")
Back to Journals » Infection and Drug Resistance » Volume 13
UPLC-MS/MS Measurement of the Effect of Isavuconazole, Itraconazole and Fluconazole on the Pharmacokinetics of Selinexor in Rats
Authors Li S , Zhang Y, Cheng Q, Xin J, Dong Z , Qiu X
Received 29 June 2020
Accepted for publication 7 August 2020
Published 14 September 2020 Volume 2020:13 Pages 3153—3161
DOI https://doi.org/10.2147/IDR.S269831
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Shuang-long Li, Yi Zhang, Qian-shi Cheng, Jun-zhe Xin, Ze-qin Dong, Xiang-jun Qiu
School of Basic Medicine, Henan University of Science and Technology, Luoyang 471023, People’s Republic of China
Correspondence: Xiang-jun Qiu
School of Basic Medicine, Henan University of Science and Technology, Luoyang 471023, People’s Republic of China
Tel +86 13698882699
Fax +86 379-64830346
Email [email protected]
Objective: An ultra performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) method for the determination of selinexor was established to investigate the effects of isavuconazole, itraconazole and fluconazole on the pharmacokinetics of selinexor in rats, respectively.
Methods: Twenty-four healthy male rats were randomly divided into four groups: group A, normal saline; group B, isavuconazole (20 mg/kg); group C, itraconazole (20 mg/kg); and group D, fluconazole (20 mg/kg). After 30 min of oral administration of normal saline, isavuconazole, itraconazole, and fluconazole, all the rats were given selinexor (8 mg/kg). The plasma concentration of selinexor was estimated by UPLC-MS/MS, and the pharmacokinetic parameters of selinexor were calculated by Drug and Statistics (DAS) 2.0 software.
Results: Under these experimental conditions, the method showed good linearity and stability. Intraday and interday accuracy and sample recovery were acceptable. Compared with group A, the Cmax, AUC(0−t) and AUC(0−∞) of selinexor in group B increased by 59.05%, 31.69%, and 31.45%; the Cmax, AUC(0−t) and AUC(0−∞) of selinexor in group C increased by 56.14%, 25.34%, and 25.08%; the Cmax, AUC(0−t) and AUC(0−∞) of selinexor in group D increased by 43.44%, 29.16%, and 31.96%, respectively. The Tmax of the experimental groups were extended, and CLz/F was also significantly reduced.
Conclusion: These results indicated that isavuconazole, itraconazole, and fluconazole have significant inhibitory effects on selinexor pharmacokinetics and increased selinexor plasma exposure in rats. Therefore, when these drugs were used in combination, clinicians should pay attention to the changes in treatment effects and the occurrence of adverse reactions caused by the drug-drug interactions.
Keywords: isavuconazole, itraconazole, fluconazole, selinexor, UPLC-MS/MS, pharmacokinetics, drug-drug interactions
Introduction
Drug-drug interactions (DDIs) refers to the fact that one drug affects the action of another, they will interact with each other, so that the clinical effect of the original drug changes.1 This can lead to treatment failure and occasionally serious clinical adverse events, which is an important issue for drug safety.2,3 About 3–26% of all admitted patients with adverse reactions are caused by DDIs.4 In clinical practice, it is often useful to understand the mechanism by which specific DDI occurs, and it can help us clarify an alternative approach to minimize or even avoid its negative effects.5,6
Selinexor (KPT-330, Figure 1A) is the world’s first oral, selective nuclear export inhibition (SINE), which functions by binding and inhibiting nuclear export protein small molecule inhibitor of Exportin-1 (XPO1).7 XPO1 is the mainly function on nuclear export proteins such as p53, p21, BRCA1/2, pRB, FOXO, c-Myc, Bcl-xL, MDM2, and so on.7,8 It can promote the accumulation of tumor suppressor proteins in the nucleus, thereby restarting and amplifying its function as a tumor inhibitors, leading to apoptosis of cancer cells.9,10 It also largely protects normal cells from damage. the US FDA approved the combination of selinexor and low-dose dexamethasone for the treatment of relapsed and refractory multiple myeloma on July 3, 2019.11 In addition, selinexor has been granted rapid approval and orphan drug status by the FDA to conduct clinical studies in patients with relapsed and refractory diffuse large B-cell lymphoma who have previously received at least two lines of treatment. Clinical trials have found that various treatments such as selinexor and low-dose dexamethasone combined with lenalidomide, pomalidomide, and daratumumab have produced good results.12–14
