")
Back to Journals » ImmunoTargets and Therapy » Volume 4
Update on use of aldesleukin for treatment of high-risk metastatic melanoma
Authors Amaria R, Reuben A, Cooper Z, Wargo JA
Received 13 January 2015
Accepted for publication 25 February 2015
Published 7 April 2015 Volume 2015:4 Pages 79—89
DOI https://doi.org/10.2147/ITT.S61590
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Michael Shurin
Rodabe N Amaria,1 Alexandre Reuben,2 Zachary A Cooper,2,3 Jennifer A Wargo2,3
1Department of Melanoma Medical Oncology, 2Department of Surgical Oncology, 3Department of Genomic Medicine, University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract: High-dose interleukin-2 has been used for the treatment of metastatic melanoma since 1998 based on data proving durable complete responses in up to 10% of treated patients. The immunomodulatory effects of this critical cytokine have been instrumental in the development of immunotherapy for melanoma and other cancers. However, with the advent of new therapies, its use as a front-line agent has come into question. Nonetheless, there is still a role for interleukin-2 as monotherapy, as well as in combination with other agents and in clinical trials. In this article, we review preclinical and clinical data regarding interleukin-2, its pharmacology and mechanism of action, its toxicity profile, and its use in ongoing and planned clinical trials. We also explore the future of this agent within the treatment landscape for melanoma.
Keywords: aldesleukin, melanoma, immunotherapy
Introduction to management of metastatic melanoma
Early-stage melanoma may often be cured by surgery, although later-stage disease is most often fatal. Until 2011, there were only two drugs approved by the US Food and Drug Administration (FDA) including dacarbazine, a chemotherapeutic agent, and aldesleukin, a cytokine therapy, also known as high-dose interleukin (IL)-2. However, since 2011, six drugs have been approved by the FDA for use in unresectable stage III and stage IV melanoma, including three agents targeting the mitogen-activated protein kinase (MAPK) pathway (two BRAF inhibitors and an MEK inhibitor) and three immune checkpoint inhibitors (which block inhibitory molecules on the surface of T lymphocytes). These new therapies have vastly changed the treatment options for patients with advanced-stage disease, and many physicians now question the utility of standard chemotherapy and IL-2 in these patients; however, these clearly still have a role which is discussed herein.
Chemotherapy
The alkylating agent dacarbazine is the only chemotherapy approved by the FDA for melanoma. Multiple prior studies have shown a 15% overall response rate with no survival benefit.1,2 Multiple other chemotherapeutic agents have been used in melanoma, including temozolomide, taxanes, and platinum-based agents, all with similarly low response rates. Use of temozolomide is reasonable for patients with metastases affecting predominantly the central nervous system. However, with the recent developments in targeted and immunotherapies for melanoma, chemotherapy is best suited for patients with rapidly growing wild-type BRAF disease or following refractoriness to BRAF blockade.
Targeted therapy
Approximately 50% of cutaneous melanomas harbor an activating mutation in the BRAF oncogene, leading to constitutive activation of the MAPK signaling pathway involved in cellular proliferation and survival.3 Use of single-agent BRAF blockade with either vemurafenib (Zelboraf®, Roche-Genentech, South San Francisco, CA, USA) or dabrafenib (Tafinlar®, GlaxoSmithKline, London, UK) results in a progression-free survival benefit of approximately 6 months and an overall survival rate of approximately 13 months.4,5 The 6-month progression-free survival benefit is indicative of the ability of melanoma to develop resistance, and studies have shown that BRAF-mutated melanoma cells can maintain MAPK signaling through RAF-independent activation of MEK, a kinase downstream of RAF in the MAPK cascade.6,7 Multiple additional mechanisms of resistance have also been noted.6–13 Targeting an additional node in the MAPK pathway by combined inhibition of BRAF and MEK (dabrafenib plus trametinib versus dabrafenib alone) achieved a higher overall response rate of 76% versus 54%, as well as a longer median progression-free survival of 9.4 months versus 5.8 months.14
Checkpoint blockade
Melanoma is known to be immunogenic and is thus the gateway solid tumor malignancy for the development of immunotherapies. Although vaccine strategies and cytokine therapy have a long history of use in melanoma, checkpoint blockade has recently emerged at the forefront of melanoma immunotherapy. Checkpoint blockade makes use of the fact that T-cell activation requires a combination of antigen-specific T-cell receptor activation as well as costimulatory activation from adjacent antigen-presenting cells or neoplastic cells. Checkpoint proteins can be either immune stimulatory or immune inhibitory. The most progress with checkpoint blockade has been achieved with the negative costimulatory molecules, ie, cytotoxic T lymphocyte antigen (CTLA)-4 and programmed death (PD)-1, that limit T-cell activation to provide negative regulatory effects and prevent excess immune activation and development of autoimmunity.15 Since T-cells in the tumor microenvironment are known to express these negative costimulatory markers, development of monoclonal antibodies to block negative regulation and thus allow for immune system activation has provided the rationale for checkpoint blockade as a cancer therapy.16
