")
Back to Journals » Journal of Inflammation Research » Volume 14
Upregulation of COX-2 and PGE2 Induced by TNF-α Mediated Through TNFR1/MitoROS/PKCα/P38 MAPK, JNK1/2/FoxO1 Cascade in Human Cardiac Fibroblasts
Authors Yang CM , Yang CC , Hsiao LD, Yu CY, Tseng HC, Hsu CK, Situmorang JH
Received 6 April 2021
Accepted for publication 28 May 2021
Published 28 June 2021 Volume 2021:14 Pages 2807—2824
DOI https://doi.org/10.2147/JIR.S313665
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Chuen-Mao Yang,1– 3 Chien-Chung Yang,4,5 Li-Der Hsiao,1 Chia-Ying Yu,1 Hui-Ching Tseng,1 Chih-Kai Hsu,1 Jiro Hasegawa Situmorang1
1Department of Pharmacology, College of Medicine, China Medical University, Taichung, 40402, Taiwan; 2Ph.D. Program for Biotech Pharmaceutical Industry, China Medical University, Taichung, 40402, Taiwan; 3Department of Post-Baccalaureate Veterinary Medicine, College of Medical and Health Science, Asia University, Wufeng, Taichung, 41354, Taiwan; 4Department of Traditional Chinese Medicine, Chang Gung Memorial Hospital at Tao-Yuan, Kwei-San, Tao-Yuan, 33302, Taiwan; 5School of Traditional Chinese Medicine, College of Medicine, Chang Gung University, Kwei-San, Tao-Yuan, 33302, Taiwan
Correspondence: Chuen-Mao Yang No. 91, Hsueh-Shih Road, Taichung, 40402, Taiwan
Tel +886-4-22053366 (ext. 2229)
Email [email protected]
Purpose: Tumor necrosis factor-α (TNF-α) has been shown to exert as a pathogenic factor in cardiac fibrosis and heart failure which were associated with the up-regulation of cyclooxygenase (COX)-2/prostaglandin E2 (PGE2) axis. However, whether TNF-α-induced COX-2/PGE2 upregulation mediated through ROS-dependent cascade remains elusive in human cardiac fibroblasts (HCFs). This study aims to address the underlying mechanisms of TNF-α-induced COX-2/PGE2 expression.
Methods: Here, we used TNF receptor neutralizing antibody (TNFR nAb), pharmacologic inhibitors, and siRNAs to dissect the involvement of signaling components examined by Western blot and ELISA in TNF-α-mediated responses in HCFs. MitoSOX Red was used to measure mitoROS generation. Isolation of subcellular fractions was performed to determine membrane translocation of PKCα. Promoter luciferase assay and chromatin immunoprecipitation (ChIP) assay were used to determine the role of transcription factor.
Results: We found that TNF-α time- and concentration-dependently upregulated COX-2 protein and mRNA expression as well as PGE2 synthesis which was attenuated by TNFR1 nAb, the inhibitor of mitochondrial ROS scavenger (MitoTEMPO), protein kinase C [(PKC)α, Gö 6976], p38 MAPK [p38 inhibitor VIII, (p38i VIII)], JNK1/2 (SP600125), or forkhead box protein O1 [(FoxO1), AS1842856], and transfection with their respective siRNAs in HCFs. TNF-α-stimulated PKCα phosphorylation was inhibited by TNFR1 nAb, MitoTEMPO, or Gö 6976. TNF-α stimulated phosphorylation of p38 MAPK and JNK1/2 was attenuated by TNFR1 nAb, MitoTEMPO, Gö 6976, and their inhibitors p38i VIII and SP600125. Moreover, TNF-α-triggered FoxO1 phosphorylation was abolished by AS1842856, TNFR1 nAb, and its upstream inhibitors MitoTEMPO, Gö 6976, p38i VIII, and SP600125. Phosphorylation of FoxO1 could enhance its interaction with the COX-2 promoter element revealed by ChIP assay, which was attenuated by AS1842856.
Conclusion: Our results suggested that TNF-α-induced COX-2/PGE2 upregulation is mediated through TNFR1-dependent MitoROS/PKCα/p38 MAPK and JNK1/2 cascade to activate FoxO1 binding with the COX-2 promoter in HCFs.
Keywords: TNF-α, COX-2, PGE2, PKC-α, mitochondrial ROS, FoxO1
Introduction
Cardiac fibrosis is an important pathological process of heart remodeling following cardiovascular diseases and cardiac injuries from the mechanical force, chemical signals, or other insults. Cardiac fibroblasts, one of the major cells in heart tissue, contribute to the process of cardiac remodeling and fibrosis by producing a large amount of fibrotic extracellular matrix and depositing collagen to repair the cardiomyocyte damage, alter electric conduction, increase vessel resistance, and cause the failure of the left ventricle.1,2 Following a cardiac injury event, the innate immune system is activated which subsequently increases pro-inflammatory cytokines release including tumor necrosis factor-α (TNF-α).3 TNF-α causes cardiomyopathy, left ventricular dysfunction, heart failure, and pulmonary edema, when it is overexpressed in human subjects.4–7 However, the detailed mechanisms of TNF-α-induced responses in cardiac fibroblasts are still unclear. Thus, in the present study, we applied human cardiac fibroblasts (HCFs) to investigate the TNF-α -mediated inflammatory responses.
Cyclooxygenase-2 (COX)-2 is a well-known enzyme that can catalyze the generation of prostanoids such as prostaglandin E2 (PGE2). PGE2 is involved in many types of diseases particularly those related to inflammation. Previous studies have demonstrated that the COX-2/PGE2 axis is upregulated in various types of cells including fibroblasts upon exposure to TNF-α.8–10 In cardiac physiology and diseases, the exact role of COX-2 remains controversial due to either protective or detrimental in the hearts reported in human beings and experimental models. However, the elevation of PGE2 level could increase proliferation and cardiac fibrosis characteristics mediated through EP1 receptor via stimulating calcium signal pathway but not EP3 and EP4 in cardiac fibroblasts.11,12 Nonetheless, the role of TNF-α in COX-2 expression and PGE2 production remains elusive in HCFs. We hypothesize that TNF-α could induce COX-2 expression and PGE2 production in HCFs.
Excessive mitochondrial reactive oxygen species (mitoROS) generation due to mitochondrial dysfunction is implicated in triggering inflammation and contributing to cardiovascular diseases.13 Since intervention targeting mitoROS could be beneficial for cardiovascular diseases.14,15 Previous studies have indicated that ROS are involved in COX-2 upregulation in various types of cells.16,17 Protein kinase C (PKC) has been suggested as a target in the strategy of heart failure and ischemic heart disorders.18,19 The previous studies have shown that PKCα is required for COX-2 expression induced by TNF-α.10,20 Our previous study also demonstrated that lysophosphatidylcholine-induced COX-2 expression via a PKCα dependent cascade.21 MAPKs were also noticed to play a role in COX-2 expression by various stimuli including TNF-α in various types of cells.22–25 Our previous study, in HCFs, has revealed that COX-2 expression/PGE2 production is mediated through forkhead box protein O1 (FoxO1) activity.21 Therefore, the present study aimed to investigate whether TNF-α induced COX-2/PGE2 axis upregulation mediated through its own receptors, mitoROS, and protein kinases to activate transcription factor FoxO1 activity. Here, we demonstrated that TNF-α exposure increases COX-2 and PGE2 levels, at least in part, mediated through TNFR1/MitoROS/PKCα/p38 MAPK and JNK1/2/FoxO1 cascade in HCFs.
Materials and Methods
Materials
DMEM/F-12 medium, fetal bovine serum (FBS), and TRIzol reagent were purchased from Invitrogen (Carlsbad, CA). Hybond C membrane and enhanced chemiluminescence (ECL) Western blotting detection system were purchased from GE Healthcare Biosciences (Buckinghamshire, England, UK). Antibodies against TNF receptor (TNFR) 1 and 2, phospho-p38 MAPK, and phospho-JNK1/2 were purchased from Cell Signaling (Danvers, MA). SP600125, p38 inhibitor (p38i) VIII, Gö6976, MitoTEMPO, and AS1842856 were purchased from Enzo Life Science (Farmingdale, NY). Monoclonal anti-COX-2 antibody was purchased from NeoMarkers (Fremont, CA). Anti-GAPDH (mouse monoclonal antibody, Cat# MCA-1D4) antibody was obtained from EnCor Biotechnology (Gainesville, FL). Enzymes and other chemicals were purchased from Sigma (St. Louis, MO).
Cell Culture and Treatment
HCFs were purchased from ScienCell Research Laboratories (San Diego, CA) and cultured in DMEM/F-12 medium supplemented with 10% FBS and antibiotics (100 U/mL penicillin G, 100 ng/mL streptomycin, and 250 ng/mL fungizone) at 37°C in a humidified 5% CO2 atmosphere. The use of the cell lines had been approved by Chang Gung University Institutional Animal Care and Use Committee (Approval Document No. CGU 16–046). When the cells reached confluence, they were treated with 0.05% trypsin/0.53 mM EDTA for 1 min at 37°C. The cell suspension was diluted with DMEM/F-12 containing 10% FBS to a concentration of 2×105 cells/mL. The cell suspension was seeded in (1 mL/well) 12-well culture plates, (2 mL/well) 6-well culture plates, and (10 mL/dish) 10-cm culture dishes, and made quiescent at confluence by incubation in serum-free DMEM/F-12 for 24 h and then treated with TNF-α. The following experiments were carried out using HCF passages from 5 to 7.
Preparation of Samples and Western Blotting Analysis
HCFs were seeded in 12-well culture plates and made quiescent at confluence by incubation in serum-free DMEM/F-12 for 24 h. The cells were incubated with or without different concentrations of TNF-α at 37°C for the indicated time intervals. When inhibitors were applied, they were added 1 h prior to the exposure to TNF-α. The cell lysates were prepared as previously described.21 Then the denatured proteins were centrifuged at 13,000 × g for 30 sec. The mixed samples (15 μL) were subjected to SDS-PAGE using a 10% running gel. Proteins were transferred to nitrocellulose membrane and the membrane was incubated successively at room temperature with 5% (w/v) BSA in TTBS [Tris-HCl 50 mM, NaCl 150 mM, 0.05% (w/v), Tween 20, pH 7.4] for 1 h. Membranes were incubated overnight at 4°C with an antibody used at a dilution of 1:500–1000 in TTBS. Membranes were washed with TTBS several times and incubated with 1:2000 dilution of an anti-mouse or anti-rabbit antibody for 1 h. Following incubation, the membranes were washed comprehensively with TTBS. The immunoreactive bands were visualized by ECL reagent. The images of the immunoblots were captured using a UVP BioSpectrum 500 imaging system (Upland, CA), and densitometry analysis was conducted using UN-SCAN-IT gel software (Orem, UT).
