")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Ursolic acid reduces the metalloprotease/anti-metalloprotease imbalance in cerebral ischemia and reperfusion injury
Received 7 January 2016
Accepted for publication 14 March 2016
Published 17 May 2016 Volume 2016:10 Pages 1663—1674
DOI https://doi.org/10.2147/DDDT.S103829
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Yanzhe Wang, Zhiyi He, Shumin Deng
Department of Neurology, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China
Background: Activators of PPARs, particularly PPARγ, may be effective neuroprotective drugs against inflammatory responses in cerebral ischemia and reperfusion injury. Ursolic acid (UA) may act as a PPARγ agonist and serve as an anti-inflammatory agent. In this study, we used a rat middle cerebral artery occlusion and reperfusion model to examine how UA acts as a neuroprotective agent to modulate the metalloprotease/anti-metalloprotease balance.
Methods: The middle cerebral artery occlusion and reperfusion model (occlusion for 2 hours followed by reperfusion for 48 hours) was induced in male Sprague Dawley rats. UA was administered intragastrically 0.5, 24, and 47 hours after reperfusion. Bisphenol A diglycidyl ether (a PPARγ antagonist) was intraperitoneally administered 1, 24.5, and 47.5 hours after reperfusion. Forty-eight hours after reperfusion, neurological deficits and infarct volume were estimated. The PPARγ level and the metalloprotease/anti-metalloprotease balance were examined by Western blotting and immunohistochemistry. The activation of MAPK signaling pathways was also assessed.
Results: UA-treated (5, 10, or 20 mg/kg) rats showed significant improvement in neurological deficit score, infarct volume, and the number of intact neurons compared with control rats (P< 0.01). Both the PPARγ protein level and the percentage of PPARγ-positive cells were increased in the UA-treated groups (P< 0.01). Compared with the control group, the UA-treated groups exhibited reduced protein levels of MMP2, MMP9, and activated MAPKs (P< 0.01) but an increased level of TIMP1 (P< 0.01). UA exerted its protective effects in a dose-dependent manner. Co-treatment with UA and bisphenol A diglycidyl ether completely abolished the UA-induced changes in PPARγ expression; however UA continued to exert a significant but partial neuroprotective effect.
Conclusion: UA can act as a PPARγ agonist to improve the metalloprotease/anti-metalloprotease balance, possibly by inhibiting the activation of the MAPK signaling pathway, thereby attenuating cerebral ischemia and reperfusion injury. Therefore, UA may serve as a novel neuroprotective therapeutic agent.
Keywords: brain ischemia, ursolic acid, peroxisome proliferator-activated receptors, matrix metalloproteases
Corrigendum for this paper has been published
Introduction
Ischemic stroke, also referred to as cerebral infarction, is a common and life-threatening disease that accounts for the majority of cerebrovascular diseases worldwide. Cerebral ischemia results from a transient or permanent local reduction in cerebral blood flow (CBF).1 Although rapid reperfusion therapy, such as thrombolysis, within the therapeutic window is widely accepted, post-reperfusion injury usually exacerbates the brain injury caused by the ischemic event.
There has been increasing effort to find effective and beneficial neuroprotective drugs to treat ischemia. Furthermore, an increasing number of researchers have shown interest in PPARs, nuclear hormone receptor family members that appear to regulate immune and inflammatory responses by inhibiting the expression of proinflammatory genes.2 After transient focal ischemia in rats, PPARγ expression and activation are reduced in the ischemic brain relative to the nonischemic brain.3 The PPARγ agonist rosiglitazone has been approved for use as a neuroprotective agent. Additionally, the recent observation that the PPARγ ligand pioglitazone attenuates ischemic neuronal damage, indicates that PPARγ activators may be effective neuroprotective drugs against ischemic injury.4 Studies have shown that PPARγ agonists help decrease excessive MMP2 and MMP9 activities to reduce the metalloprotease/anti-metalloprotease imbalance that is associated with emphysema in chronic obstructive pulmonary disease.5 Experiments have shown that metalloproteases perform a harmful function that is related to the severity of ischemic brain injury.
Ursolic acid (UA, 3b-hydroxy-urs-12-ene-28-oic acid), a natural pentacyclic triterpenoid, is one of the main components of certain medicinal plants. UA has a broad spectrum of biological activities, such as anti-inflammatory, anti-oxidative, antitumor, antirheumatic, and antiviral activities.6 UA may act as a PPARγ agonist to suppress ovalbumin-induced airway inflammation in a mouse model of allergic asthma.7 UA can also attenuate subarachnoid hemorrhage injury by decreasing the expression of inflammatory mediators, such as MMP9.8
In the present study, we used a middle cerebral artery occlusion and reperfusion (MCAO/R) rat model together with UA and bisphenol A diglycidyl ether (BADGE) treatment to examine how UA acts as a neuroprotective agent in a PPARγ-dependent manner. We investigated whether UA altered the MCAO/R-induced metalloprotease/anti-metalloprotease imbalance and explored the molecular mechanism underlying this imbalance.
Materials and methods
Animals
All experiments were conducted in accordance with the guidelines of the Research Ethics Committee of China Medical University. All experimental protocols involving animals were performed according to the guidelines of the National Institute of Health Guide for the Care and Use of Laboratory Animals. A total of 162 male Sprague Dawley rats were purchased from Liaoning Changsheng Biotechnology Company (Benxi, People’s Republic of China) and housed under a 12-hour day/12-hour night cycle with free access to food and water. Rats weighing between 250 and 280 g were randomly divided into nine groups: 1) sham group (n=18): rats underwent the same surgical procedures as MCAO/R except for filament insertion and received the vehicle; 2) control group (n=18): rats underwent surgical procedures for MCAO/R and received the vehicle; 3) MCAO/R +5 mg/kg UA intragastrically (ig) group (n=18); 4) MCAO/R +10 mg/kg UA ig group (n=18); 5) MCAO/R +20 mg/kg UA ig group (n=18); 6) MCAO/R +5 mg/kg UA ig +30 mg/kg BADGE intraperitoneally (ip) group (n=18); 7) MCAO/R +10 mg/kg UA ig +30 mg/kg BADGE ip group (n=18); 8) MCAO/R +20 mg/kg UA ig +30 mg/kg BADGE ip group (n=18); and 9) MCAO/R +30 mg/kg BADGE ip group (n=18).
UA (purity ≥95.0%, Sigma-Aldrich Co., St Louis, MO, USA) in distilled water containing 0.5% Tween-80 (ddH2O/0.5% Tween-80) was administered via oral gavage 0.5, 24, and 47 hours after reperfusion. BADGE (Sigma-Aldrich Co.) in ddH2O/0.5% Tween-80 was administered ip 1, 24.5, and 47.5 hours after reperfusion. The sham and control groups received the ddH2O/0.5% Tween-80 vehicle ig at the same time that the other treatment groups received UA or BADGE.