Two randomized Phase III clinical trials tested the anti-CTLA-4 monoclonal antibody, ipilimumab (Yervoy®, Bristol-Myers Squibb, New York, NY, USA), in patients with melanoma. The initial trial randomized 676 patients with pretreated, unresectable stage III or IV melanoma to treatment with either ipilimumab dosed at 3 mg/kg, the HLA-A*0201-restricted gp100 peptide vaccine, or a combination of ipilimumab and the vaccine. Although the overall response rate to ipilimumab in either treatment arm was low (10.9%), ipilimumab-treated patients had improved overall survival compared with those treated with vaccine alone (10.0 months versus 6.4 months).17 Similar results were reported in a second randomized controlled trial comparing ipilimumab plus dacarbazine with dacarbazine alone in 502 patients with treatment-naïve advanced melanoma. This study utilized a higher dose of ipilimumab at 10 mg/kg. Response rates were 15% in the group treated with ipilimumab and dacarbazine compared with 10% for the dacarbazine alone arm.18 Overall survival of ipilimumab-treated patients improved by 2 months compared with the control arm. Recently, a pooled overall survival analysis of eleven different ipilimumab clinical trials showed a 3-year overall survival of 22%, confirming the durability of responses even though the overall response rate are low.19
Treatment with ipilimumab is associated with immune-mediated adverse events such as diarrhea or colitis, dermatitis, and pruritus. The dermatitis and pruritus are usually self-limiting and are easily treated with supportive measures; however, development of colitis and colon perforation can be life-threatening if not aggressively treated with steroids. Endocrinopathies, such as hypothyroidism, hyperthyroidism, or hypophysitis, can be appreciated, so there is a long-term requirement for hormonal replacement.17,18 Significantly more toxicity is seen with high-dose ipilimumab, with grade 3 or 4 adverse events observed in 56% of patients treated at a dose of 10 mg/kg.18
PD-1 blockade using the monoclonal antibodies pembrolizumab (Keytruda®, Merck Sharpe and Dohme, Whitehouse Station, NJ, USA) and nivolumab (Opdivo®, Bristol-Myers Squibb) has been found to be both safe and effective, leading to approval of both drugs by the FDA in 2014. In a pooled analysis of 411 melanoma patients treated with the anti-PD-1 antibody MK-3475 (pembrolizumab) with over 6 months of follow-up data, the overall response rate was 40% in ipilimumab-naïve patients and 28% in ipilimumab-refractory patients. Median progression-free survival was 24 weeks and median overall survival had not been reached at the time of analysis. Pembrolizumab is very well tolerated, with 12% of patients experiencing a grade 3 or 4 adverse event attributed to the drug, but only 4% of patients having to discontinue treatment due to adverse events.20 The most commonly appreciated adverse events are fatigue, nausea, and pruritus. Diarrhea and colitis are relatively rare, although fatal cases of pneumonitis have been reported.21,22 Pembrolizumab was approved by the FDA in September 2014 for patients with melanoma refractory to ipilimumab and BRAF-directed therapy if applicable. In December 2014, nivolumab received FDA approval for the same indication. The initial Phase I trial enrolled 296 patients with either advanced melanoma or other solid tumors, including non-small cell lung cancer, prostate cancer, renal cell carcinoma, or colorectal cancer. The overall response rate was 28% in the melanoma cohort, with durable responses lasting longer than 1 year in half of responding patients.21 Nivolumab has also been tested in the first-line setting for patients without BRAF-mutated disease. In one such trial, 418 patients were randomized to either nivolumab or dacarbazine. The overall response rate was 40% in the nivolumab-treated group compared with 14% in the dacarbazine group. The 1-year overall survival rate was 79% for the nivolumab-treated patients and 40% for the dacarbazine-treated patients, with a hazard ratio for death of 0.42.22
Combination checkpoint blockade represents a very potent although potentially toxic modality of treatment. In a Phase I trial of 86 patients with unresectable stage III or IV melanoma treated with either concurrent or sequential ipilimumab and nivolumab, concurrent CTLA-4 and PD-1 blockade achieved a higher overall response rate of 40%, with 53% of patients achieving a complete response or partial response at the maximum doses tested, and 31% of responders demonstrating tumor regression of 80% or more, even with bulky disease.23 Grade 3 or 4 adverse events did occur at a higher frequency than with monotherapy in 53% of patients, but the majority of events were reversible with appropriate supportive management. Further study is necessary to determine the role of this combination in the melanoma treatment landscape.