Total RNA Extraction and Real-Time PCR Analysis
Total RNA was extracted with TRIzol according to the manufacturer’s instructions. First-strand cDNA synthesis was performed with 5 μg of total RNA using Oligo(dT)15 as primers in a final volume of 20 μL [25 ng/μL Oligo(dT)15, 0.5 mM dNTPs, 10 mM DTT, 2 units/μL RNase inhibitor, and 10 units/μL of Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA)]. The synthesized cDNAs were used as templates for PCR reaction using Q-Amp™ 2x screening fire Taq master mix (Bio-Genesis Technologies, Taipei, Taiwan) and primers for the target genes. Real-time PCR was performed with the TaqMan (GeneDireX®, San Diego, CA) gene expression assay system, using primer and probe mixes for COX-2 and endogenous GAPDH control genes. PCRs were performed using a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA). The relative amount of the target gene was calculated using 2(Ct test gene-Ct GAPDH) (Ct = threshold cycle). All experiments were performed in triplicate.
The cDNA obtained from 0.5 μg total RNA was used as a template for PCR amplification. Oligonucleotide primers were designed based on GenBank entries for human COX-2 and GAPDH. The following primers were used for amplification reaction:
COX-2
Forward primer: 5′-CAAACTGAAATTTGACCCAGAACTAC-3′;
Reverse primer: 5′-ACTGTTGATAGTTGTATTTCTGGTCATGA-3′;
Probe: 5′-AACACCCTCTATCACTGGCATCCCCTTC-3′.
GAPDH
Forward primer: 5′-GCCAGCCGAGCCACAT-3′;
Reverse primer: 5′-CTTTACCAGAGTTAAAAGCAGCCC-3′;
Probe: 5′- CCAAATCCGTTGACTCCGACCTTCA-3′.
Transfection and Promoter Luciferase Assay
Gene expression is derived from gene activation through various transcription activators and co-activators. In order to investigate TNF-α-induced COX-2 gene activity, COX-2-luciferase plasmid (COX-2-luci-plasmid) was constructed. COX-2-luci-plasmid was cloned with −459 to +9 of human COX-2 promoter region into pGL3-basic vector, as previously described.21 COX-2-luci-plasmid was transfected into HCFs together with β-galactosidase plasmid using the Lipofectamine reagent according to the manufacturer’s instructions. To assess promoter activity, after treatment with TNF-α, cells were collected and disrupted by sonication in lysis buffer (25 mM Tris, pH 7.8, 2 mM EDTA, 1% Triton X-100, and 10% glycerol). After centrifugation, aliquots of the supernatants were tested for luciferase activity using the luciferase assay system (Promega, Madison, WI) according to the manufacturer’s instructions. Firefly luciferase activities were standardized for β-galactosidase activity.
Measurement of PGE2 Release
The growth-arrested cells were treated with 15 ng/mL TNF-α for the indicated time intervals. The media were collected to analyze PGE2 levels by using an ELISA kit as specified by the manufacturer (Cayman Chemicals).
Determination of MitoROS
HCFs were seeded in 6-well culture plates with coverslips, shifted to serum-free DMEM/F-12, and then incubated with 15 ng/mL TNF-α for the indicated time intervals in the presence or the absence of pharmacological inhibitors for 1 h prior to TNF-α exposure. Then, cells were incubated in DMEM/F12 medium containing 5 μM of MitoSOX Red at 37°C for 10 min. The fluorescence signals were recorded (excitation/emission: 510 nm/570 nm) using a fluorescence plate reader (Synergy HT1; BioTek, Winooski, VT). To visualize mitoROS generation, the cells were washed thrice with medium, and their fluorescence intensity was determined by a fluorescence microscopy with a rhodamine filter (Axiovert 200M; Carl Zeiss, Thornwood, NY) and quantified using ImageJ software (1.41v; US National Institutes of Health).
Transient Transfection with siRNAs
SMARTpool RNA duplexes corresponding to JNK2 (HSS108550) and p38α (HSS102352, HSS102353, HSS175313) siRNA were purchased from Invitrogen Life Technologies (Carlsbad, CA), and PKCα (SASI_Hs01_00018816), JNK1 (SASI_Hs02_00319556), FoxO1FoxO1 (SASI_Hs01_0076732), and scramble siRNA were obtained from Sigma-Aldrich (St. Louis, MO). HCFs cultured onto 12-well plates at 70–80% confluence, transient transfection of siRNAs was carried out using Lipofectamine 2000 transfection reagent. Briefly, siRNA (100 nM) was formulated with Lipofectamine 2000 transfection reagent according to the manufacturer’s instruction. The transfection complex was diluted into 900 μL of DMEM/F12 medium and added directly to the cells. The cells were washed with PBS and maintained in serum-free DMEM/F-12 medium for 24 h before treatment with TNF-α.
Isolation of Subcellular Fractions
HCFs were seeded in 10 cm dishes and reached 90% confluence, they were shifted to serum-free DMEM/F-12 medium for 24 h, and then incubated with 15 ng/mL TNF-α for the indicated time intervals. The subcellular fractions were prepared using a NE-PER nuclear and cytoplasmic extraction kit according to the manufacturer’s instructions (Thermo Scientific, Rockford, IL). The protein concentrations of samples were determined by BCA assay. Samples from these fractions (200 μL protein) were denatured, subjected to SDS-PAGE using a 12% (w/v) running gel, and determined as described above.
ChIP Assay
To detect the association of transcription factors with human COX-2 promoter, ChIP analysis was performed, as previously described.21 Protein-DNA complexes were fixed by 1% formaldehyde in DMEM/F-12 medium and the reaction was terminated with 125 mM glycine. The sample was lysed, immune-precipitated, washed, and eluted. The enrichment of specific DNA and input DNA (as an internal control) were subjected to PCR amplification. The primer sequences were: FoxO1 forward primer 5′-AAGACATCTGGCGGAAACC-3′ and reverse primer 5′-ACAATTGGTCGCTAACCGAG-3′, which were specifically designed from the COX-2 promoter region (–300 to +2). qPCR was performed using Luna Universal qPCR master mix kit (M3003; New England BioLabs) on a StepOnePlus™ real-time PCR system (Applied Biosystems, Foster City, CA).
Statistical Analysis of Data
Statistical analysis was performed by using GraphPad Prism Program 6.0 software (GraphPad, San Diego, CA). We used one-way ANOVA followed by Dunnett’s post hoc test when comparing more than two groups of data or nonparametric Kruskal–Wallis test, followed by Dunn’s post hoc test when comparing multiple independent groups and ANOVA normality assumptions not met. P values of 0.05 were considered to be statistically significant. Post hoc tests were run only if F achieved P < 0.05 and there was no significance in the homogeneity of variance. All the data were expressed as the mean ± SEM, at least three individual experiments (n= number of independent cell culture preparations). Error bars were omitted when they fell within the dimensions of the symbols.
Results
TNF-α Induces COX-2 Expression and PGE2 Production in HCFs
To determine the effects of TNF-α on expression of COX-2 protein and gene in HCFs, the cells were incubated with various concentrations of TNF-α for the indicated time intervals before being harvested for analyses. As shown in Figure 1A, TNF-α induced COX-2 protein expression in a time- and concentration-dependent manner. There was a significant increase response being observed within 6 h, when the cells were exposed to 15 ng/mL TNF-α. The maximal response was obtained within 16 h, and slightly declined within 24 h. GAPDH expression was served as an internal control to reveal equivalent loading amounts of protein. To further examine whether the effects of TNF-α on COX-2 expression were involved at the level of transcription, mRNA expression of COX-2 was determined by real-time PCR (Figure 1B). TNF-α-induced cox-2 mRNA was significantly increased within 2 h, reached a maximum response within 4 h, and sustained for 6 h during the period of observation. To further investigate the effect of TNF-α on COX-2 expression at transcriptional level, COX-2 promoter activity was determined by using a firefly luciferase gene as a reporter of promoter activity (Figure 1C). TNF-α stimulated COX-2 promoter activity and reached a maximum within 1 h. To observe whether TNF-α-increased the amount of COX-2 protein was accompanied by a corresponding production of PGE2, the culture media were collected to measure PGE2 levels using an ELISA kit (Figure 1D). Addition of TNF-α to HCFs induced a significant increase in PGE2 production within 16 h and reached a maximum within 24 h. These results suggested that TNF-α-induced COX-2 expression accounts for increased PGE2 production in HCFs.