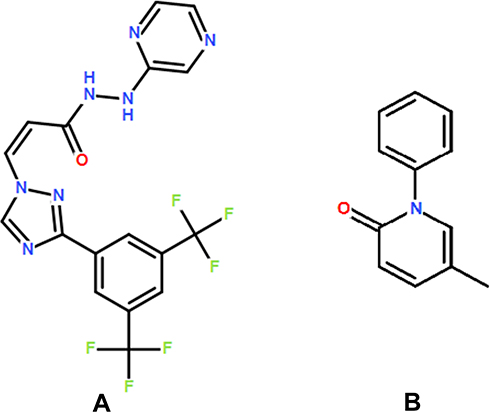 |
Figure 1 The chemical structure of selinexor (A) and pirfenidone (IS, (B)). |
Adults take selinexor (80 mg) as a single oral dose and reach Cmax within two to four hours.15 There is no clinical significance in the pharmacokinetic effect of taking medicine with high fat diet. The t1/2 of single oral selinexor in cancer patients is six to eight hours.16 Selinexor is mainly metabolized by CYP3A4, multiple UDP-glucuronosyltransferases and glutathione S-transferases (GSTs).16 Isavuconazole, itraconazole and fluconazole are commonly used clinical antifungal drugs, and mainly metabolized by CYP450 in the liver, especially by CYP3A4. Due to the possibility of fungal infection in cancer patients, antifungal drugs are combined clinically. Because they are metabolized by CYP450, and DDIs may occur.
Interactions between drugs can lead to decrease in the bioavailability of drugs, resulting in treatment failure or increased likelihood of adverse drug reactions, and even produce toxicity. There are no reports about the combined use of selinexor and conazole drugs. In order to prevent adverse drug events, we established an UPLC-MS/MS method to determine the concentration of selinexor in rat plasma, and observed the effects of isavuconazole, itraconazole and fluconazole on the pharmacokinetics of selinexor, respectively.
Materials and Methods
Chemicals Materials
Selinexor, isavuconazole, itraconazole, fluconazole, and pirfenidone (IS, Figure 1B) were obtained from Sigma (St Louis, MO, USA), and the purity exceeds 98%. Acetonitrile and methanol were HPLC grade and purchased from Merck Company. Formic acid was analytical grade and purchased from Sigma. Other chemicals were analytical grade.
Animal Experiments
All experimental procedures were approved by the Animal Ethics Committee of Henan University of Science and Technology. Sprague Dawley rats weighing 200±20 g were purchased from the Experimental Animal Center of Henan University of Science and Technology (Luoyang, China) and the Animal certificate was SCXK (Hubei) 2007–0001. The experiment was approved according to the Laboratory animals-guidelines for ethical review of welfare (issued by the General Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China, and the Standardization Administration of China; GB/T 35,892–2018). Twenty-four healthy male rats were randomly divided into four groups: group A, normal saline; group B, isavuconazole (20 mg/kg); group C, itraconazole (20 mg/kg); and group D, fluconazole (20 mg/kg). After 30 min of oral administration of normal saline, isavuconazole, itraconazole, and fluconazole, all the rats were orally given selinexor (8 mg/kg), and 300 µL of blood was collected from the tail vein of each rat and put into 1.5 mL heparinized tubes at 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 9, 12, 12, 24, and 48 h. The blood samples were centrifuged at 4000 g for 10 min, and the supernatant was separated and frozen at −20°C until analysis.
Sample Processing
In a 1.5 mL EP tube, accurately 50 μL of plasma and 10 μL of IS working solution (50 ng/mL) was added, and then mixed for 15 seconds. Two hundred microliters of acetonitrile was added and vortexed for 1.0 min. The mixture was centrifuged at 13,000 g for 15 min, and 2 μL of the supernatant was taken into a UPLC-MS/MS system for detection.
Instrumentation and Conditions
Selinexor and IS were separated on an Acquity BEH C18 column (2.1×50 mm, 1.7 μm) and the flow rate was 0.40 mL/min. The mobile phase was 0.1% formic acid (A) and acetonitrile (B). The gradient program was as follows: 0.00–0.50 min, 90% A; 0.50–1.00 min, 90→10% A; 1.00–2.00 min, 10% A; 2.00–2.10 min, 10→90% A; 2.10–3.00 min, 90% A. Mass spectrometry were measured by XEVO TQD triple quadrupole mass spectrometer, and the data was acquired using Masslynx V4.1 software. The multiple reaction monitoring modes of transitions as quantitative analysis were m/z 443.95→333.90 for selinexor, and m/z 185.98→91.97 for IS. The analysis time of each sample was three minutes, and the temperature of the autosampler and column were 4°C and 40°C, respectively. The cone voltage and collision energy were 20 V and 25 eV for selinexor, and were 30 V and 15 eV for IS, respectively.