IL-2 receptor pathways and melanoma
IL-2 is a central cytokine governing the immune system through pleiotropic effects mediated by promoting expansion of both cytotoxic T-cells (Tconvs) and regulatory T-cells (Tregs). The capacity of IL-2 to promote the suppressive capability of Tregs further confirms the antagonism of its effects.
IL-2 and IL-2R expression
IL-2 is produced by many types of cells, although mainly by activated effector T lymphocytes. The IL-2 receptor (IL-2R), however, is expressed by all T lymphocytes as well as by numerous other types of immune cells. It is composed of three subunits that may be assembled in many combinations, each with its own affinity for IL-2. The α-chain (CD25) is detected mainly on regulatory and effector T-cells, although it may be expressed at lower levels by other cell types, such as endothelial cells, Langerhans cells, and fibroblasts. The β-chain (CD122) is expressed mainly on effector and memory T-cells as well as on natural killer cells. Finally, the γ-chain (CD132) presents an evenly distributed expression pattern in most immune cell types.24
The CD25 chain of the IL-2R may bind IL-2 on its own, although this type of binding provides a weak affinity (Kd 10−8) and results in lack of intracellular signaling.25 On the other hand, the IL-2R may also be composed of the CD122/CD132 heterodimer, which possesses an intermediate affinity for IL-2 (Kd 10−9) or by all three subunits for optimal binding (Kd 10−11).25 Importantly, the CD122 and CD132 subunits of the IL-2R are the only subunits capable of intracellular signaling.26,27
Signaling pathways
Signaling through the IL-2R occurs following binding of IL-2 to CD25, which induces a structural change in the CD25 subunit and recruitment of the CD122 and CD132 subunits. This results in optimal binding when all three subunits are coexpressed. Subsequent signaling may occur in one or all of three pathways including phosphatidylinositol-4 5-bisphosphate 3-kinase (PI3K), MAPK, and STAT5.
In the first and second pathways, the CD132 subunit recruits JAK3, which then activates JAK1 in association with CD122. Phosphorylation of tyrosine 338 (Y338) allows recruitment of the adaptor protein SHC, which results in signaling through the MAPK and PI3K pathways and induces proliferation and cell survival signals.28–30
In the second pathway, phosphorylation of Y392 and Y510 on CD122 recruits the transcription factor STAT5, which causes STAT5-dependent gene synthesis such as expression of the transcription factor FoxP3 and the IL2-Rα subunit CD25 (Figure 1). Phosphorylation of all three tyrosines (Y338, Y392, Y510) on CD122 is required for optimal T-cell proliferation.30,31
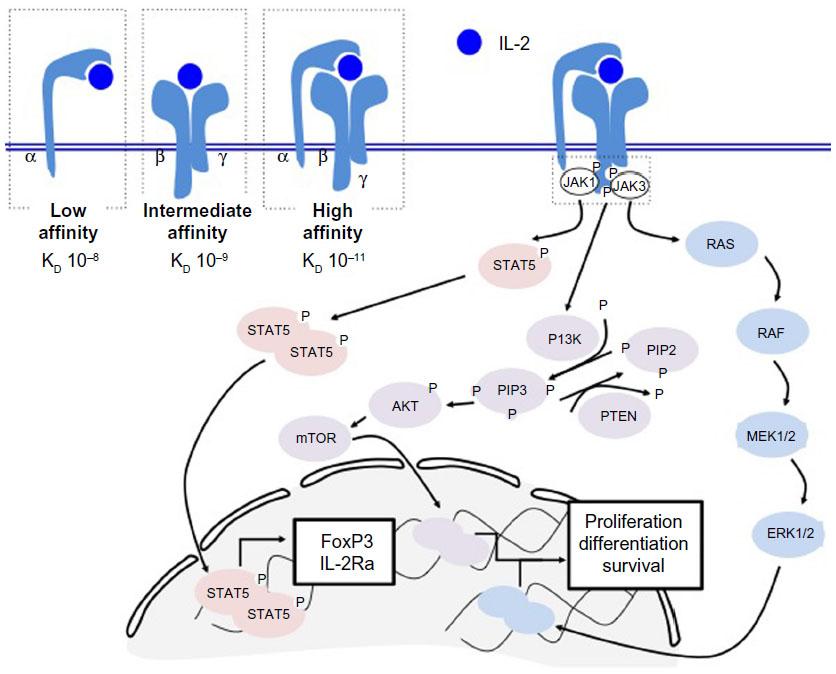 | Figure 1 IL-2R signaling triggers three distinct pathways. The IL-2R is comprised of three subunits that may be assembled in three different combinations (top left). The low affinity form contains only CD25 (IL-2Rα), the intermediate affinity form contains subunits CD122 and CD132 (IL-2Rβ and IL-2Rγ), and the high affinity form contains subunits CD25, CD122, and CD132. Importantly, signaling occurs through the CD122 and CD132 subunits. Upon binding to IL-2, the IL-2R recruits JAK1 and JAK3, which cause downstream signaling through the STAT5, phosphatidylinositol-4 5-bisphosphate 3-kinase, or mitogen-activated protein kinase pathways and result in expression of the FoxP3 transcription factor, or proliferation signals. |
Importantly, IL-2 signaling in Tconvs results in signaling through the STAT5, PI3K, and MAPK pathways, whereas only STAT5 signaling is induced in Tregs due to activation of phosphatase and tensin homolog and subsequent inhibition of the MAPK-PI3K pathway, a feature not present in Tconvs, which rather downregulate phosphatase and tensin homolog upon antigen stimulation. Finally, production of IL-2 induces expression of BLIMP-1, which, in turn, represses production of IL-2, and thus serves as a negative feedback mechanism for secretion of IL-2.32