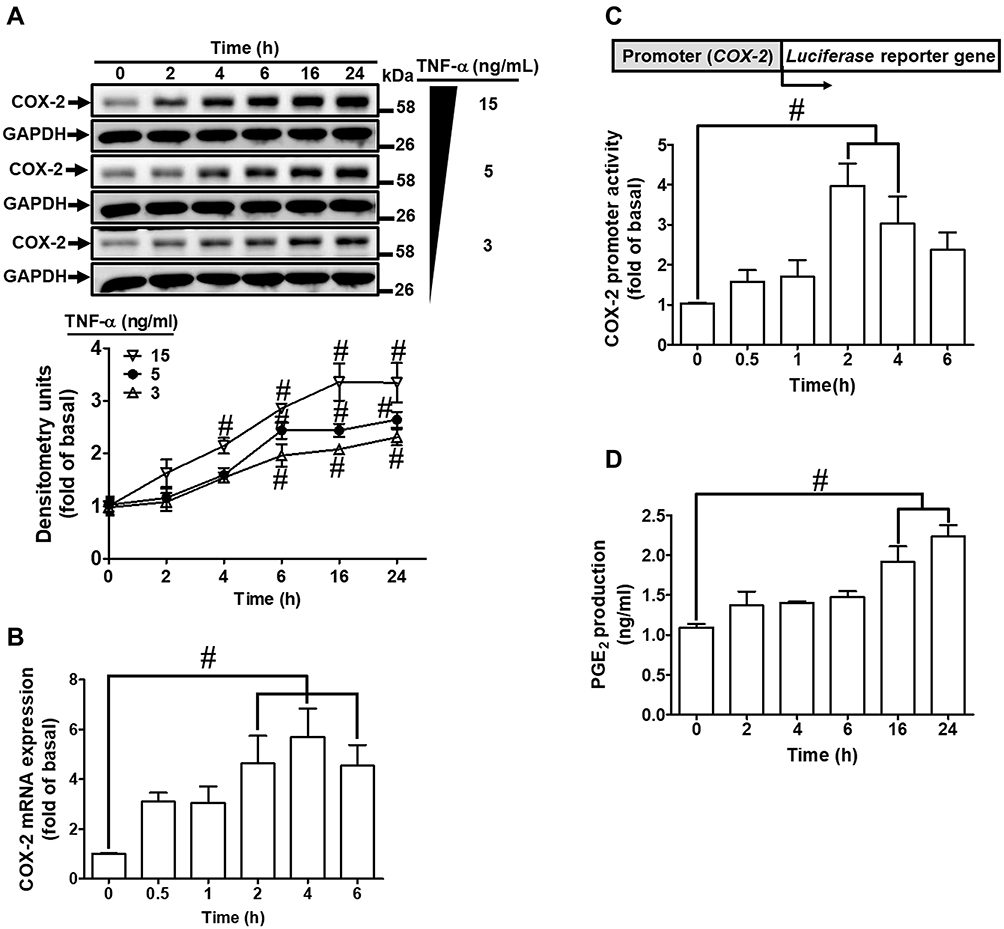 |
Figure 1 TNF-α-induced COX-2 expression and PGE2 production in HCFs. (A) The cells were incubated with different concentrations of TNF-α (3, 5, and 15 ng/mL) for the indicated time intervals (2, 4, 6, 16, and 24 h). The levels of COX-2 and GAPDH protein were determined by Western blot. (B) The cells were incubated with 15 ng/mL TNF-α for the indicated time intervals (0.5, 1, 2, 4, and 6 h). The levels of cox-2 and gapdh mRNA were analyzed by real-time PCR. (C) The cells were co-transfected with COX-2-luci-plasmid and β-galactosidase plasmid, and then incubated with 15 ng/mL TNF-α for the indicated time intervals (0.5, 1, 2, 4, and 6 h). The promoter activity was determined in the cell lysates using a promoter assay kit. (D) The media of 15 ng/mL TNF-α from (A) were saved to determine the levels of PGE2 synthesis using a PGE2 ELISA kit. Data are expressed as the mean ± S.E.M. of three independent experiments. #p<0.05, as compared with the control. |
TNF-α-Induced COX-2 Expression via TNFR1 in HCFs
To investigate which type of TNFRs is involved in TNF-α-induced COX-2 expression, HCFs were pretreated with either TNF receptor 1 or 2 neutralized antibody (TNFR1 or 2 nAb) for 1 h and then incubated with TNF-α (15 ng/mL) for 16 h. As shown in Figure 2A, pretreatment of HCFs with TNFR1 nAb significantly inhibited the TNF-α-induced COX-2 expression in a concentration-dependent manner, but not with TNFR2 nAb (Figure 2B). TNF-α-induced cox-2 mRNA expression was also inhibited by pretreatment with TNFR1 nAb (Figure 2C), but not with TNFR2 nAb (Figure 2D) revealed by real-time PCR. These results suggested that TNF-α-induced COX-2 expression is mediated through TNFR1 in HCFs.
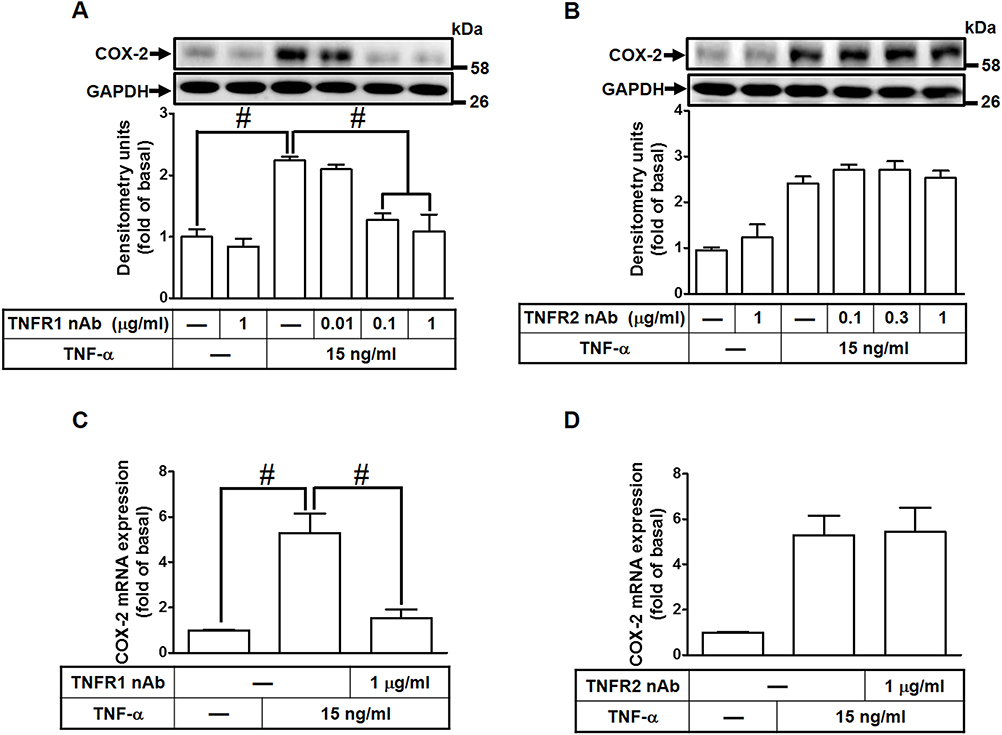 |
Figure 2 TNF-α-induced COX-2 expression and PGE2 production mediated through TNFR1 in HCFs. (A, B) The cells were pretreated with various concentrations of either (A) TNFR1 or (B) TNFR2 neutralized antibody for 1 h and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2 and GAPDH protein were determined by Western blot. (C, D) The cells were pretreated with either (C) TNFR1 nAb or (D) TNFR2 nAb for 1 h and then incubated with TNF-α (15 ng/mL) for 4 h. The levels of cox-2 and gapdh mRNA were analyzed by real-time PCR. Data are expressed as the mean ± S.E.M. of three independent experiments. #p<0.05, as compared with the control. |
MitoROS Generation Induced by TNF-α mediates COX-2 and PGE2 Upregulation
Previous studies have shown that TNF-α exposure alters mitochondrial integrity and causes mitochondrial DNA damage which is mediated by mitoROS elevation.26,27 To determine whether TNF-α increases mitoROS generation, HCFs were pretreated without or with MitoTEMPO for 1 h, and then treated with TNF-α for the indicated time intervals. As shown in Figure 3A, TNF-α (15 ng/mL) induced mitoROS generation which was attenuated by pretreatment with MitoTEMPO (100 nM) or TNFR1 nAb (1 μg/mL) during the period of observation. To investigate whether mitoROS is involved in COX-2 upregulation, various concentrations of MitoTEMPO (10, 30, 100 nM) were applied before addition of TNF-α. As shown in Figure 3B, COX-2 protein expression induced by TNF-α was inhibited by MitoTEMPO in a concentration-dependent manner. The levels of cox-2 mRNA expression and COX-2 promoter activity were also reduced by MitoTEMPO pretreatment (Figure 3C). In addition, we found that PGE2 generation was returned to the basal level following MitoTEMPO pretreatment in HCFs (Figure 3D). These results suggested that mitoROS generation induced by TNF-α mediates COX-2 and PGE2 upregulation in HCFs.
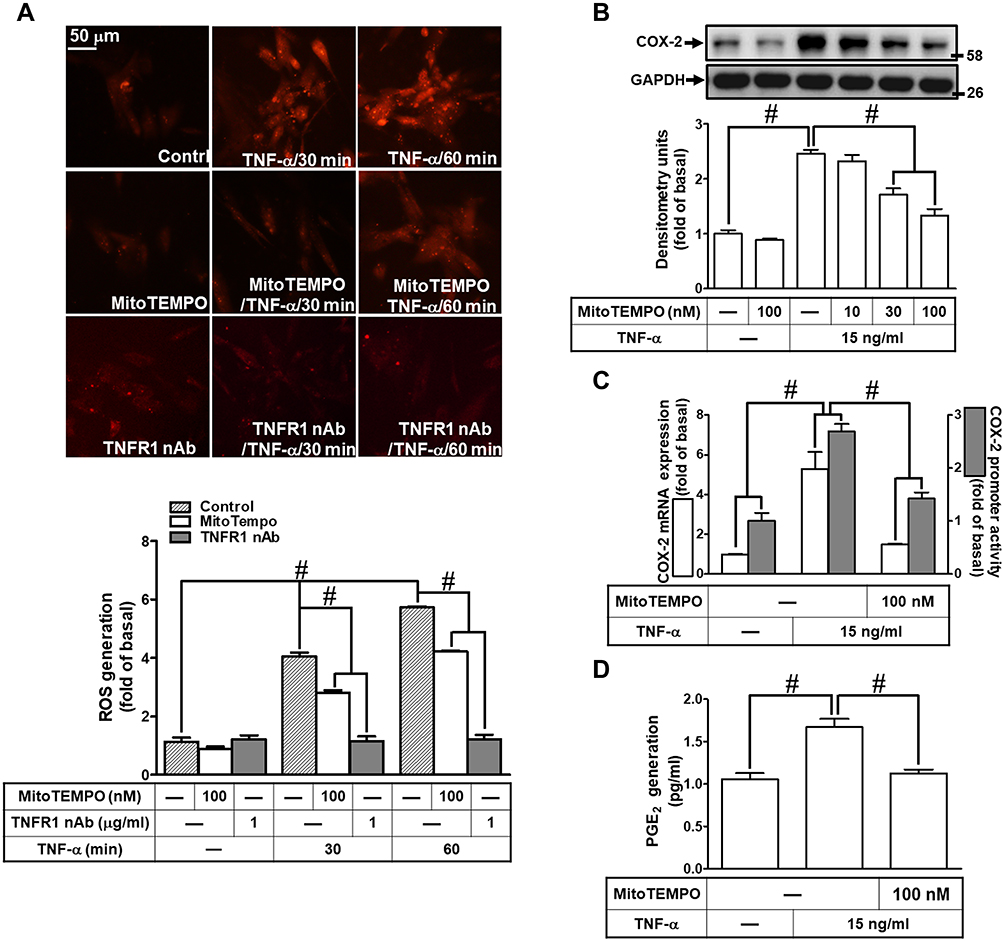 |
Figure 3 MitoROS generation induced by TNF-α mediates COX-2 and PGE2 upregulation. (A) HCFs were pretreated without or with 100 nM MitoTEMPO or TNFR1 nAb (1 μg/mL) for 1 h, and then incubated with 15 ng/mL TNF-α for the indicated time intervals (30 and 60 min). MitoROS production was determined by MitoSOX Red staining (scale bar = 50 µm). The fluorescence unit of MitoSOX Red was measured using a fluorescent microplate reader. (B) HCFs were pretreated with various concentrations of MitoTEMPOL (10, 30, and 100 nM) for 1 h and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2 and GAPDH protein were determined by Western blot. (C) The cells were pretreated with 100 nM MitoTEMPO for 1 h and then incubated with TNF-α (15 ng/mL) for 4 h. The levels of cox-2 and gapdh mRNA were analyzed by real-time PCR (open bars). Cells were co-transfected with COX-2-luci-plasmid along with a β-galactosidase plasmid, sequentially pretreated with 100 nM MitoTEMPO for 1 h, and then incubated with 15 ng/mL TNF-α for 2 h. Promoter activity was determined in the cell lysates using a promoter assay kit (solid bars). (D) The media saved from (A) were used to determine the levels of PGE2. Data are expressed as the mean ± S.E.M. of three independent experiments. #p<0.05, as compared with the control. |
Involvement of PKCs in COX-2 Expression Induced by TNF-α
Several studies have shown that PKC activation plays an important role in the cellular functions modulated by TNF-α.28 To address the possible involvement of PKC activation in the TNF-α-induced COX-2 expression, the cells were pretreated with a pharmacological inhibitor of PKCα (Gö6976) for 1 h and then incubated with 15 ng/mL of TNF-α for 16 h. As shown in Figure 4A, pretreatment of HCFs with Gö6976 significantly attenuated the TNF-α-induced COX-2 expression in a concentration-dependent manner. TNF-α-induced cox-2 mRNA expression and COX-2 promoter activity were also inhibited by pretreatment with Gő6976, revealed by real-time PCR and promoter activity assay, respectively (Figure 4B), suggesting that PKCα is involved in COX-2 expression induced by TNF-α in HCFs. To ensure the role of PKCα in COX-2 expression, as shown in Figure 4C, transfection with PKCα siRNA down-regulated the PKCα protein expression and also attenuated the TNF-α-induced COX-2 expression. To further investigate the role of PKCα activation in the TNF-α-induced COX-2 expression, we determined the effect of TNF-α on membrane translocation of PKCα in HCFs, revealed by Western blotting using an anti-PKCα antibody. We found that TNF-α stimulated translocation of PKCα from cytosol to membrane in a time-dependent manner (Figure 4D). To further observe whether PKCα phosphorylation participates in the TNF-α-induced COX-2 expression in HCFs, activation of this kinase was assayed by Western blotting using an antibody specific for the phosphorylated, active form of PKCα. We found that TNF-α stimulated a time-dependent phosphorylation of PKCα, which was attenuated by Gö6976 (Figure 4E). In addition, pretreatment with either TNFR1 nAb or MitoTEMPO also attenuated TNF-α-stimulated PKCα phosphorylation (Figure 4E), indicating that PKCα was a downstream component of TNFR1 and mitoROS in HCFs. We also found that TNF-α-induced PGE2 production was inhibited by Gö6976 (Figure 4F). These results suggested that TNFR1/mitoROS-dependent activation of PKCα is involved in COX-2 expression and PGE2 production induced by TNF-α in HCFs.