Preparation of the Standard Solutions
Ten milligrams of selinexor and IS were accurately weighed respectively in two different 10 mL volumetric flask, and added methanol to the scale. Through gradient dilution, various working solutions of calibration curve and quality control (QC) were obtained. The final concentration of the calibration curve includes the following points: 1, 5, 10, 50, 100, 200, 500, and 1000 ng/mL. If the concentration of the sample exceeds the linear interval, the sample needs to be diluted before testing. QC sample in plasma concentrations of selinexor were set at 2.5, 200, and 800 ng/mL. All solutions were stored at 4°C in a refrigerator.
Method Validation
According to the guidelines of the US FDA, the specificity, linearity, precision, accuracy, recovery, and stability of the experimental method were verified.17
Statistical Analysis
DAS software (version 2.0) was used to analyze and calculate the pharmacokinetic parameters of selinexor, and the results were expressed as mean ±SD. The data were processed using SPSS 18.0 statistical software. P values were calculated using Independent sample t-test, and P<0.05 was considered statistically significant (P<0.05; P<0.01).
Results
Method Validation
Specificity
Typical chromatogram of selinexor and IS were shown in Figure 2, including blank samples, blank samples spiked with selinexor and IS, and plasma samples after oral selinexor. The experimental results showed that this method had selectivity, and no obvious endogenous substance interferences were found. The mean retention times of selinexor and IS were 1.43 and 1.27 min, respectively.