Mode of action
High-dose (HD) IL-2 has been shown to be highly efficacious in improving survival in patients with metastatic melanoma. However, its failures also suggest that immunosuppressive mechanisms may counteract tumor clearance following administration of IL-2.
Antitumor mechanisms
IL-2 promotes the activation and expansion of Tconv CD4 and CD8 T lymphocytes through PI3K/MAPK signaling and enhances bcl-2 expression and induction of cyclins required for cell cycle progression and survival.28 Upon administration of IL-2, CD8 and CD4 T-cells expand exponentially, which further enhances tumor reactivity and clearance and promotes the generation of terminally differentiated CD4 and CD8 Tconvs. IL-2 also controls production of interferon-gamma (IFN-γ), which in turn increases the expression of major histocompatibility complex molecules in the immediate environment and thereby enhances antigen presentation and recognition of tumor cells.33 In addition to promoting the proliferation and differentiation of effector T lymphocytes, IL-2 has also been shown to induce expression of granzymes A, B, and C, as well as perforin, all of which are proteases that are crucial in mediating the cytotoxic potential of Tconvs in response to tumor antigens.34 Further, administration of IL-2 promotes generation of both Th1 and Th2 cells via transcription factors Tbet and Gata3,24 respectively, with a Th1 response being preferable for optimal tumor clearance.35 Treatment with IL-2 may also result in induction of vascular permeability through activation of endothelial cells. However, this activation is indirect, as it is entirely dependent on the presence of peripheral blood mononuclear cells and thus is likely due to the production of IL-2-induced cytokines, ie, IL-1, tumor necrosis factor, lymphotoxin, and IFN-γ, which affect vascular permeability, as IL-2-activated peripheral blood mononuclear cell supernatants cause endothelial cell activation when isolated.36 Finally, IL-2 stimulates production of chemokines, such as macrophage inflammatory protein-1β, macrophage chemoattractant protein-1, and granulocyte-macrophage colony-stimulating factor, by monocytes and macrophages early on after infusion,36 which suggests the main mechanism responsible for the efficacy of IL-2 may lie in activation of monocytes and macrophages and increased recruitment of Tconvs into tumors. This hypothesis is supported by the rapid decrease in circulating T-cells in blood following infusion of IL-2, suggesting that T-cells are homing to tumors since IL-2 does not affect peripheral blood mononuclear cell numbers in vitro.36
Protumor mechanisms
On the other hand, IL-2 has not been entirely efficacious in combating metastatic melanoma, and is well known to promote development and survival of Tregs. In fact, studies in IL-2R knockouts have demonstrated that, unexpectedly, severe autoimmunity rather than immunosuppression occurs in mice, suggesting that Tregs may be most dependent on IL-2 for survival.37,38 To this effect, higher expression of CD25 by Tregs allows better binding and uptake of IL-2 compared with Tconvs, thereby behaving as cytokine sinks and depriving Tconvs of much needed circulating IL-2 concentrations.39 Finally, the initial expansion of Tconvs following IL-2 exposure may be short-lived, as T-cells responding to IL-2 may undergo apoptosis upon re-encountering their cognate antigen.40 The short half-life of IL-2 in vivo may further hamper tumor clearance.41
All in all, treatment with aldesleukin activates both antitumor and protumor mechanisms and cell types that interact to mediate tumor outcome. Based on the clinical data thus far, it appears that the immunosuppressive mechanisms induced may ultimately dominate IL-2-mediated antitumor responses in the context of metastatic melanoma (Figure 2).