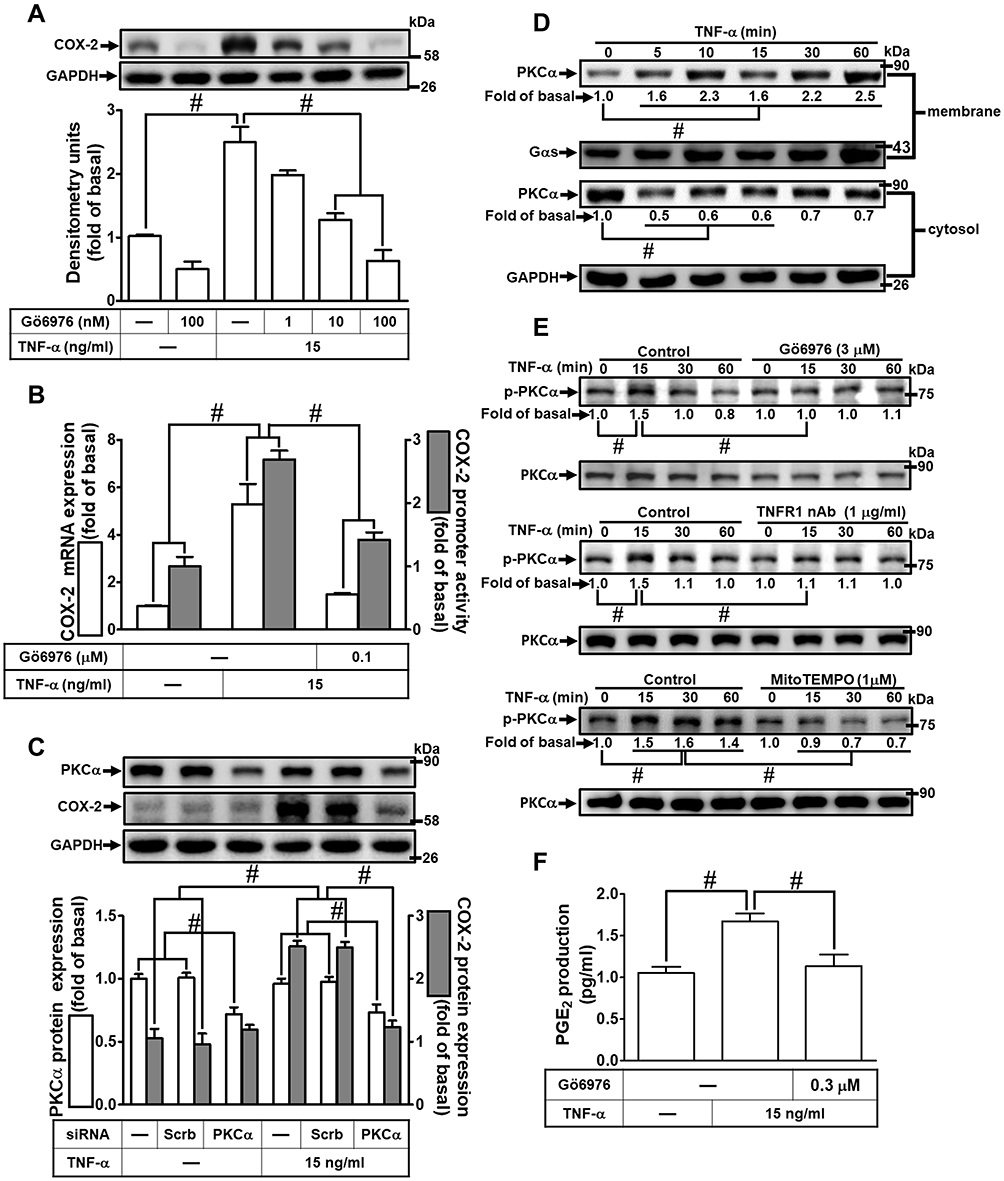 |
Figure 4 PKCα was involved in TNF-α-induced COX-2 expression and PGE2 production in HCFs. (A) The cells were pretreated with various concentrations of Gö6976 (1, 10, and 100 nM) for 1 h and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2 and GAPDH protein were determined by Western blot. (B) The cells were pretreated with 100 nM Gö6976 for 1 h and then incubated with 15 ng/mL TNF-α for 4 h. The levels of cox-2 and gapdh mRNA were analyzed by real-time PCR (open bars). Cells were co-transfected with COX-2-luci-plasmid along with a β-galactosidase plasmid, pretreated with 100 nM Gö6976 for 1 h, and then incubated with 15 ng/mL TNF-α for 1 h. Promoter activity was determined in the cell lysates using a promoter assay kit (solid bars). (C) The cells were transfected with scrambled or PKCα siRNA and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2, PKCα, and GAPDH proteins were analyzed by Western blot. (D) The cells were incubated with 15 ng/mL TNF-α for the indicated time intervals (5, 10, 15, 30, and 60 min). The cytosolic and membrane fractions were analyzed by Western blot to determine the levels of PKCα with Gαs and GAPDH as membrane and cytosolic internal control, respectively. (E) The cells were pretreated without or with 3 μM Gö6976, 1 μg/mL TNFR1 nAb, or 1 μM MitoTEMPO for 1 h and then incubated with 15 ng/mL TNF-α for the indicated time intervals (15, 30, and 60 min). The levels of phospho-PKCα and total PKCα proteins were analyzed by Western blot. (F) The media saved from (A) were used to determine the levels of PGE2. Data are expressed as the mean ± S.E.M. of three independent experiments. #p<0.05, as compared with the control. |
TNF-α Induces COX-2 Expression via p38 MAPK Phosphorylation
To determine whether p38 MAPK is involved in the TNF-α-induced COX-2 expression in HCFs, a p38 MAPK inhibitor VIII (p38i VIII) was used. As shown in Figure 5A, pretreatment with p38i VIII resulted in a significant attenuation of the TNF-α-induced COX-2 expression in a concentration-dependent manner revealed by Western blotting. Moreover, TNF-α-induced cox-2 mRNA expression and COX-2 promoter activity were also inhibited by p38i VIII, revealed by real-time PCR and promoter activity, respectively (Figure 5B), suggesting that p38 MAPK is involved in COX-2 expression induced by TNF-α in HCFs. To ensure the role of p38 MAPK in COX-2 expression, as shown in Figure 5C, transfection with p38 MAPK siRNA down-regulated the p38 MAPK protein expression and also attenuated the TNF-α-induced COX-2 expression. To further investigate whether p38 MAPK phosphorylation participates in TNF-α-induced COX-2 expression in HCFs, activation of this kinase was assayed by Western blotting using an antibody specific for the phosphorylated, active form of p38 MAPK. We found that TNF-α stimulated a time-dependent phosphorylation of p38 MAPK which was attenuated by p38i VIII (Figure 5D). In addition, pretreatment with TNFR1 nAb, Gö6976, or MitoTEMPO also attenuated TNF-α-stimulated p38 MAPK phosphorylation (Figure 5D). TNF-α-stimulated PKCα phosphorylation was not significantly inhibited by p38i VIII (Figure 5D). Further, TNF-α-induced PGE2 production was also inhibited by p38i VIII (Figure 5E). These results suggested that TNF-α-induced COX-2 expression and PGE2 production is mediated through TNFR1/mitoROS/PKCα-dependent activation of p38 MAPK in HCFs.