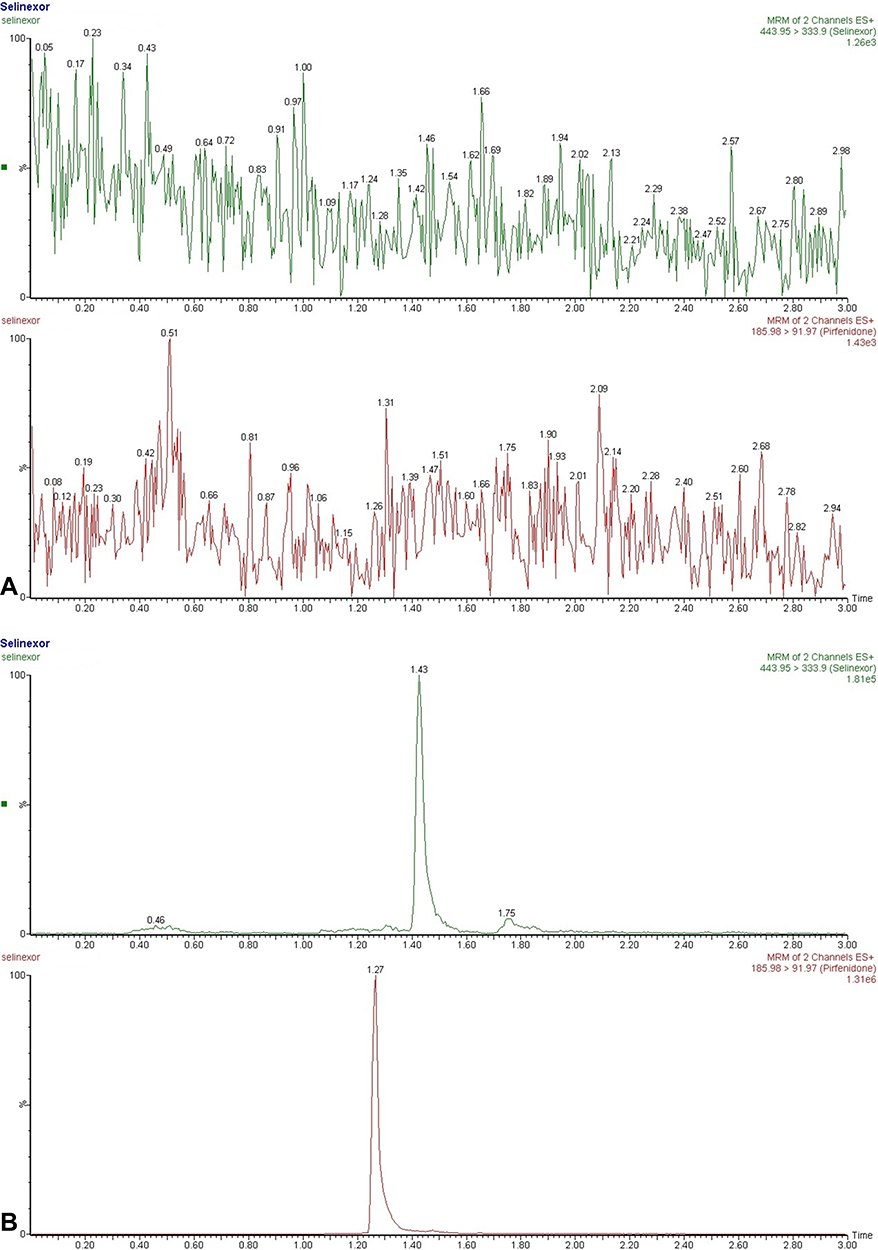 |
Figure 2 Continued. |
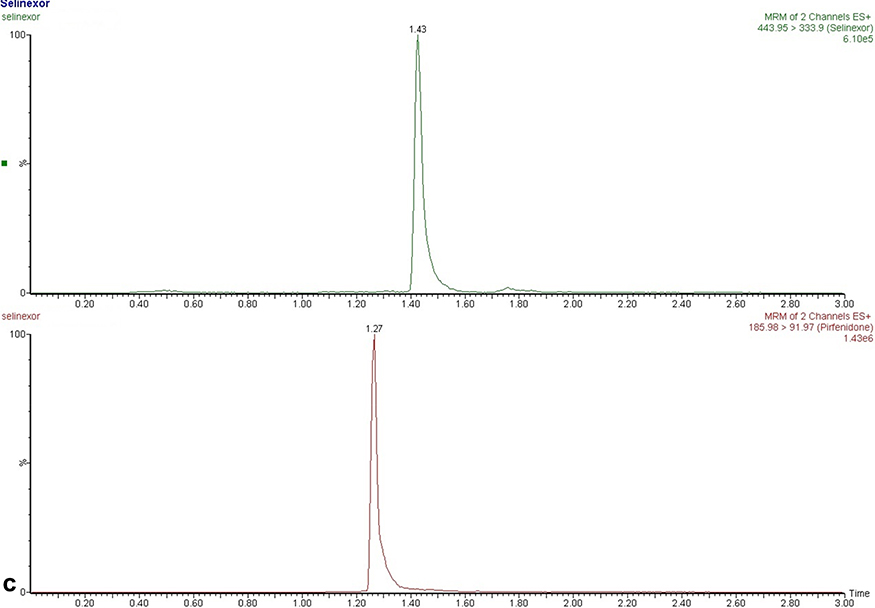 |
Figure 2 Representative chromatograms in positive ion mode. (A) A blank plasma sample; (B) a blank plasma sample spiked with selinexor and IS; (C) a rat plasma sample three hours after oral administration of selinexor (8 mg/kg). |
Linearity and Carryover
By analyzing the dosing calibration samples for three days, the calibration curve was constructed and verified. Selinexor plasma samples with concentrations of 1, 5, 10, 50, 100, 200, 500, and 1000 ng/mL were prepared, and the chromatograms were recorded after protein precipitation. The peak area ratio of selinexor and IS was y, and the plasma concentration of selinexor was x, and standard curves were fitted by weighted (1/χ2) least squares linear regression. The lowest point of the linear range was LLOQ, and the selinexor linearity and LLOQ results were shown in Table 1. The detection limit was sufficient to meet the requirements of the study.
 |
Table 1 Regression Equation, Linearity Range, Correlation Coefficients and LLOQ of Selinexor |
The carryover test was performed by adding IS (50 ng/mL) or selinexor (1000 ng/mL) to the blank plasma and then injecting a blank sample. In the blank sample, each analyte should be less than 20% of the LLOQ. The carryover test results indicate that no residual analytes were detected by the analyzer when the sample was injected at the next injection. Carryover did not affect the determination of selinexor.