Forty-eight hours after reperfusion, the 18 rats in each group were randomly divided into three subgroups by a researcher who was unaware of their neurological deficits. For each group, six rats were decapitated to obtain fresh brain tissue samples for biochemical analyses. Six rats were used for 2,3,5-triphenyltetrazolium chloride (TTC) staining to determine the infarct volume; and six rats were given the fixative perfusion for histological preparation and analysis.
Experimental MCAO/R model
Surgical procedures for MCAO/R were performed on the rats using the previously described intracranial suture method.9–12 Rats were anesthetized with 10% chloral hydrate (350 mg/kg). Body temperature was monitored and maintained at 36.5°C–37.5°C using a heating pad and heating lamp during the operation. Briefly, after a 2 cm midline skin incision was created, the right common carotid artery, external carotid artery, and internal carotid artery were isolated. Then, a 5 cm nylon monofilament (diameter 0.26 mm) with a rounded tip coated with silicon (Jialing Biotechnology Company, Guangzhou, People’s Republic of China) was inserted into the right internal carotid artery through the broken end of the external carotid artery to block the origin of the MCA (approximately 18±2 mm) with 10 mm of the filament left outside the body. Cerebral ischemia caused by the placement of the intraluminal suture was maintained for 120 minutes; afterward, the suture was removed to achieve reperfusion. Rats in the sham group underwent the same surgical procedures except for filament insertion. CBF was monitored during all operative procedures. Success of the MCAO/R model was defined as a minimum 80% decrease in CBF during MCAO and a return to at least 80% of the original CBF after reperfusion based on CBF monitoring.
Neurological deficit analysis
Neurological deficits were scored 48 hours after reperfusion by other investigators, who were blinded to the experimental groups (n=18, per group). The scoring criteria for neurological deficits were as follows; this evaluation method was described previously:12–14
- 0 points: no neurological deficit;
- 1 point: difficulty in fully extending the contralateral forelimb;
- 2 points: unable to fully extend the contralateral forelimb;
- 3 points: circling to the contralateral side;
- 4 points: no consciousness or ambulation.
The higher the neurological deficit score, the more severe the impairment of motor function.
Infarct volume measurement
Infarct volume was assessed 48 hours after reperfusion (n=6, each group). As described in detail previously,15 rats were deeply anesthetized with chloral hydrate (400 mg/kg), and coronary brain slices (thickness: 2 mm) were stained using 0.5% TTC (Sigma-Aldrich Co.). The stained slices were photographed and quantified using Image Pro Plus 6.0.
To compensate for the effect of postischemic edema on the volume of injury, the lesion volumes were corrected using the following formula:
|
|

Nissl staining and immunohistochemical staining
Brain samples from each animal (n=6, each group) were sectioned into three slices, starting 3 mm from the anterior tip of the frontal lobe in the coronal plane. The first, second, and third slices from front to back were 3, 4, and 3 mm thick, respectively. The second slices were embedded in paraffin, cut into 5 μm sections, and then mounted on poly-L-lysine-coated slides. Nissl staining was performed using 1% cresyl violet (Sigma-Aldrich Co.) solution. We used rat antibodies against PPARγ (1:50; Abcam PLC, Cambridge, UK), MMP2 (1:50; Abcam PLC), MMP9 (1:50; Abcam PLC), and TIMP1 (1:50; Abcam PLC) to determine the immunolocalization of each respective protein. Primary antibodies were recognized using a biotinylated anti-rabbit or anti-mouse secondary antibody at room temperature for 1 hour and detected using a streptavidin-peroxidase kit (Maixin, Fuzhou, People’s Republic of China) at room temperature for 1 hour. Images were captured using a visible microscope objective lens. For images obtained using a 40×10 optical microscope, we used Image-Pro Plus 6.0 for analysis.
In Nissl-stained sections, only intact neurons were counted. In order to ensure that the intact neurons were counted at the same coronal level, we collected ten 5 μm coronal sections of the dorsal hippocampus (-3.3 mm to -4.5 mm from bregma). The number of intact neurons in one section was averaged from three non-overlapping fields at the same site of ischemic cortex, and six rats in each group were quantified. Cells in which PPARγ had translocated from the cytoplasm to the nucleus were considered PPARγ-positive cells. The PPARγ-positive cells were identified, counted, and analyzed in each section used for immunochemistry. The investigators were blinded to the experimental groups during analysis. The average percentage of PPAR-positive cells in each rat brain section was calculated. For each section, three sequential non-overlapping fields in the ischemic cortical region receiving blood supply from the MCA were randomly selected and observed. Three sections of each rat brain sample and six rats in each group were examined. To verify signal specificity for PPARγ, MMP2, MMP9, and TIMP1, control sections were incubated with only primary or secondary antibody in parallel with the experimental samples. Positive staining was not observed in any control section.
Western blot analysis
Proteins were isolated from the ischemic portion of the cerebral cortex of each animal (n=6 per group) for Western blot analysis. As previously described in detail, protein samples were separated via 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA USA). The membrane was incubated with the following primary rat antibodies overnight at 4°C: anti-PPARγ (1:500; Abcam PLC), anti-MMP2 (1:500; Abcam PLC), anti-MMP9 (1:500; Abcam PLC), anti-TIMP1 (1:500; Abcam PLC), anti-di-phosphorylated pERK1/2 (1:500; Abcam PLC), anti-pJNK (1:500; Abcam PLC), or anti-pp38 (1:500; Abcam PLC). After the membrane was washed three times, it was incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Zhongshan-Golden Bridge, Beijing, People’s Republic of China) (1:2,000 dilution) for 2 hours at room temperature. To confirm equal loading, the membranes were analyzed for GADPH expression using an anti-GADPH antibody (1:500 dilution, Santa Cruz Biotechnology Inc., Dallas, TX, USA). The mean density and area of each band was quantified using ImageJ.
Statistical analysis
All data were expressed as the mean values ± standard deviation and analyzed via one-way analysis of variance using SPSS 20.0 (IBM Corporation, Armonk, NY, USA). P<0.05 was defined as a significant difference. The neurological deficit scores for different groups were compared using the Kruskal–Wallis test. When the Kruskal–Wallis test showed a significant difference, the Mann–Whitney U-test with Bonferroni correction was applied.
Results
Effects of UA on neurological deficits and general condition
Compared with the sham group, the control group of rats displayed significant motor deficits (Figure 1A). Rats treated with UA (5, 10, or 20 mg/kg) showed significant improvements in the general condition and increases in neurological deficit scores after treatment compared with the control rats, and BADGE co-treatment partly but significantly abolished the improvements caused by UA treatment (Figure 1A). The protective effect of UA on neurological deficits was dose dependent. With increasing concentration of UA, the neurological deficit score and general condition were improved, and the median neurological deficit score was decreased.