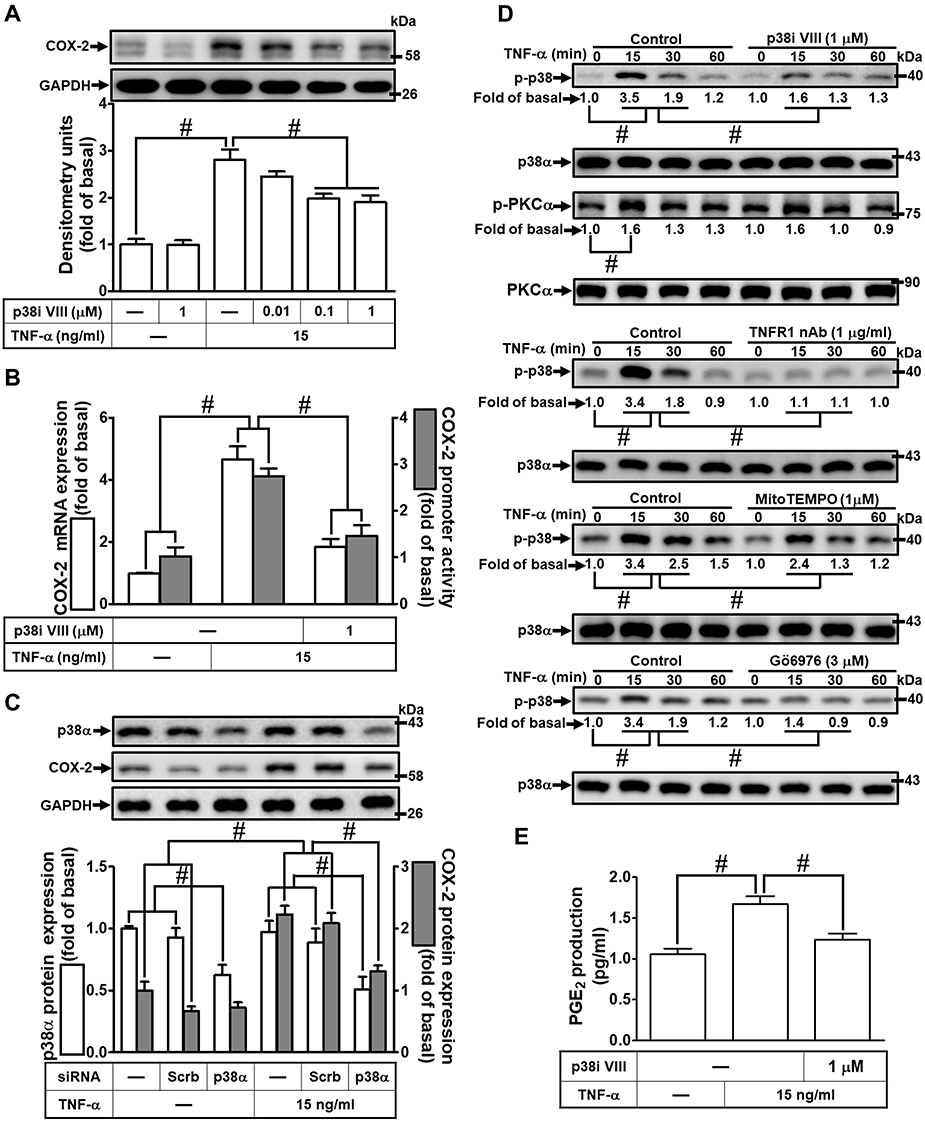 |
Figure 5 Involvement of p38 MAPK in TNF-α-induced COX-2 expression and PGE2 production in HCFs. (A) The cells were pretreated with various concentrations of p38 inhibitor VIII (p38i VIII; 0.01, 0.1, and 1 μM) for 1 h and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2 and GAPDH protein were determined by Western blot. (B) The cells were pretreated with 1 μM p38i VIII for 1 h and then incubated with 15 ng/mL TNF-α for 4 h. The levels of cox-2 and gapdh mRNA were analyzed by real-time PCR (open bars). Cells were co-transfected with COX-2-luci-plasmid along with a β-galactosidase plasmid, pretreated with 1 μM p38i VIII for 1 h, and then incubated with 15 ng/mL TNF-α for 2 h. Promoter activity was determined in the cell lysates using a promoter assay kit (solid bars). (C) The cells were transfected with scrambled or p38 MAPK siRNA and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2, p38 MAPK, and GAPDH proteins were analyzed by Western blot. (D) The cells were pretreated with 1 μM p38i VIII, 1 μg/mL TNFR1 nAb, 3 μM Gö6976, or 1 μM MitoTEMPO for 1 h and then incubated with 15 ng/mL TNF-α for the indicated time intervals (15, 30, and 60 min). The levels of phospho-p38 MAPK, phospho-PKCα, total p38 MAPK, and total PKCα proteins were analyzed by Western blot. (E) The media saved from (A) were used to determine the levels of PGE2. Data are expressed as the mean ± S.E.M. of three independent experiments. #p<0.05, as compared with the control. |
TNF-α Induces COX-2 Expression via JNK1/2 Phosphorylation
To determine whether activation of JNK1/2 is involved in the TNF-α-induced COX-2 expression in HCFs, a pharmacological inhibitor of JNK1/2 (SP600125) was used. As shown in Figure 6A, pretreatment of HCFs with SP600125 effectively blocked the TNF-α-induced COX-2 expression in a concentration-dependent manner revealed by Western blotting. TNF-α-induced cox-2 mRNA expression and COX-2 promoter activity were also inhibited by pretreatment with SP600125, revealed by real-time PCR and promoter activity assay, respectively (Figure 6B), suggesting that JNK1/2 is involved in COX-2 expression induced by TNF-α in HCFs. To ensure the role of JNK1/2 in COX-2 expression as shown in Figure 6C, transfection with JNK1/2 siRNA down-regulated the JNK1/2 protein expression and attenuated TNF-α-induced COX-2 expression. To further investigate whether JNK1/2 phosphorylation participates in TNF-α-induced COX-2 expression in HCFs, activation of this kinase was assayed by Western blotting using an antibody specific for the phosphorylated, active forms of JNK1/2. We found that TNF-α stimulated a time-dependent phosphorylation of JNK1/2 which was attenuated by SP600125 (Figure 6D). In addition, pretreatment with TNFR1 nAb, Gö6976, or MitoTEMPO also attenuated TNF-α-stimulated JNK1/2 phosphorylation (Figure 6D). TNF-α-stimulated PKCα phosphorylation was not significantly inhibited by SP600125 (Figure 6D). Further, TNF-α-induced PGE2 production was also inhibited by SP600125 (Figure 6E). These results suggested that TNFR1/mitoROS/PKCα-dependent activation of JNK1/2 is involved in COX-2 expression and PGE2 production induced by TNF-α.
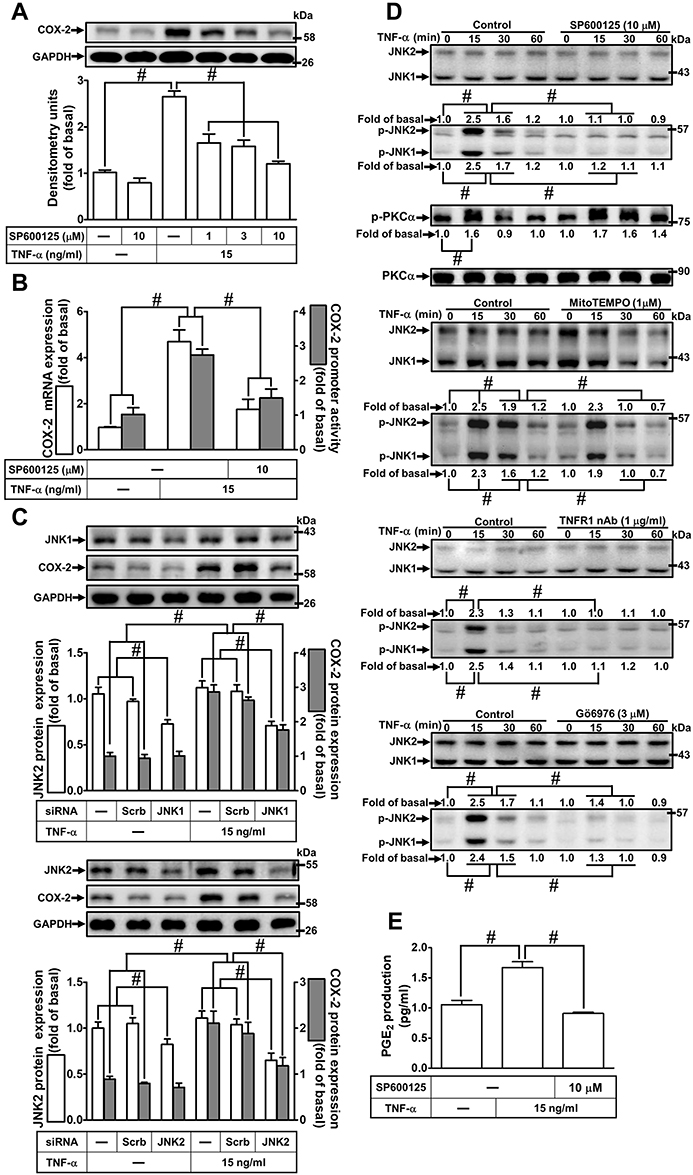 |
Figure 6 Involvement of JNK1/2 in TNF-α-induced COX-2 expression and PGE2 production in HCFs. (A) The cells were pretreated with various concentrations of SP600125 (1, 3, and 10 μM) for 1 h and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2 and GAPDH protein were determined by Western blot. (B) The cells were pretreated with 10 μM SP600125 for 1 h and then incubated with 15 ng/mL TNF-α for 4 h. The levels of cox-2 and gapdh mRNA were analyzed by real-time PCR (open bars). The cells were co-transfected with COX-2-luci-plasmid along with a β-galactosidase plasmid, pretreated with 10 μM SP600125 for 1 h, and then incubated with 15 ng/mL TNF-α for 2 h. Promoter activity was determined in the cell lysates using a promoter assay kit (solid bars). (C) The cells were transfected with scrambled, JNK1, or JNK2 siRNA, individually, and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2, JNK2, JNK1, and GAPDH proteins were analyzed by Western blot. (D) The cells were pretreated with 10 μM SP600125, 1 μg/mL TNFR1 nAb, 3 μM Gö6976, or 1 μM MitoTEMPO for 1 h and then incubated with 15 ng/mL TNF-α for the indicated time intervals (15, 30, and 60 min). The levels of phospho-JNK1/2, phospho-PKCα, total JNK1/2, and total PKCα proteins were analyzed by Western blot. (E) The media saved from (A) were used to determine the levels of PGE2. Data are expressed as the mean ± S.E.M. of three independent experiments. #p<0.05, as compared with the control. |
FoxO1 is Involved in TNF-α-Induced COX-2 Expression in HCFs
The promoter region of COX-2 possesses serial binding elements for recognition of transcription factors including FoxO1. Thus, we examined whether activation of FoxO1 is required for COX-2 expression induced by TNF-α in HCFs, a pharmacological inhibitor of FoxO1 (AS1842856) was used. As shown in Figure 7A, pretreatment of HCFs with AS1842856 significantly attenuated the TNF-α-induced COX-2 protein expression. Moreover, TNF-α-induced cox-2 mRNA and COX-2 promoter activity were also inhibited by pretreatment with AS1842856, revealed by real-time PCR and promoter activity assay, respectively (Figure 7B), suggesting that FoxO1 is involved in COX-2 expression induced by TNF-α in HCFs. To ensure the role of FoxO1 in COX-2 expression, as shown in Figure 7C, transfection with FoxO1 siRNA down-regulated the FoxO1 protein expression and attenuated the TNF-α-induced COX-2 expression. To further investigate whether FoxO1 phosphorylation participates in TNF-α-induced COX-2 expression in HCFs, activation of this kinase was assayed by Western blotting using an antibody specific for the phosphorylated, active form of FoxO1. We found that TNF-α induced a time-dependent phosphorylation of FoxO1 which was attenuated by AS1842856 (Figure 7D). In addition, pretreatment with TNFR1 nAb, Gö6976, p38i VIII, SP600125, or MitoTEMPO also attenuated the TNF-α-stimulated FoxO1 phosphorylation (Figure 7D), indicating that FoxO1 was a downstream component of TNFR1, mitoROS, PKCα, p38 MAPK and JNK1/2 in HCFs. We further investigate the binding activity of FoxO1 with the COX-2 promoter following TNF-α exposure. We revealed that TNF-α exposure enhanced FoxO1 binding activity with the COX-2 promoter which was attenuated by AS1842856 pretreatment (Figure 7E). In addition, TNF-α-induced PGE2 production was also inhibited by AS1842856 (Figure 7F). These results suggested that TNFR1/mitoROS/PKCα/p38 MAPK and JNK1/2-dependent activation of FoxO1 is involved in COX-2 expression and PGE2 production induced by TNF-α in HCFs.