Precision and Accuracy
The precision and accuracy results were shown in Table 2. The precision and accuracy of selinexor was analyzed by using QC samples at concentrations of 1, 2.5, 200, and 800 ng/mL, each concentration was repeated six times. The test was performed on the same day, and the intraday precision and accuracy was calculated. The continuous measurement was performed for three days, and the intraday precision and accuracy was calculated. The precision was expressed as relative standard deviation (RSD%≤15%), and accuracy was expressed as relative error (RE%≤±15%). The results showed that the method was reliable, accurate and reproducible.
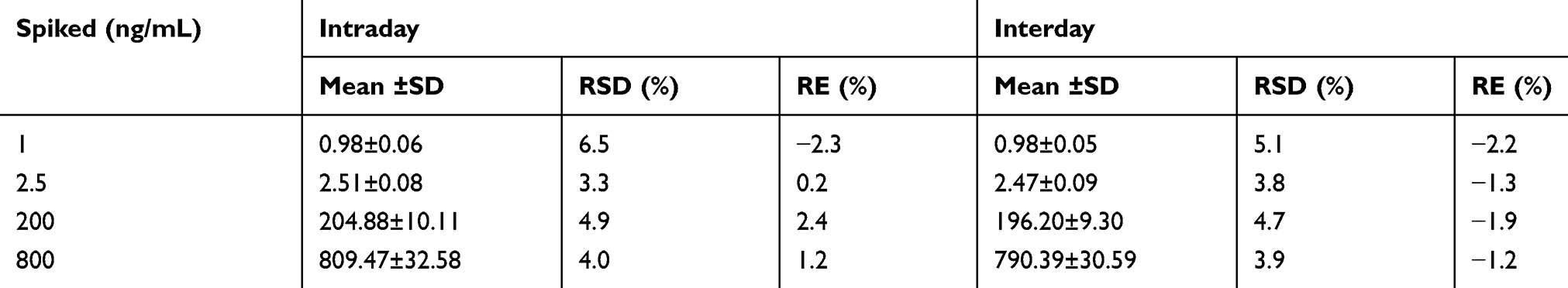 |
Table 2 Precision and Accuracy of Selinexor in Rat Plasma (n=6) |
Recovery and Matrix Effect (ME)
The recovery and ME results were shown in Table 3. The recovery and ME of selinexor were repeated six times at concentrations of 2.5, 200, and 800 ng/mL, respectively. The recovery was calculated by comparing the peak area of the conventionally pretreated QC sample with the corresponding blank plasma after extraction. The ME was evaluated by comparing the peak area ratio of the analyte in the sample after extraction to the corresponding water substitution sample. It could be seen from the results that this method had good recovery and ME.
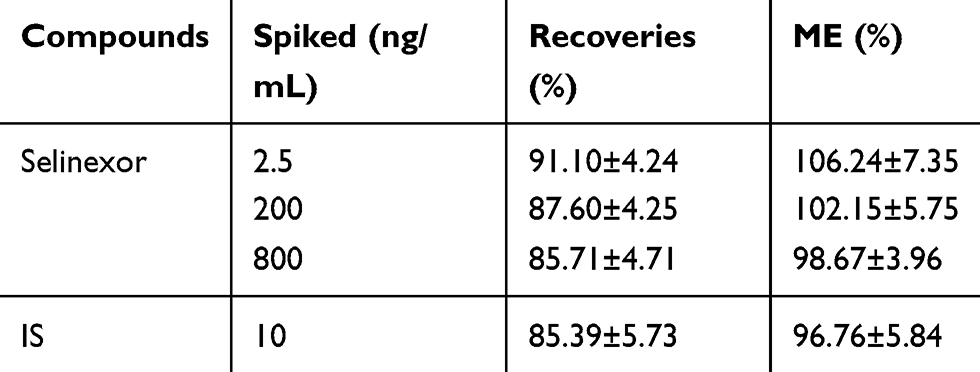 |
Table 3 The Recoveries and ME of Selinexor and is in Rat Plasma (n=6, Mean ±SD) |
Stability
The stability results of plasma samples under different conditions were shown in Table 4. The stability was obtained by repeating six times at concentrations of 2.5, 200, and 800 ng/mL, respectively. The QC samples were placed at room temperature for four hours, 4°C for 24 h, three cycles of freezing and thawing (−20~25°C), four weeks storage at −20°C to examine the stability. Under the above four conditions, the RE value was less than 10% and the RSD value was less than 15%. It could be seen from the results that the QC sample had good stability.