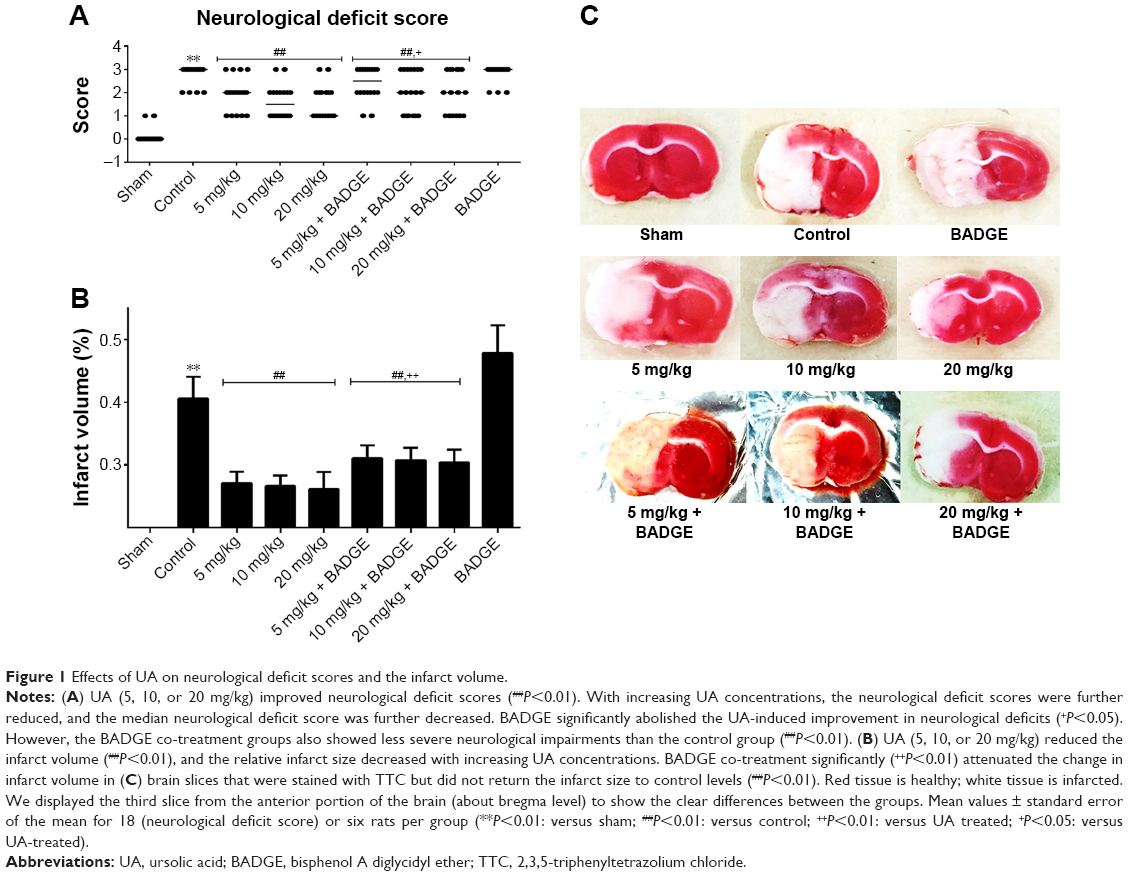 | Figure 1 Effects of UA on neurological deficit scores and the infarct volume. |
Effect of UA on infarct volume
No infarction was observed in the sham group (Figure 1B and C). The infarct volume was clearly smaller in the UA-treated rats (5, 10, or 20 mg/kg) than in the control rats, and UA decreased the infarct volume in a dose-dependent manner (Figure 1B and C). However, BADGE co-treatment partly but significantly diminished the UA-induced reduction in infarct volume (Figure 1B and C).
Effect of UA on MCAO/R-induced histological changes in the brain
UA treatment (5, 10, or 20 mg/kg) significantly alleviated the histological damage to the rat brain (Figure 2). We used Nissl staining to observe neurons in the ischemic core in three sequential non-overlapping fields at the same coronal level and the same site of cortex. The number of intact neurons was decreased after MCAO/R, and the number of injured neurons exhibiting cell shrinkage was increased (Figure 2). UA treatment at 5–20 mg/kg increased the number of intact neurons in a dose-dependent manner, whereas BADGE co-treatment partly but significantly blocked this improvement (Figure 2).
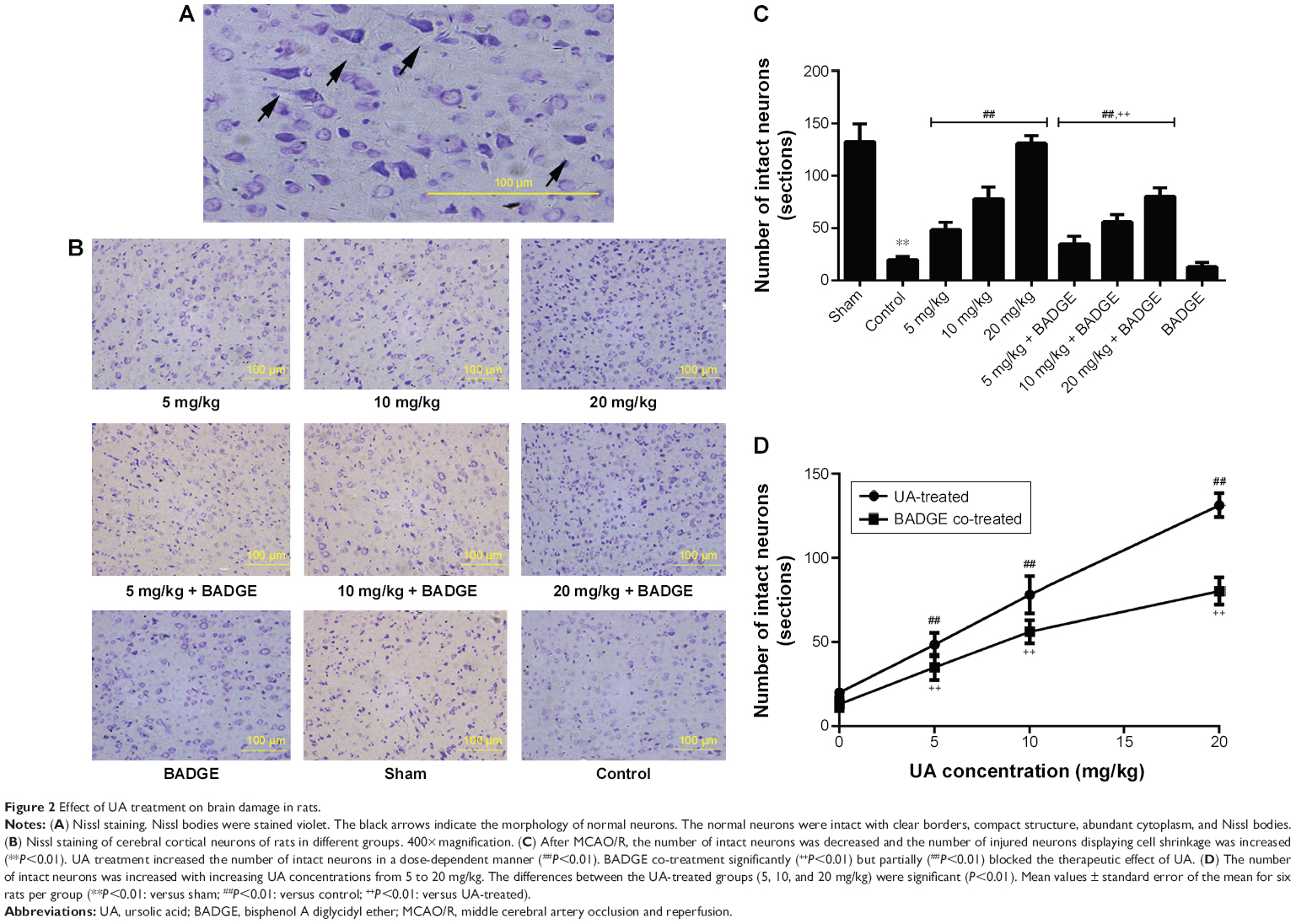 | Figure 2 Effect of UA treatment on brain damage in rats. |
Effect of UA on PPARγ
Brain cells in which PPARγ had translocated to the nucleus were considered PPARγ-positive cells. Fewer PPARγ-positive cells were present in the control group than in the sham group (Figure 3). UA treatment (5, 10, or 20 mg/kg) increased the percentage of PPARγ-positive cells in the injured region (Figure 3). These results demonstrated that UA significantly increased PPARγ immunoreactivity. Semi-quantitative analysis