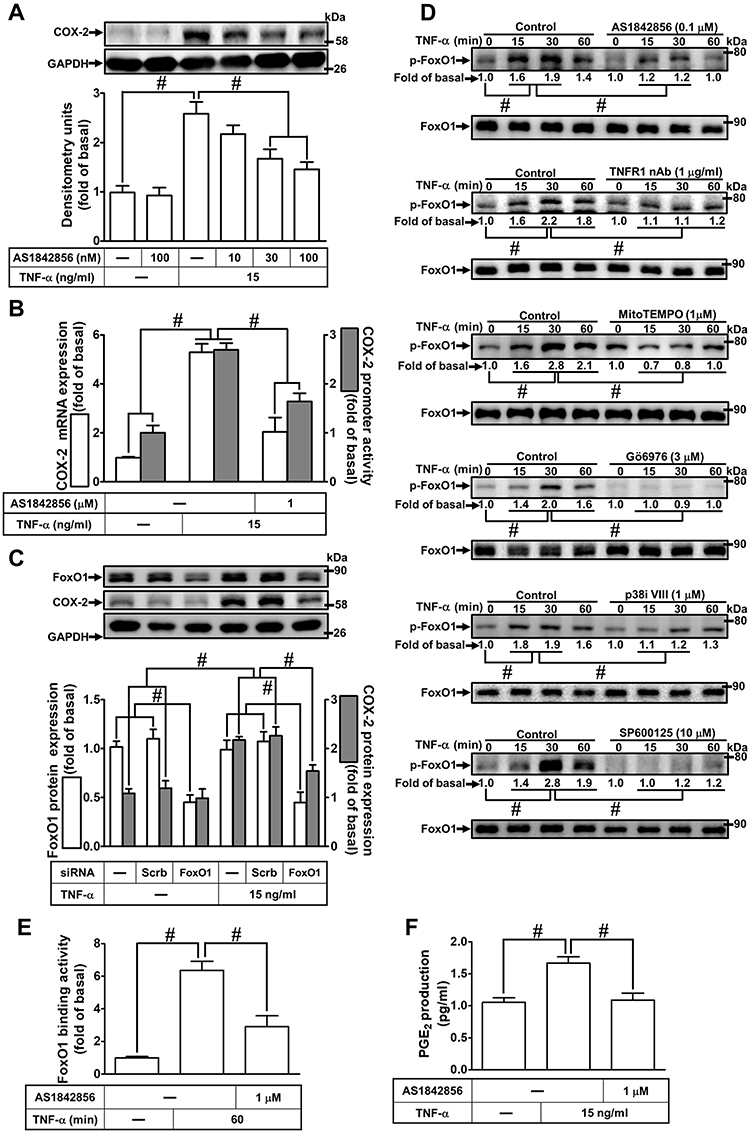 |
Figure 7 Involvement of FoxO1 in TNF-α-induced COX-2 expression and PGE2 production in HCFs. (A) The cells were pretreated with various concentrations of AS1842856 (10, 30, and 100 nM) for 1 h and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2 and GAPDH protein were determined by Western blot. (B) The cells were pretreated with 1 μM AS1842856 for 1 h and then incubated with 15 ng/mL TNF-α for 4 h. The levels of cox-2 and gapdh mRNA were analyzed by real-time PCR (open bars). Cells were co-transfected with COX-2-luci-plasmid along with a β-galactosidase plasmid, sequentially pretreated with 1 μM AS1842856 for 1 h, and then incubated with 15 ng/mL TNF-α for 2 h. Promoter activity was determined in the cell lysates using a promoter assay kit (solid bars). (C) The cells were transfected with scrambled or FoxO1 siRNA and then incubated with 15 ng/mL TNF-α for 16 h. The levels of COX-2, FoxO1, and GAPDH proteins were analyzed by Western blot. (D) The cells were pretreated with 0.1 μM AS1842856, 1 μg/mL TNFR1 nAb, 3 μM Gö6976, 1 μM MitoTEMPO, 1 μM p38i VIII, or 10 μM SP600125 for 1 h and then incubated with 15 ng/mL TNF-α for the indicated time intervals (15, 30, and 60 min). The levels of phospho-FoxO1 and total FoxO1 proteins were analyzed by Western blot. (E) The cells were pretreated with 0.1 μM AS1842856 and then incubated with 15 ng/mL TNF-α for 60 min. The DNA binding ability of FoxO1 was detected by chromatin immunoprecipitation assay. (F) The media saved from (A) were used to determine the levels of PGE2. Data are expressed as the mean ± S.E.M. of three independent experiments. #p<0.05, as compared with the control. |
Discussion
Cardiac fibrosis occurs in association with coronary heart diseases and myocardial infarction which ultimately lead to heart failure. Elevated PGE2 level is one of the key factors that contribute to cardiac fibrosis initiation by stimulating cardiac fibroblast migration and proliferation.11,12,29 The increase in PGE2 level is closely associated with COX-2 upregulation as it catalyzes the formation of PGE2. The signaling of several pro-inflammatory cytokines such as TNF-α has been known to induce COX-2-mediated PGE2 elevation in human gingival fibroblasts, human lung epithelial cells, and human tracheal smooth muscle cells.8–10 Moreover, TNF-α has been involved in cardiovascular diseases where its activation has resulted in vascular dysfunction, cardiac remodeling, and heart failure.4 In the present study, we elucidated that the mechanisms by which TNF-α induces COX-2 overexpression and PGE2 elevation are mediated through TNFR1-dependent MitoROS/PKCα/p38 MAPK and JNK1/2 cascade to activate FoxO1 binding with the COX-2 promoter in HCFs (Figure 8).
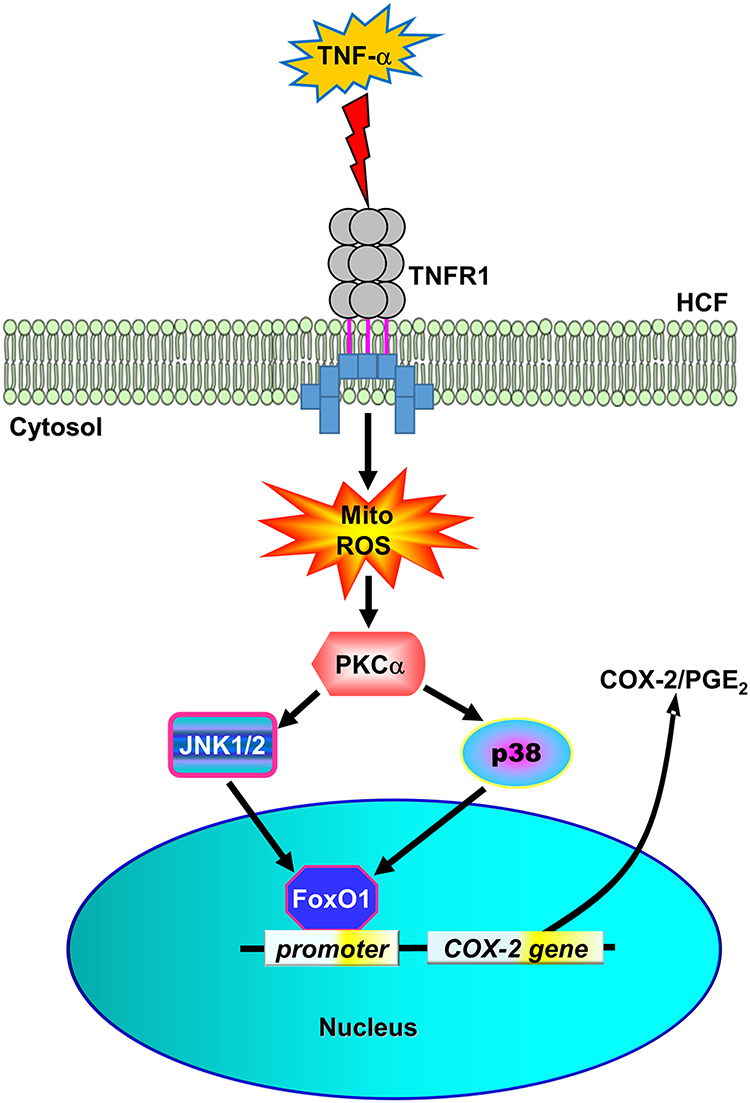 |
Figure 8 The schematic signaling pathways involved in TNF-α-induced COX-2 expression and PGE2 production in HCFs. TNF-α activated TNFR1 to stimulate mitoROS generation, in turn, activating PKCα/p38 MAPK and JNK1/2 cascade-dependent FoxO1 activity and binding with COX-2 promoter leading to COX-2 expression and PGE2 production in HCFs. |
TNF-α is a key regulator in the initiation and potentiation of inflammation due to its role in the immune system that contributes to innate and adaptive immunity.30 Deregulation of TNF-α expression and related-signalings may trigger pathogenesis associated with tissue damage and chronic inflammation. Indeed, increased levels of TNF-α were identified in patients with autoimmune and degenerative diseases. An early study indicated that circulating levels of TNF-α are increased in patients with end-stage cardiac disease.6 These findings have evoked a large effort to investigate the effects of TNF-α in the failing heart. Thus, several lines of evidence have shown that increased expression of TNF-α is involved in injured and failing hearts including animal experiments and human patients.7,31 In fibroblasts, TNF-α may lead to extracellular matrix (ECM) degradation mediated through disrupting the balance between matrix metalloproteinases (MMPs) and their inhibitors.32 TNF-α binds to the two kinds of type I transmembrane receptors TNFR1 and TNFR2 that in their extracellular domains, contain four cysteine-rich domains (CRD). These cause TNF-α to induce opposing action in the immune system. TNF-α-mediated proinflammatory functions are most predominantly mediated through TNFR1 signaling. In contrast, in immunity, TNFR2 expression is mostly to suppress the development of autoimmune responses via regulation of immune response signaling in regulatory T cells (Tregs).33 In the present study, we demonstrated that TNF-α-induced COX-2 upregulation is most predominantly mediated through a TNFR1-related pathway, because pretreatment with TNFR1 nAb attenuated COX-2 induction but not with TNF2 nAb in HCFs. This finding is consistent with previous studies suggesting that the effect of TNF-α on COX-2 expression is mediated via TNFR1.34,35 These results may result from different binding affinity between TNF-α and two types of TNFR. A study has indicated that TNFR1 with a higher affinity (Kd = 1.9 × 10–11 M) than TNFR2 (Kd = 4.2 × 10–10 M) binds with TNF-α.36 These results suggest that TNF-α-induced COX-2/PGE2 elevation in cardiac fibroblasts is initiated via TNFR1 activity and its downstream signaling cascades and possibly involved in the development of cardiovascular disorders.37
ROS are involved in the pathogenesis of disease development, which induce modifications in subcellular organelles such as mitochondria, nucleus, sarcoplasmic reticulum, and sarcolemma through oxidative modification of most intracellular macromolecules (such as DNA, proteins, or lipids). Thus, an increased production of ROS has already been found to participate in several cardiac diseases such as myocardial fibrosis,38,39 heart failure,40 and myocardial infarction.41 Although global oxidative stress comes from different resources, mitochondrial compartment contributes to the majority of intracellular ROS about 90%. Mitochondrial dysfunction triggers excessive ROS generation and simultaneously induces irreversible damage to mitochondria due to mitochondria are also a vulnerable target of ROS, which occurs during the development of cardiovascular diseases.42 Growing evidence suggests that mitochondrial ROS production plays a crucial role in many diabetes-related cardiovascular complications and cardiac hypertrophy.43,44 It has been noted that TNFR1 activation positively affects ROS and reactive nitrogen species generation.17,45 By utilizing MitoTEMPO, a mitochondria-targeted ROS scavenger, we demonstrated that TNF-α-induced mitoROS was decreased along with COX-2 expression and PGE2 production. These results revealed the role of mitoROS in the TNF-α-induced COX-2 expression and PGE2 production. These results are consistent with previous studies demonstrating that ROS are an important factor for COX-2 upregulation in various types of cells.16,17