 |
Table 4 The Stability of Selinexor and is in Rat Plasma (n=6) |
Stock Solutions Stability
The stock solutions stability of selinexor and IS were shown in Table 5. Each QC sample was repeated six times. The room temperature stability was evaluated by comparing the stock solution stored at room temperature for 12 h with the remainder of the stock solution stored in a −20°C refrigerator. The freezing stability was evaluated by comparing the newly prepared stock solution with the stock solution stored in a −20°C refrigerator for three weeks. It could be seen from the results that the sample stability was good.
 |
Table 5 The Stock Solution Stability of Selinexor and is in Rat Plasma (n=6) |
Pharmacokinetic Study
If the concentration of the sample exceeds the linear interval, the sample needs to be diluted before testing. The pharmacokinetic parameters of selinexor for four groups included Tmax, t1/2, Cmax, AUC, MRT, CLz/F were shown in Table 6. The mean concentration-time curves of selinexor in rat plasma were shown in Figure 3.
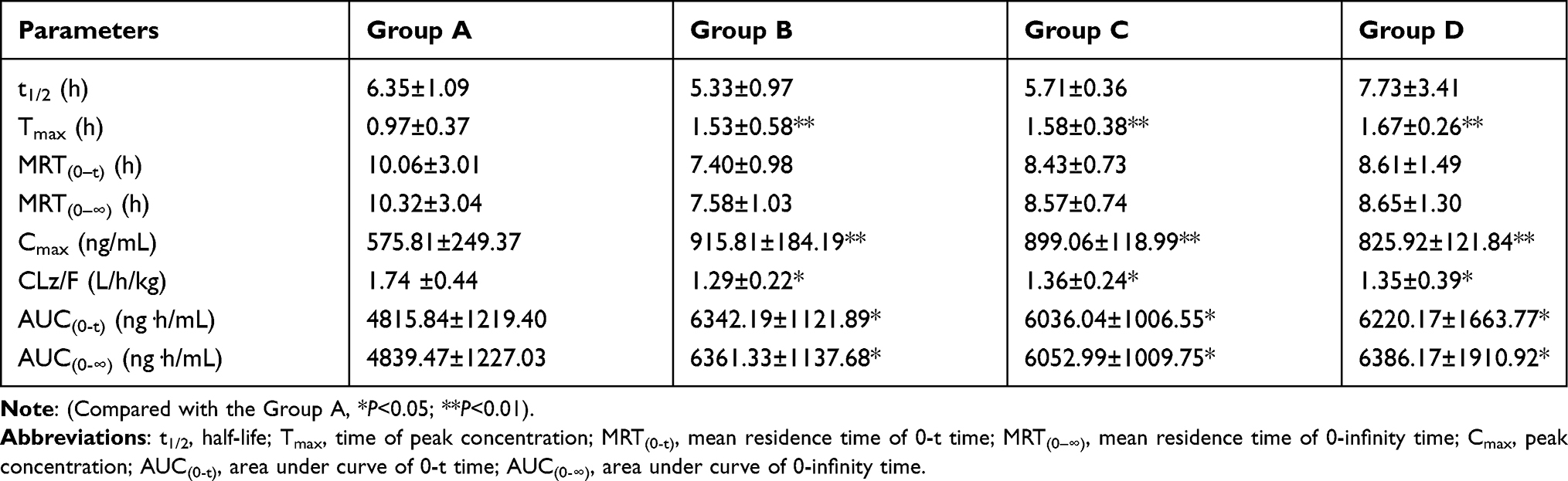 |
Table 6 Pharmacokinetic Parameters of Selinexor After Oral Administration to 8 mg/kg Selinexor (n=6, Mean ±SD) |
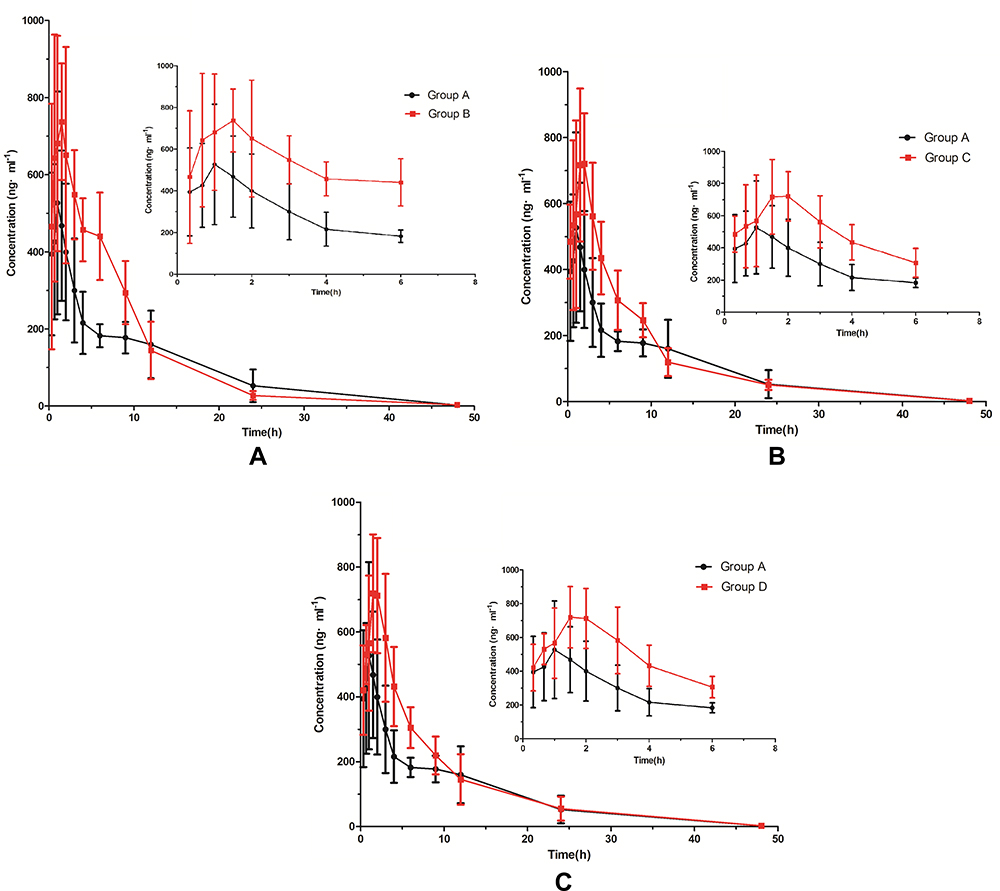 |
Figure 3 The mean plasma concentration-time curve of selinexor (zoomed one to six hours pharmacokinetic profile). Group A, normal saline; Group B, isavuconazole (20 mg/kg); Group C, itraconazole (20 mg/kg); and Group D, fluconazole (20 mg/kg) (n=6). |
Discussion