of immunohistochemistry produced the same results as Western blot analysis. Cerebral ischemia and reperfusion injury significantly decreased the PPARγ protein level, as indicated by a comparison of the control group with the sham group (Figure 3). The results of Western blot and immunochemical analyses showed that UA treatment increased the PPARγ protein level in a dose-dependent manner. Co-treatment with BADGE completely inhibited the UA-induced change in the PPARγ protein level, and 30 mg/kg BADGE abolished the PPARγ-agonistic effect of UA (5, 10, and 20 mg/kg), consequently reducing PPARγ protein expression to control levels (Figure 3).
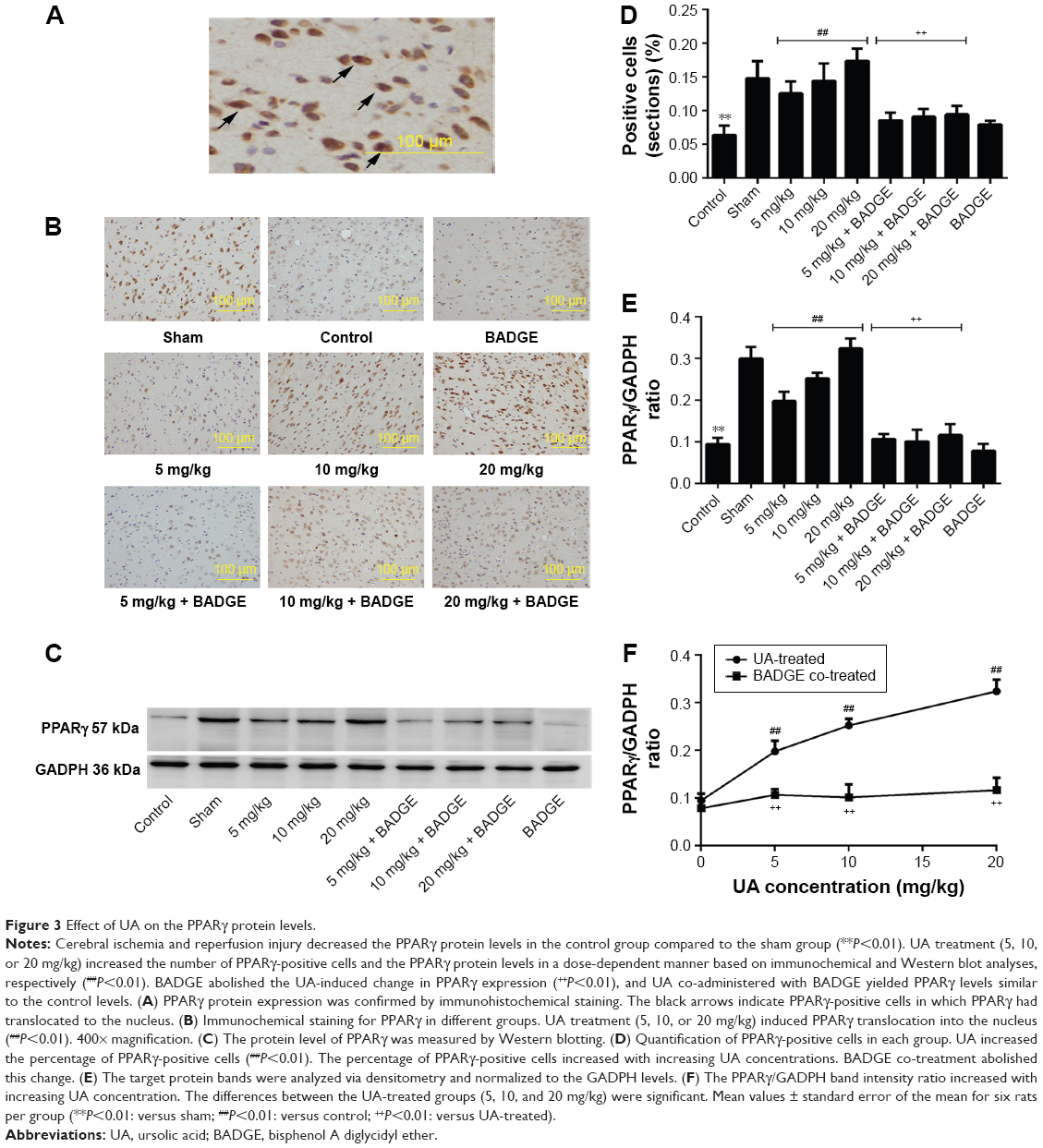 | Figure 3 Effect of UA on the PPARγ protein levels. |
Effect of UA on the metalloprotease/anti-metalloprotease balance
Cerebral ischemia and reperfusion increased the protein levels of MMP2 and MMP9 and attenuated the protein level of TIMP1, as indicated by a comparison of the control group with the sham group (Figures 4 and 5). UA treatment (5, 10, or 20 mg/kg) significantly decreased the MMP2 and MMP9 protein levels and increased the TIMP1 protein level. Moreover, we distinguished the active form of MMP9 from its inactive form. UA impaired both the protein and enzymatic activity levels of MMP9. With increasing UA concentrations, the metalloprotease/anti-metalloprotease balance was further restored. This dose-dependent effect was apparent based on both Western blot and immunochemical analyses. Comparison of the UA-treated groups and the BADGE co-treatment groups revealed that the UA-induced reductions in the MMP2 and MMP9 levels, and the increase in the TIMP1 level was partly but significantly blocked by BADGE (Figures 4 and 5).