PKC plays an important role in many human cardiovascular diseases, including ischaemic heart disease18 and heart failure.46 In particular, PKCα, one of the PKC isozymes has emerged as an important protein kinase contributing to cardiac dysfunction, cardiac fibrosis, and even heart failure.47,48 PKCα can cause decreased cardiac contractility, the force of myofilaments, and uncoupling of β-adrenergic receptors in the pathology of heart failure.19,49 Although there are not consistent results to be concluded in PKC as a therapeutic target for cardiovascular diseases, PKC inhibitors have been considered as a potential strategy for cardiovascular diseases.50,51 Several lines of evidence indicated that various stimuli including TNF-α might activate PKCα leading to upregulation of COX-2 expression and PGE2 release in various types of cells.20,52 Our results also consistently showed that pretreatment with Gö6976 (PKCα inhibitor) or transfection with PKCα siRNA attenuated COX-2 expression induced by TNF-α. Under normal status, inactive PKCα is distributed in the cytosol, upon stimulation, PKCα can be activated and translocated to the cell membrane and subsequently activates its downstream signaling components.53 As expected, following TNF-α exposure, we observed the elevated protein expression of PKCα in the cell membrane as early as 5 min until the end of time intervals observed. In contrast, PKCα expression in the cytosol was simultaneously decreased during the first 15 min of TNF-α exposure. Our data demonstrated that PKCα activation and translocation following TNF-α exposure occurred at a relatively early event. Our results revealed that pretreatment with TNFR1 nAb or MitoTEMPO decreased PKCα phosphorylation, indicating that TNFR1/mitoROS is required for PKCα phosphorylation in HCFs.
Previous studies have shown that p38 MAPK and JNK1/2 signaling cascades mediate TNFR1 responses elicited by TNF-α in heart diseases.37 TNF-α has been demonstrated to induce COX-2 expression mediated through p38 MAPK activity in various types of cells.24,25 Consistently, we revealed that TNF-α-induced COX-2 and PGE2 upregulation were mediated by p38 MAPK signaling pathway, which was attenuated by pretreatment with p38i VIII or transfection with p38 siRNA. JNK1/2 was also involved in COX-2 upregulation induced by various mediators in different types of cells.22,23 Our results also suggested that JNK1/2 plays a crucial role in COX-2 expression and PGE2 production induced by TNF-α in HCFs, which was inhibited by pretreatment with JNK1/2 inhibitor SP600125 or transfection with JNK1/2 siRNA. In addition, our data suggested that PKCα is required for p38 MAPK and JNK1/2 activation, verified by that the levels of both p38 MAPK and JNK1/2 phosphorylation were attenuated by pretreatment with TNFR1 nAb, mitoROS inhibitor MitoTEMP, or PKCα inhibitor Gö6976. In contrast, pretreatment with either p38i VIII or SP600125 had no significant effect on PKCα phosphorylation stimulated by TNF-α. Thus, our results further revealed that in HCFs, both p38 MAPK and JNK1/2 are activated by TNF-α through a TNFR1-dependent mitoROS/PKCα signaling pathway leading to COX-2 expression and PGE2 production.
The FoxO1 transcription factor plays an important role in the widely physiological processes, including metabolism, proliferation, apoptosis, stress, and inflammation by regulating its target genes.54 FoxO1 has been shown to play a crucial role in the development of cardiac hypertrophy due to regulating the expression of target genes in the myocardium that is involved in calcium homeostasis, autophagy, cardiac growth/protein synthesis, and cell apoptosis.54 FoxO1 is activated upon exposure to various inflammatory mediators such as sphingosine 1‐phosphate,55 lysophosphatidylcholine,56 and LPS.57 Besides, TNF-α has been demonstrated to activate FoxO1 in both in vitro and in vivo studies.58 In our previous studies, FoxO1 has been demonstrated to be involved in COX-2 expression.55,56 In the present study, we clarified that TNF-α-induced COX-2 and PGE2 upregulation is, at least in part, dependent on FoxO1 activity. To our best knowledge, this is the first time to reveal that FoxO1 is involved in the TNF-α-induced COX-2 expression in HCFs. In addition, this result was verified by ChIP assay showing that the TNF-α-triggered binding activity of FoxO1 to COX-2 promoter was blocked by pretreatment with FoxO1 inhibitor AS1842856. Moreover, our results showed that FoxO1 activity was inhibited by pretreatment with TNFR1 nAb, MitoTEMPO, Gö6976, p38i VIII, or SP600125 in HCFs challenged with TNF-α.
Conclusions
In this study, we depicted the mechanisms by which TNF-α induced COX-2 upregulation and PGE2 release in HCFs are, at least partially, mediated through TNFR1/MitoROS/PKCα/JNK1/2 and p38 MAPK-dependent FoxO1 activity. The elucidation of the mechanism of TNF-α-induced COX-2/PGE2 production in HCFs provides us with a better understanding of the pathogenesis of TNF-α in heart disorders and potential strategies in therapies for cardiovascular diseases.
Abbreviations
BSA, Bovine serum albumin; DMEM, Dulbecco’s modified Eagle’s medium; ECL, Enhanced Chemiluminescence; ELISA, Enzyme-linked immunosorbent assay; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; HO-1, Heme oxygenase-1; HCFs, Heart cardiac fibroblasts; JNK, c-Jun N-terminal kinase; MAPK, Mitogen-activated protein kinase; PBS, Phosphate-buffered saline; PKC, Protein kinase C; ROS, Reactive oxygen species; SDS-PAGE, Sodium dodecyl sulfate polyacrylamide gel electrophoresis; siRNA, Small interfering RNA; Tris-HCl, Tris[hydroxymethyl]aminomethane; TTBS, Tween-Tris-buffered saline.