HPLC-UV, HPLC-Flu and LC-MS/MS had been widely used in the detection of drug concentration in biological samples. However, the method of detecting selinexor using the UPLC-MS/MS method is rarely reported. In this study, a new method was developed for the detection of selinexor. The lowest detection concentration was 1 ng/mL, which had high sensitivity. This method has short analysis time (three min). The internal standard is pirfenidone because it is stable and easy to obtain, and it has good separation from selinexor, good peak shape, and similar retention times. The endogenous substances in rat plasma samples did not affect the determination of selinexor and IS concentrations.
DDIs have been shown to be an important cause of adverse drug events.18 Drug interactions in the field of cancer are likely to become more and more common, because the number of new treatment options for anticancer drugs has grown exponentially, and they have the potential to extend the life expectancy of cancer patients.19 Compared with other causes of adverse drug reactions, DDIs were usually more predictable and preventable.
The experimental results (Table 6) showed that after the combined used of isavuconazole, itraconazole, and fluconazole, the pharmacokinetic parameters of selinexor had changed significantly, the elimination behavior of selinexor in rats might be affected. The Cmax, AUC(0−t) and AUC(0−∞) of selinexor in group B were 59.05%, 31.69%, and 31.45% higher than group A, respectively. The Cmax, AUC(0−t) and AUC(0−∞) of selinexor in group C were 56.14%, 25.34%, and 25.08% higher than group A, respectively. The Cmax, AUC(0−t) and AUC(0−∞) of selinexor in group D were 43.44%, 29.16%, and 31.96% higher than group A, respectively. The Tmax of the experimental groups were extended, and CLz/F was also significantly reduced. These results indicated that isavuconazole, itraconazole and fluconazole had significant inhibitory effects on the pharmacokinetics of selinexor and increase selinexor plasma exposure in rats.