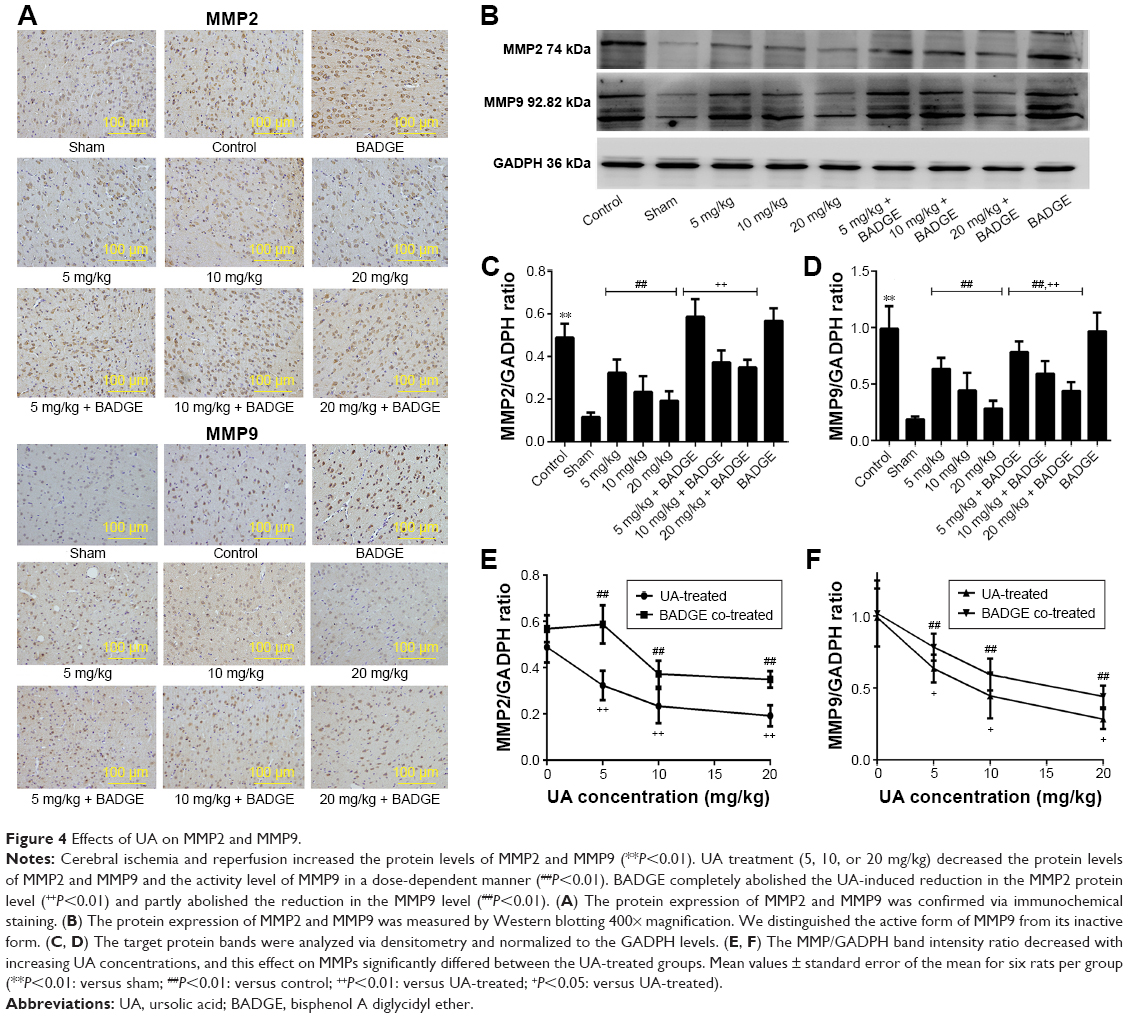 | Figure 4 Effects of UA on MMP2 and MMP9. |
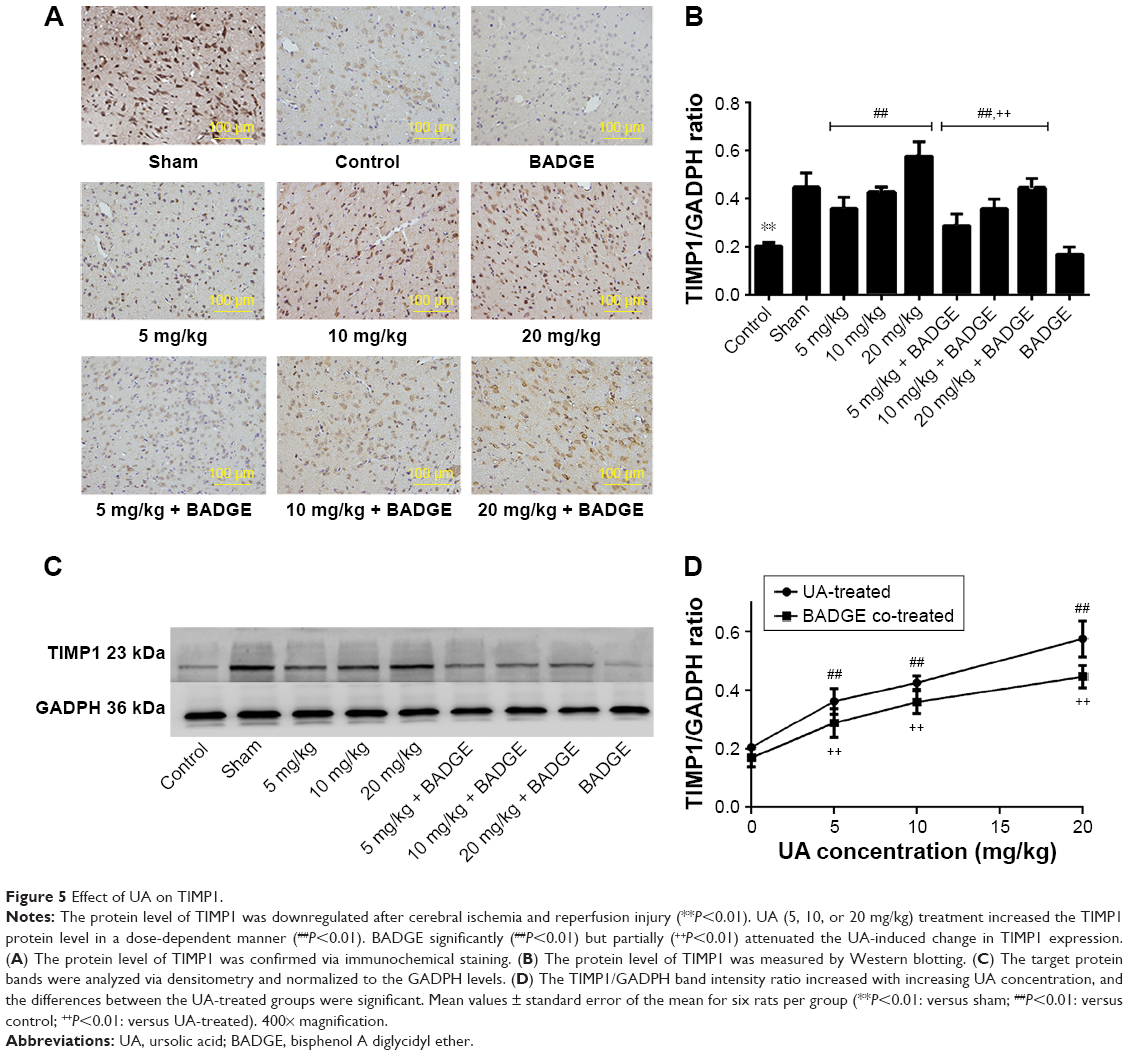 | Figure 5 Effect of UA on TIMP1. |
Effect of UA on the MAPK signaling pathway
We examined the effect of UA on the MAPK pathway to identify the mechanism by which UA regulates the metalloprotease/anti-metalloprotease balance. Western blot analysis showed significantly higher levels of pp38 MAPK, pERK1/2, and pJNK1/2 in the control group than in the sham group. UA treatment (5, 10, or 20 mg/kg) significantly decreased the pERK1/2, pJNK1/2, and pp38 protein levels and suppressed the activation of the MAPK signaling pathway in a dose-dependent manner. Comparison of the UA-treated and BADGE co-treatment groups indicated that the BADGE co-treatment partly but significantly blocked the UA-induced changes in MAPK activities (Figure 6).