Data Sharing Statement
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Ministry of Science and Technology, Taiwan [Grant numbers: MOST108-2320-B-039-061, MOST109-2320-B-039-061, MOST109-2813-C-039-029-B, and MOST108-2320-B-182-014]; China Medical University, Taiwan [Grant number: CMU109-MF-09]; Chang Gung Medical Research Foundation, Taiwan [Grant numbers: CMRPG5F0203, CMRPG5J0142, and CMRPG5J0143].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Humeres C, Frangogiannis NG. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl Sci. 2019;4(3):449–467. doi:10.1016/j.jacbts.2019.02.006
2. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225(3):631–637. doi:10.1002/jcp.22322
3. Bartekova M, Radosinska J, Jelemensky M, Dhalla NS. Role of cytokines and inflammation in heart function during health and disease. Heart Fail Rev. 2018;23(5):733–758. doi:10.1007/s10741-018-9716-x
4. Schumacher SM, Naga Prasad SV. Tumor necrosis factor-α in heart failure: an updated review. Curr Cardiol Rep. 2018;20(11):117. doi:10.1007/s11886-018-1067-7
5. Sun M, Chen M, Dawood F, et al. Tumor necrosis factor-α mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115(11):1398–1407. doi:10.1161/CIRCULATIONAHA.106.643585
6. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323(4):236–241. doi:10.1056/nejm199007263230405
7. Torre-Amione G, Kapadia S, Lee J, et al. Tumor necrosis factor-α and tumor necrosis factor receptors in the failing human heart. Circulation. 1996;93(4):704–711. doi:10.1161/01.cir.93.4.704
8. Lin CC, Hsiao LD, Chien CS, Lee CW, Hsieh JT, Yang CM. Tumor necrosis factor-α-induced cyclooxygenase-2 expression in human tracheal smooth muscle cells: involvement of p42/p44 and p38 mitogen-activated protein kinases and nuclear factor-κB. Cell Signal. 2004;16(5):597–607. doi:10.1016/j.cellsig.2003.10.002
9. Nakao S, Ogtata Y, Shimizu E, Yamazaki M, Furuyama S, Sugiya H. Tumor necrosis factor alpha (TNF-α)-induced prostaglandin E2 release is mediated by the activation of cyclooxygenase-2 (COX-2) transcription via NFκB in human gingival fibroblasts. Mol Cell Biochem. 2002;238(1/2):11–18. doi:10.1023/a:1019927616000
10. Chen CC, Sun YT, Chen JJ. TNF-α-induced cyclooxygenase-2 expression in human lung epithelial cells: involvement of the phospholipase C-γ2, protein kinase C-α, tyrosine kinase, NF-κB-inducing kinase, and I-κB kinase 1/2 pathway. J Immunol. 2000;165(5):2719–2728. doi:10.4049/jimmunol.165.5.2719
11. Ma Y, Yue Z, Zhang B, et al. Calcium signal pathway is involved in prostaglandin E2 induced cardiac fibrosis in cardiac fibroblasts. J Pharm Pharm Sci. 2018;21(1):326–339. doi:10.18433/jpps29322
12. Harding P, LaPointe MC. Prostaglandin E2 increases cardiac fibroblast proliferation and increases cyclin D expression via EP1 receptor. Prostaglandins Leukot Essent Fatty Acids. 2011;84(5–6):147–152. doi:10.1016/j.plefa.2011.01.003
13. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. 2019;51(12):1–13. doi:10.1038/s12276-019-0355-7
14. Ye Y, Li J, Yuan Z. Effect of antioxidant vitamin supplementation on cardiovascular outcomes: a meta-analysis of randomized controlled trials. PLoS One. 2013;8(2):e56803. doi:10.1371/journal.pone.0056803
15. Escobales N, Nunez RE, Jang S, et al. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J Mol Cell Cardiol. 2014;77:136–146. doi:10.1016/j.yjmcc.2014.10.009
16. Onodera Y, Teramura T, Takehara T, Shigi K, Fukuda K. Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Bio. 2015;5:492–501. doi:10.1016/j.fob.2015.06.001
17. Zhao Y, Usatyuk PV, Gorshkova IA, et al. Regulation of COX-2 expression and IL-6 release by particulate matter in airway epithelial cells. Am J Respir Cell Mol Biol. 2009;40(1):19–30. doi:10.1165/rcmb.2008-0105OC
18. Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. 2006;70(2):222–230. doi:10.1016/j.cardiores.2006.02.015
19. Liu Q, Molkentin JD. Protein kinase Cα as a heart failure therapeutic target. J Mol Cell Cardiol. 2011;51(4):474–478. doi:10.1016/j.yjmcc.2010.10.004
20. Huang WC, Chen JJ, Inoue H, Chen CC. Tyrosine phosphorylation of I-κB kinase α/β by protein kinase C-dependent c-Src activation is involved in TNF-α-induced cyclooxygenase-2 expression. J Immunol. 2003;170(9):4767–4775. doi:10.4049/jimmunol.170.9.4767
21. Tseng HC, Lin CC, Hsiao LD, Yang CM. Lysophosphatidylcholine-induced mitochondrial fission contributes to collagen production in human cardiac fibroblasts. J Lipid Res. 2019;60(9):1573–1589. doi:10.1194/jlr.RA119000141
22. Kitanaka T, Nakano R, Kitanaka N, et al. JNK activation is essential for activation of MEK/ERK signaling in IL-1β-induced COX-2 expression in synovial fibroblasts. Sci Rep. 2017;7(1):39914. doi:10.1038/srep39914
23. Wu G, Luo J, Rana JS, Laham R, Sellke FW, Li J. Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovasc Res. 2006;69(2):512–519. doi:10.1016/j.cardiores.2005.09.019
24. Hobbs SS, Goettel JA, Liang D, et al. TNF transactivation of EGFR stimulates cytoprotective COX-2 expression in gastrointestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301(2):G220–9. doi:10.1152/ajpgi.00383.2010
25. Saini S, Liu T, Yoo J. TNF-α stimulates colonic myofibroblast migration via COX-2 and Hsp27. J Surg Res. 2016;204(1):145–152. doi:10.1016/j.jss.2016.04.034
26. Kim JJ, Lee SB, Park JK. TNF-α-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. 2010;17(9):1420–1434. doi:10.1038/cdd.2010.19
27. Suematsu N, Tsutsui H, Wen J, et al. Oxidative stress mediates tumor necrosis factor-α-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107(10):1418–1423. doi:10.1161/01.cir.0000055318.09997.1f
28. Sanchez-Bautista S, Nicolas FE. Recent patents concerning modulators of protein kinase C. Recent Pat DNA Gene Seq. 2013;7(1):74–81. doi:10.2174/1872215611307010011
29. Kassem KM, Clevenger MH, Szandzik DL, Peterson E, Harding P. PGE2 reduces MMP-14 and increases plasminogen activator inhibitor-1 in cardiac fibroblasts. Prostaglandins Other Lipid Mediat. 2014;113–115:62–68. doi:10.1016/j.prostaglandins.2014.09.002
30. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–756. doi:10.1038/nri1184
31. Kapadia S, Lee J, Torre-Amione G, Birdsall HH, Ma TS, Mann DL. Tumor necrosis factor-α gene and protein expression in adult feline myocardium after endotoxin administration. J Clin Invest. 1995;96(2):1042–1052. doi:10.1172/jci118090
32. Li YY, Feng YQ, Kadokami T, et al. Myocardial extracellular matrix remodeling in transgenic mice overexpressing tumor necrosis factor α can be modulated by anti-tumor necrosis factor α therapy. Proc Natl Acad Sci U S A. 2000;97(23):12746–12751. doi:10.1073/pnas.97.23.12746
33. Chen X, Subleski JJ, Kopf H, Howard OZ, Männel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+ CD25+ FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol. 2008;180(10):6467–6471. doi:10.4049/jimmunol.180.10.6467
34. Wen X, Chen X, Liang X, et al. The small molecule NSM00191 specifically represses the TNF-α/NF-small ka, CyrillicB axis in foot and ankle rheumatoid arthritis. Int J Biol Sci. 2018;14(12):1732–1744. doi:10.7150/ijbs.24232
35. Aoki T, Fukuda M, Nishimura M, Nozaki K, Narumiya S. Critical role of TNF-α-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol Commun. 2014;2:34. doi:10.1186/2051-5960-2-34
36. Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proce National Acad Sci. 1998;95(2):570–575. doi:10.1073/pnas.95.2.570
37. Rolski F, Błyszczuk P. Complexity of TNF-α signaling in heart disease. J Clin Med. 2020;9. doi:10.3390/jcm9103267
38. Hermida N, Michel L, Esfahani H, et al. Cardiac myocyte β3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling. Eur Heart J. 2018;39(10):888–898. doi:10.1093/eurheartj/ehx366
39. Li X, Zhang W, Cao Q, et al. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discovery. 2020;6(1):80. doi:10.1038/s41420-020-00316-9
40. Dai D-F, Hsieh EJ, Liu Y, et al. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93(1):79–88. doi:10.1093/cvr/cvr274
41. Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88(5):529–535. doi:10.1161/01.RES.88.5.529
42. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochimica et Biophysica Acta. 2017;1863(5):1066–1077. doi:10.1016/j.bbadis.2016.11.010
43. Jiménez-González S, Marín-Royo G, Jurado-López R, et al. The Crosstalk between cardiac lipotoxicity and mitochondrial oxidative stress in the cardiac alterations in diet-induced obesity in rats. Cells. 2020;9(2):451. doi:10.3390/cells9020451
44. Sverdlov AL, Elezaby A, Qin F, et al. Mitochondrial reactive oxygen species mediate cardiac structural, functional, and mitochondrial consequences of diet‐induced metabolic heart disease. J Am Heart Assoc. 2016;5(1):e002555. doi:10.1161/JAHA.115.002555
45. Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26(4):249–261. doi:10.1016/j.tcb.2015.12.002
46. Ferreira JCB, Brum PC, Mochly-Rosen D. βIIPKC and εPKC isozymes as potential pharmacological targets in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51(4):479–484. doi:10.1016/j.yjmcc.2010.10.020
47. Singh RM, Cummings E, Pantos C, Singh J. Protein kinase C and cardiac dysfunction: a review. Heart Fail Rev. 2017;22(6):843–859. doi:10.1007/s10741-017-9634-3
48. Song X, Qian X, Shen M, et al. Protein kinase C promotes cardiac fibrosis and heart failure by modulating galectin-3 expression. Biochim Biophys Acta. 2015;1853(2):513–521. doi:10.1016/j.bbamcr.2014.12.001
49. Koide Y, Tamura K, Suzuki A, et al. Differential induction of protein kinase C isoforms at the cardiac hypertrophy stage and congestive heart failure stage in Dahl salt-sensitive rats. Hypertension Res. 2003;26(5):421–426. doi:10.1291/hypres.26.421
50. Marrocco V, Bogomolovas J, Ehler E, et al. PKC and PKN in heart disease. J Mol Cell Cardiol. 2019;128:212–226. doi:10.1016/j.yjmcc.2019.01.029
51. Liu Q, Molkentin JD. Protein kinase Cα as a heart failure therapeutic target. J Mol Cell Cardiol. 2011;51(4):474–478. doi:10.1016/j.yjmcc.2010.10.004
52. Giroux M, Descoteaux A. Cyclooxygenase-2 expression in macrophages: modulation by protein kinase C-α. J Immunol. 2000;165(7):3985–3991. doi:10.4049/jimmunol.165.7.3985
53. Nakashima S. Protein kinase C alpha (PKC α): regulation and biological function. J Biochem. 2002;132(5):669–675. doi:10.1093/oxfordjournals.jbchem.a003272
54. Yu W, Chen C, Cheng J. The role and molecular mechanism of FoxO1 in mediating cardiac hypertrophy. ESC Heart Failure. 2020;7(6):3497–3504. doi:10.1002/ehf2.13065
55. Hsu CK, Lin CC, Hsiao LD, Yang CM. Mevastatin ameliorates sphingosine 1-phosphate-induced COX-2/PGE2-dependent cell migration via FoxO1 and CREB phosphorylation and translocation. Br J Pharmacol. 2015;172(22):5360–5376. doi:10.1111/bph.13326
56. Tseng HC, Lin CC, Wang CY, Yang CC, Hsiao LD, Yang CM. Lysophosphatidylcholine induces cyclooxygenase-2-dependent IL-6 expression in human cardiac fibroblasts. Cell Mol Life Sci. 2018;75(24):4599–4617. doi:10.1007/s00018-018-2916-7
57. Su D, Coudriet GM, Hyun Kim D, et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1β production in macrophages. Diabetes. 2009;58(11):2624–2633. doi:10.2337/db09-0232
58. Alikhani M, Alikhani Z, Graves DT. FOXO1 functions as a master switch that regulates gene expression necessary for tumor necrosis factor-induced fibroblast apoptosis. J Biol Chem. 2005;280(13):12096–12102. doi:10.1074/jbc.M412171200
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.