics.bwh.harvard.edu/pph2/), but the RYR2 variant was also considered probably damaging, and diagnosis of Opitz G/BBB syndrome mostly relied on the fact that the MID1 variant could explain the phenotype.
Patients 4, 6, and 8 carried de novo variants. The variants in GABBR2 and in CDK13 had not been described in the searched databases (GnomAD, dbSNP, and ClinVar) and are therefore novel, while the variant in DDX3X had already been described in ClinVar and classified as probably pathogenic.
Patient 4 harbored VUS in GABBR2. Variants in this gene have been described recently, and the associated phenotype has not been consolidated in OMIM, but they emerge as important contributors for epileptic encephalopathies,24–27 in accordance with our patient clinical phenotype.
Patient 6 carried a known pathogenic variant in DDX3X; the disorder associated with this gene, x-linked mental retardation, is clinically variable and includes other symptoms in addition to cognitive impairment, such as hearing loss, which is present in this patient. On the other hand, polycystic kidney disease, exhibited by the patient, has not been reported in DDX3X mutation carriers. Although mutations in DDX3X have only recently been reported, it is estimated that they are responsible for 1%–3% of idiopathic ID in females.28
Patient 8, carrier of a likely pathogenic variant in CDK13, exhibited ID and dysmorphic features commonly associated with CDK13 mutations, including hypertelorism, telecanthus, inverse epicanthal folds, broad nasal bridge, and low-set, posteriorly rotated ears. She did not present heart defects and seizures, which are frequently found among patients carrying CDK13 mutations. A recent paper by Hamilton et al shows that mutation in CDK13 results in syndromic ID, with or without congenital heart disease and seizures.28
In Patient 5, we identified potential pathogenic variants in more than one candidate gene. She was a compound heterozygote for variants in NALCN, a gene associated with recessive syndromic hypotonia and psychomotor retardation. This would have been considered as the probable cause of the phenotype if she was not a compound heterozygote for loss of function variants in VPS13B, associated with Cohen syndrome. The patient presented many clinical signs of Cohen syndrome in addition to ID, including low birth weight (2300 g), short stature (<5 centile), microcephaly (<<2 centile), palpebral ptosis, myopia, decreased visual acuity, optic atrophy, and neutropenia, but loss of function of both VPS13B alleles would have led to Cohen syndrome diagnosis even if phenotypic data were not available. It is disturbing to realize that the NALCN variants, which were predicted to be damaging/likely damaging by SIFT and PolyPhen-2, respectively, would likely be considered responsible for the phenotype in the absence of the VPS13B variants. However, we cannot exclude the possibility that the NALCN variants contribute to the patient’s phenotype.
In the last few years, exome sequencing has become an important clinical tool in genetic diagnosis, at least in developed countries. An issue on the procedure is whether only the exome of the proband should be sequenced or sequencing the trio (probands plus parents) would be more cost-effective. Obviously, the latter approach costs three times more, and the magnitude of the increase in diagnostic yield is not clear, depending on the criteria of patient referral. Using the trio analyses of exome sequencing, we identified the probable genetic cause of the clinical phenotype in five out of eight patients with sporadic syndromic ID, which resulted in a diagnostic rate of 62%. It is important to note that some rare variants that were considered as candidate for the phenotypes were later excluded with the release of a database of variants in the Brazilian Population (http://abraom.ib.usp.br/), being relatively common variants among Brazilians.18
In the two patients with the diagnosis of recessive disorders, the causative mutations would have been identified even if only the proband exomes had been sequenced. However, in the three patients with dominant disorders, parental exome sequencing was instrumental to reach the conclusion that the de novo variants were likely pathogenic. A recent publication from the Deciphering Developmental Disorders Study on the exomes of 4,293 families reported damaging de novo mutations in 42% of the cohort.29 These results clearly show that parental exome sequencing is fundamental for efficient diagnosis in isolated cases.
Acknowledgments
This work was supported by scholarships from the Brazilian National Council for Scientific and Technological Development (CNPq—306879/2014-0; CR) and grants from the São Paulo Research Foundation (FAPESP—2012/50981-5 and 2013/08028-1). We thank our colleagues from the University of São Paulo who contributed with ideas and expertise for the research and, of course, our collaborators from other department of the University of São Paulo and AC Camargo Cancer center for all the help during this work. We would like to especially thank those who took time to read and improve this paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Topper S, Ober C, Das S. Exome sequencing and the genetics of intellectual disability. Clin Genet. 2011;80(2):117–126. | ||
Moeschler JB, Shevell M; American Academy of Pediatrics Committee on Genetics. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics. 2006;117(6):2304–2316. | ||
Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11:161–187. | ||
Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17(1):9–18. | ||
Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42(12):1109–1112. | ||
Çalışkan M, Chong JX, Uricchio L, et al. Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum Mol Genet. 2011;20(7):1285–1289. | ||
Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478(7367):57–63. | ||
Philips AK, Sirén A, Avela K, et al. X-exome sequencing in Finnish families with intellectual disability – four novel mutations and two novel syndromic phenotypes. Orphanet J Rare Dis. 2014;9:49. | ||
Bissar-Tadmouri N, Donahue WL, Al-Gazali L, Nelson SF, Bayrak-Toydemir P, Kantarci S. X chromosome exome sequencing reveals a novel ALG13 mutation in a nonsyndromic intellectual disability family with multiple affected male siblings. Am J Med Genet A. 2014;164A(1):164–169. | ||
Volk A, Conboy E, Wical B, Patterson M, Kirmani S. Whole-exome sequencing in the clinic: lessons from six consecutive cases from the clinician’s perspective. Mol Syndromol. 2015;6(1):23–31. | ||
Monroe GR, Frederix GW, Savelberg SM, et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet Med. 2016;18(9):949–956. | ||
Chérot E, Keren B, Dubourg C, et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: experience of 2 clinical units and 216 patients. Clin Genet. 2018;93(3):567–576. | ||
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. | ||
DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. | ||
Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. | ||
Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. | ||
Lek M, Karczewski KJ, Minikel EV, et al; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. | ||
Naslavsky MS, Yamamoto GL, de Almeida TF, et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat. 2017;38(7):751–763. | ||
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. | ||
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. | ||
Shihab HA, Gough J, Mort M, Cooper DN, Day IN, Gaunt TR. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum Genomics. 2014;8:11. | ||
Richards S, Aziz N, Bale S, et al; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. | ||
Appenzeller S, Balling R, Barisic N, et al; EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2014;95(4):360–370. | ||
Hamdan FF, Myers CT, Cossette P, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. 2017;101(5):664–685. | ||
Yoo Y, Jung J, Lee YN, et al. GABBR2 mutations determine phenotype in Rett syndrome and epileptic encephalopathy. Ann Neurol. 2017;82(3):466–478. | ||
Snijders Blok L, Madsen E, Juusola J, et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am J Hum Genet. 2015;97(2):343–352. | ||
Hamilton MJ, Caswell RC, Canham N, et al. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J Med Genet. 2018;55(1):28–38. | ||
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433–438. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.