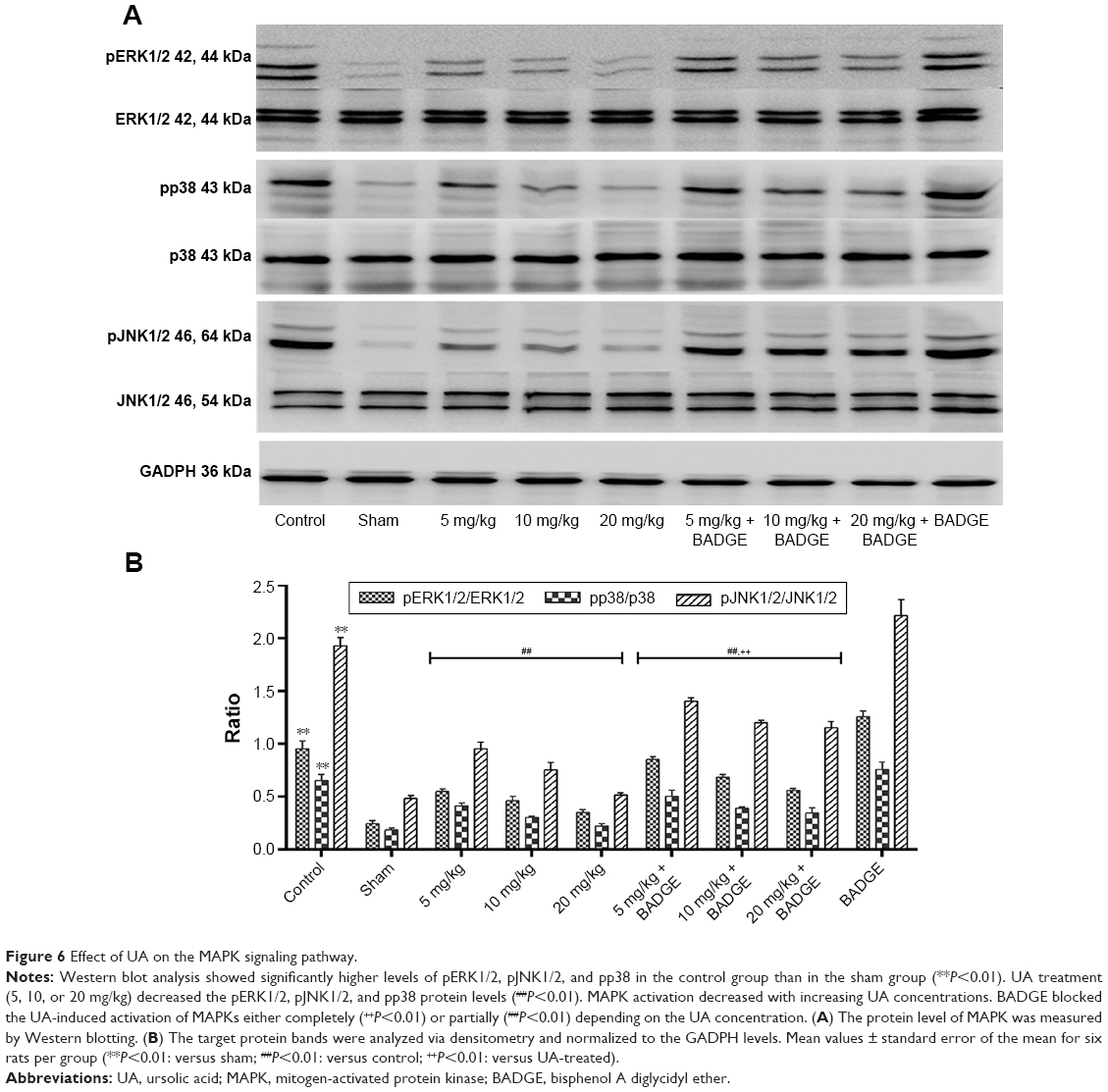 | Figure 6 Effect of UA on the MAPK signaling pathway. |
Discussion
UA, a natural pentacyclic triterpenoid, has already been revealed to be neuroprotective in several studies. Recently, UA has been reported to protect the brain against ischemia and reperfusion injury via its anti-oxidative and anti-inflammatory effects after MCAO in mice.16 UA also protected neurons by attenuating the brain inflammatory response to improve cognitive dysfunction in chronic (but not acute) brain injuries such as neurodegenerative diseases that are induced by inflammatory responses.4,17–19 In our study, we found that UA reduced neurological deficits and decreased infarct volume when administered ig at a dose of 5, 10, or 20 mg/kg after reperfusion. Consistent with previous studies, our study indicated that UA was an effective therapeutic agent against cerebral ischemia and reperfusion injury. Furthermore, several studies have suggested that UA attenuated ischemia and reperfusion injury in regions other than the brain such as the heart and the kidney.20–24 UA may also hold potential as a therapeutic agent to prevent muscle ischemic injury; this evidence may provide new insight into the effects of UA on ischemic vascular diseases.24
As widely acknowledged, abrupt reoxygenation due to the rapid restoration of blood flow is frequently associated with an exacerbation of tissue injury and an intense inflammatory response that is triggered by inflammatory mediators, such as MMPs.25,26 Studies have shown that the activity of MMPs, particularly MMP9, is markedly increased after infarction and is involved in infarct size growth.27,28,42 We found that cerebral ischemia and reperfusion injury activated MMP2 and MMP9 and inhibited TIMP1 in the control group compared to the sham group, and the observed metalloprotease/anti-metalloprotease imbalance paralleled the ischemic volume. Our observation supports the proposal that the metalloprotease/anti-metalloprotease balance plays a central role in brain injury after transient ischemia, and that the anti-inflammatory effect of UA might underlie its neuroprotective activity. Recent evidence has further shown that the infarct size was reduced in MMP9-deficient mice compared with control mice, and this finding suggests that excessive activation of MMP9 is deleterious to the brain and that MMP9 inhibitors may serve as therapeutic agents for ischemia and reperfusion injury.29,30
The nuclear hormone receptor PPARγ plays a vital role in a diverse range of biological processes, including the prevention of acute inflammation. Several studies have demonstrated that pharmacological PPARγ activation suppressed the inflammatory response in cerebral ischemia,31,32,43 and this finding suggests that PPARγ might be critical in cerebral ischemia and reperfusion injury. In our study, we observed that the PPARγ protein levels and the percentage of PPARγ-positive cells were increased after UA treatment in a dose-dependent manner, and these results demonstrated that UA enhanced PPARγ function. Our study provided compelling evidence that UA activated PPARγ; this finding was consistent with the previous observation that UA isolated from Chromolaena odorata acts as PPARγ agonist.33 In addition, BADGE, a PPARγ antagonist, abolished the agonistic effect of UA on PPARγ. Co-treatment with UA and BADGE resulted in PPARγ levels similar to the control levels, and the changes in MMP2, MMP9, and TIMP1 expression were significantly diminished by BADGE co-treatment. Taken together, our results clearly showed that UA acted as a PPARγ agonist to reduce the metalloprotease/anti-metalloprotease imbalance that could suppress cerebral ischemia and reperfusion injury. In line with previous studies, this PPARγ agonist could protect against ischemia and reperfusion injury by reducing the activities of MMPs in other organs. In our study, we have shown for the first time that UA can act as a PPARγ agonist to attenuate cerebral ischemia and reperfusion injury by restoring the balance between MMPs and TIMP. Nevertheless, BADGE co-treatment completely abolished the UA-induced change in PPARγ expression. However, UA continued to have a clear influence on MMPs and TIMP activities, and this result indicated that UA might affect the metalloprotease/anti-metalloprotease imbalance via alternative mechanisms.