Although there are certain differences in metabolism in different species, the results of animal experiments can provide a reference for clinical medication. Therefore, when conazole drugs and selinexor are used together in clinic, the dosage of the drug should be adjusted to ensure the therapeutic effect to avoid the occurrence of adverse reactions.
Conclusion
In this study, a sensitive, rapid and stable UPLC-MS/MS method was established to detect selinexor, and this method was successfully applied to the pharmacokinetic study. Isavuconazole, itraconazole, and fluconazole had significant inhibitory effects on the pharmacokinetics of selinexor and increase selinexor plasma exposure in rats. Azole antifungal drugs could significantly affect the pharmacokinetics of selinexor. Therefore, when these drugs were used in combination, clinicians should pay attention to the changes in treatment effects and the occurrence of adverse reactions caused by the interaction between the drugs.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author Xiang-jun Qiu upon request.
Acknowledgments
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Busari AA, Oreagba IA, Oshikoya KA, Kayode MO, Olayemi SO. High risk of drug-drug interactions among hospitalized patients with kidney diseases at a Nigerian Teaching Hospital: a call for action. Niger Med J. 2019;60(6):317–325. doi:10.4103/nmj.NMJ_2_19
2. Pirmohamed M. Drug-drug interactions and adverse drug reactions: separating the wheat from the chaff. Wien Klin Wochenschr. 2010;122(3–4):62–64. doi:10.1007/s00508-010-1309-1
3. Leone R, Magro L, Moretti U, et al. Identifying adverse drug reactions associated with drug-drug interactions: data mining of a spontaneous reporting database in Italy. Drug Saf. 2010;33(8):667–675. doi:10.2165/11534400-000000000-00000
4. Dechanont S, Maphanta S, Butthum B, Kongkaew C. Hospital admissions/visits associated with drug-drug interactions: a systematic review and meta-analysis. Pharmacoepidemiol Drug Saf. 2014;23(5):489–497. doi:10.1002/pds.3592
5. Scripture CD, Figg WD. Drug interactions in cancer therapy. Nat Rev Cancer. 2006;6(7):546–558. doi:10.1038/nrc1887
6. Riechelmann RP, Saad ED. A systematic review on drug interactions in oncology. Cancer Invest. 2006;24(7):704–712. doi:10.1080/07357900601063766
7. Abdul Razak AR, Mau-Soerensen M, Gabrail NY, et al. First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol. 2016;34(34):4142–4150. doi:10.1200/JCO.2015.65.3949
8. Gounder MM, Zer A, Tap WD, et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol. 2016;34(26):3166–3174. doi:10.1200/JCO.2016.67.6346
9. Lapalombella R, Sun QX, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120(23):4621–4634. doi:10.1182/blood-2012-05-429506
10. Yoshimura M, Ishizawa J, Ruvolo V, et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma. Cancer Sci. 2014;105(7):795–801. doi:10.1111/cas.12430
11. FDA grants accelerated approval to selinexor for multiple myeloma. Case Med Res. 2019.
12. Chen C, Kotb R, Sebag M, et al. Selinexor shows synergy in combination with pomalidomide and low dose dexamethasone in patients with relapsed/refractory multiple myeloma. Blood. 2016;128(22). doi:10.1182/blood-2016-06-724161
13. Bahlis NJ, Sutherland H, White D, et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood. 2018;132(24):2546–2554. doi:10.1182/blood-2018-06-858852
14. ClinicalTrials.gov. Selinexor and backbone treatments of multiple myeloma patients (STOMP).
15. Karyopharm Therapeutics Inc. XPOVIO™ (selinexor) tablets, for oral use: US prescribing information. 2019.
16. Syed YY. Selinexor: first global approval. Drugs. 2019;79(13):1485–1494. doi:10.1007/s40265-019-01188-9
17. US Food and Drug Administration. Guidance for Indus-Try: Bioanalytical Method Validation. Rockville, MD, USA: US Department of Health and Human Services, US FDA, Center for Drug Evaluation and Research; 2018.
18. Toivo TM, Mikkola JA, Laine K, Airaksinen M. Identifying high risk medications causing potential drug-drug interactions in outpatients: a prescription database study based on an online surveillance system. Res Social Adm Pharm. 2016;12(4):559–568. doi:10.1016/j.sapharm.2015.09.004
19. Riechelmann RP, Del Giglio A. Drug interactions in oncology: how common are they? Ann Oncol. 2009;20(12):1907–1912. doi:10.1093/annonc/mdp369
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.