As a common family of inflammatory mediators in the brain, MMPs have been shown to be increasingly dependent on activation of the MAPK signaling pathway under numerous inflammatory conditions. Activated MAPK is known to be involved in inflammatory mediator production in cerebral ischemia and reperfusion injury.34 According to more recent findings, the metalloprotease/anti-metalloprotease imbalance is reduced via a mechanism involving MAPK inhibition under many pathologic conditions.35–37 In the present study, we evaluated the protein levels of MAPKs (JNK1/2, p38 MAPK, and ERK1/2) after MCAO/R and UA treatment. The levels of pp38, pERK1/2, and pJNK1/2 were elevated in the control group compared with the sham group, and UA inhibited p38, ERK1/2, and JNK1/2 activation in a dose-dependent manner. These results suggest that the suppressive effect of UA on the MMP levels after MCAO/R may be related to its reduction of MAPK pathway activation, which could be involved in the neuroprotective effects of UA. Our findings provide new evidence that decreasing MAPK pathway activation, which further affected the metalloprotease/anti-metalloprotease imbalance, could be neuroprotective in cerebral ischemia and reperfusion injury. In addition, BADGE significantly reduced the UA-induced effects on MAPK activation, and this finding indicates that PPARγ plays an important role in regulating MMPs, probably through the MAPK pathway.
Previous studies have demonstrated that an intact form of UA was available to the brain after oral intake, and the detection of UA in the brain suggested that UA can penetrate the brain–blood barrier.38,39 These findings support the possibility of using UA to treat cerebral ischemia and reperfusion injury. Moreover, clinical studies showed that liposomes have been utilized as a drug delivery system to overcome the poor solubility of UA to enhance the bioavailability of this drug.40,41
Our study has only examined whether UA acts as a PPARγ agonist to maintain the metalloprotease/anti-metalloprotease balance, thus attenuating cerebral ischemia and reperfusion injury. However, when BADGE co-treatment returned PPARγ expression to the control levels, the neuroprotective effect of UA was only partly attenuated. Because UA performs several biological functions, MMPs could be modulated via the multiple pathways in which UA may participate. For example, UA was reported to decrease MMP9 expression by regulating the TLR4 signaling pathway in experimental subarachnoid hemorrhage models.8 Further study is necessary to clarify the mechanism by which UA reduces the metalloprotease/anti-metalloprotease imbalance in cerebral ischemia and reperfusion injury, and the other biological activities by which UA exerts its outstanding neuroprotective effect.
Conclusion
In summary, the results from our rat focal cerebral ischemia and reperfusion model suggests that UA, a PPARγ agonist, maintained the metalloprotease/anti-metalloprotease balance, possibly by inhibiting the MAPK signaling pathway activation, to attenuate cerebral ischemia and reperfusion injury. These mechanistic insights may support the use of UA as a novel neuroprotective therapeutic agent.
Disclosure
The authors have no conflicts of interest or disclosures regarding the results presented in this paper.
References
Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics – 2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–e322. | ||
Kostadinova R, Wahli W, Michalik L. PPARs in diseases: control mechanisms of inflammation. Curr Med Chem. 2005;12(25):2995–3009. | ||
Victor NA, Wanderi EW, Gamboa J, et al. Altered PPARgamma expression and activation after transient focal ischemia in rats. Eur J Neurosci. 2006;24(6):1653–1663. | ||
Wang YJ, Lu J, Wu DM, et al. Ursolic acid attenuates lipopolysaccharide-induced cognitive deficits in mouse brain through suppressing p38/NF-kappaB mediated inflammatory pathways. Neurobiol Learn Mem. 2011;96(2):156–165. | ||
Hou G, Yin Y, Han D, Wang QY, Kang J. Rosiglitazone attenuates the metalloprotease/anti-metalloprotease imbalance in emphysema induced by cigarette smoke: involvement of extracellular signal-regulated kinase and NFkappaB signaling. Int J Chron Obstruct Pulmon Dis. 2015;10:715–724. | ||
Ikeda Y, Murakami A, Ohigashi H. Ursolic acid: an anti- and pro-inflammatory triterpenoid. Mol Nutr Food Res. 2008;52(1):26–42. | ||
Kim SH, Hong JH, Lee YC. Ursolic acid, a potential PPARgamma agonist, suppresses ovalbumin-induced airway inflammation and Penh by down-regulating IL-5, IL-13, and IL-17 in a mouse model of allergic asthma. Eur J Pharmacol. 2013;701(1–3):131–143. | ||
Zhang T, Su J, Guo B, Zhu T, Wang K, Li X. Ursolic acid alleviates early brain injury after experimental subarachnoid hemorrhage by suppressing TLR4–mediated inflammatory pathway. Int Immunopharmacol. 2014;23(2):585–591. | ||
Jiang M, Li J, Peng Q, et al. Neuroprotective effects of bilobalide on cerebral ischemia and reperfusion injury are associated with inhibition of pro-inflammatory mediator production and down-regulation of JNK1/2 and p38 MAPK activation. J Neuroinflammation. 2014;11:167. | ||
Tan Z, Turner RC, Leon RL, et al. Bryostatin improves survival and reduces ischemic brain injury in aged rats after acute ischemic stroke. Stroke. 2013;44(12):3490–3497. | ||
Sun J, Tong L, Luan Q, et al. Protective effect of delayed remote limb ischemic postconditioning: role of mitochondrial K(ATP) channels in a rat model of focal cerebral ischemic reperfusion injury. J Cereb Blood Flow Metab. 2012;32(5):851–859. | ||
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. | ||
Yonemori F, Yamaguchi T, Yamada H, Tamura A. Evaluation of a motor deficit after chronic focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18(10):1099–1106. | ||
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–476. | ||
Tsubokawa T, Jadhav V, Solaroglu I, Shiokawa Y, Konishi Y, Zhang JH. Lecithinized superoxide dismutase improves outcomes and attenuates focal cerebral ischemic injury via antiapoptotic mechanisms in rats. Stroke. 2007;38(3):1057–1062. | ||
Li L, Zhang X, Cui L, et al. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013;1497:32–39. | ||
Yoon JH, Youn K, Ho CT, Karwe MV, Jeong WS, Jun M. p-Coumaric acid and ursolic acid from Corni fructus attenuated beta-amyloid(25–35)-induced toxicity through regulation of the NF-kappaB signaling pathway in PC12 cells. J Agric Food Chem. 2014;62(21):4911–4916. | ||
Hong SY, Jeong WS, Jun M. Protective effects of the key compounds isolated from Corni fructus against beta-amyloid-induced neurotoxicity in PC12 cells. Molecules. 2012;17(9):10831–10845. | ||
Lu J, Wu DM, Zheng YL, et al. Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-kappaB pathway activation. Cereb Cortex. 2010;20(11):2540–2548. | ||
Chen J, Ko KM. Ursolic-acid-enriched Herba Cynomorii extract protects against oxidant injury in H9c2 cells and rat myocardium by increasing mitochondrial ATP generation capacity and enhancing cellular glutathione redox cycling, possibly through mitochondrial uncoupling. Evid Based Complement Alternat Med. 2013;2013:924128. | ||
Radhiga T, Rajamanickam C, Senthil S, Pugalendi KV. Effect of ursolic acid on cardiac marker enzymes, lipid profile and macroscopic enzyme mapping assay in isoproterenol-induced myocardial ischemic rats. Food Chem Toxicol. 2012;50(11):3971–3977. | ||
Senthil S, Sridevi M, Pugalendi KV. Protective effect of ursolic acid against myocardial ischemia induced by isoproterenol in rats. Toxicol Mech Methods. 2007;17(1):57–65. | ||
Senthil S, Chandramohan G, Pugalendi KV. Isomers (oleanolic and ursolic acids) differ in their protective effect against isoproterenol-induced myocardial ischemia in rats. Int J Cardiol. 2007;119(1):131–133. | ||
Lee AW, Chen TL, Shih CM, et al. Ursolic acid induces allograft inflammatory factor-1 expression via a nitric oxide-related mechanism and increases neovascularization. J Agric Food Chem. 2010;58(24):12941–12949. | ||
Shichita T, Sakaguchi R, Suzuki M, Yoshimura A. Post-ischemic inflammation in the brain. Front Immunol. 2012;3:132. | ||
Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17(11):1391–1401. | ||
Morancho A, Rosell A, Garcia-Bonilla L, Montaner J. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann N Y Acad Sci. 2010;1207:123–133. | ||
Maddahi A, Chen Q, Edvinsson L. Enhanced cerebrovascular expression of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 via the MEK/ERK pathway during cerebral ischemia in the rat. BMC Neurosci. 2009;10:56. | ||
Svedin P, Hagberg H, Savman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007;27(7):1511–1518. | ||
Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20(12):1681–1689. | ||
Han L, Cai W, Mao L, et al. Rosiglitazone promotes white matter integrity and long-term functional recovery after focal cerebral ischemia. Stroke. 2015;46(9):2628–2636. | ||
Gliem M, Klotz L, van Rooijen N, Hartung HP, Jander S. Hyperglycemia and PPARgamma antagonistically influence macrophage polarization and infarct healing after ischemic stroke. Stroke. 2015;46(10):2935–2942. | ||
Zhang ML, Irwin D, Li XN, et al. PPARgamma agonist from Chromolaena odorata. J Nat Prod. 2012;75(12):2076–2081. | ||
Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22(6):631–647. | ||
Massaro M, Scoditti E, Carluccio MA, et al. Dipyridamole decreases inflammatory metalloproteinase-9 expression and release by human monocytes. Thromb Haemost. 2013;109(2):280–289. | ||
Deng X, Zhong Y, Gu L, Shen W, Guo J. MiR-21 involve in ERK-mediated upregulation of MMP9 in the rat hippocampus following cerebral ischemia. Brain Res Bull. 2013;94:56–62. | ||
Choi HJ, Park YR, Nepal M, et al. Inhibition of osteoclastogenic differentiation by Ikarisoside A in RAW 264.7 cells via JNK and NF-kappaB signaling pathways. Eur J Pharmacol. 2010;636(1–3):28–35. | ||
Peng W, Ding F, Jiang YT, Peng YK. Bioavailability and activity of natural food additive triterpenoids as influenced by protein. J Agric Food Chem. 2014;62(10):2271–2283. | ||
Udeani GO, Zhao GM, Geun Shin Y, et al. Pharmacokinetics and tissue distribution of betulinic acid in CD-1 mice. Biopharm Drug Dispos. 1999;20(8):379–383. | ||
Qian Z, Wang X, Song Z, et al. A phase I trial to evaluate the multiple-dose safety and antitumor activity of ursolic acid liposomes in subjects with advanced solid tumors. Biomed Res Int. 2015;2015:809714. | ||
Yang G, Yang T, Zhang W, Lu M, Ma X, Xiang G. In vitro and in vivo antitumor effects of folate-targeted ursolic acid stealth liposome. J Agric Food Chem. 2014;62(10):2207–2215. | ||
Reuter B, Rodemer C, Grudzenski S, et al. Effect of simvastatin on MMPs and TIMPs in human brain endothelial cells and experimental stroke. Transl Stroke Res. 2015;6(2):156–159. | ||
Zuhayra M, Zhao Y, von Forstner C, et al. Activation of cerebral peroxisome proliferator-activated receptors gamma (PPARgamma) reduces neuronal damage in the substantia nigra after transient focal cerebral ischaemia in the rat. Neuropathol Appl Neurobiol. 2011;37(7):738–752. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.