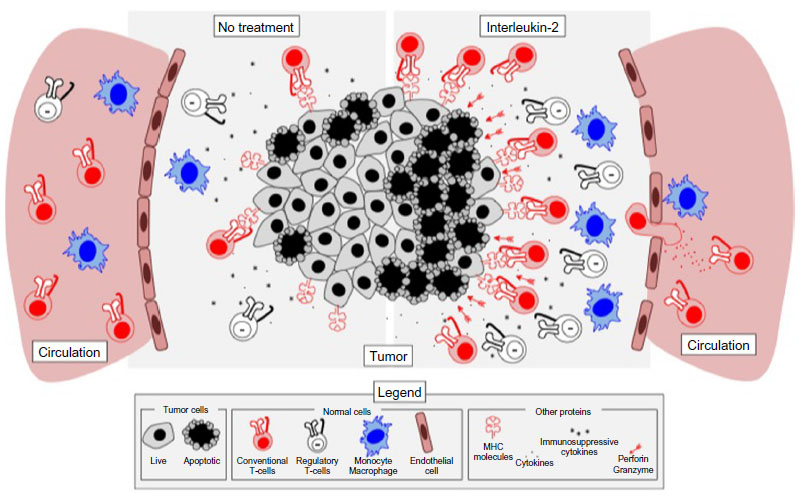 | Figure 2 IL-2 therapy promotes both immune activation and immune suppression in melanoma tumors. The tumor microenvironment is highly immunosuppressive and can contain limited amounts of infiltrating T lymphocytes (left). Upon treatment with IL-2 (right), effector T-cells are expanded and recruited into tumors, along with regulatory T-cells that bind IL-2 more efficiently and thus deplete it from the microenvironment. IL-2 also results in activation of monocytes and increases in production of interferon-gamma, which may result in increased antigen presentation and recognition of tumor cells by infiltrating T-cells. Finally, IL-2 causes increases in granzymes A, B, and C, as well as in perforin, which allow for better cytotoxicity against tumor cells upon recognition by antigen-specific T-cells. IL-2 therapy has also been shown to be associated with increased vascular leakage, a side effect linked to both efficacy and toxicity of IL-2 therapy. |
Clinical efficacy, safety, and tolerability
Aldesleukin as a single agent
The seminal publication describing the efficacy of high-dose IL-2 assessed 270 patients with metastatic melanoma treated in eight separate clinical trials between 1985 and 1993. Doses of 600,000 or 720,000 IU/kg every 8 hours up to 14 doses with courses repeated after a 6–9-day rest were utilized, with a second course being administered every 6–12 weeks in stable or responding patients. The overall response rate was 16%, with 6% of patients achieving a complete response and 10% of patients achieving a partial response. The minimal duration of response for the 10% of partial responders was approximately 5.9 months. The median duration of response was not achieved for the patients who achieved a complete response. The median survival for all 270 patients was 11.4 months, with 15 patients surviving over 5 years at the time of writing. In patients achieving a complete response, the responses were long-lasting, with responses over 40 months in duration observed at the time of writing. Multiple prognostic factors were analyzed, and the only strong correlations were that better responses were more likely in patients with better baseline performance status. The objective response rate was higher in patients who were treatment-naïve, although responses were seen in patients who had received prior systemic chemotherapy or immunotherapy. Treatment responses were appreciated in virtually every organ involved, although responses were less vigorous in patients with hepatic metastases. Durable responses were more likely to be appreciated in patients with lymph node, lung, or skin involvement.42
Toxicity
Toxicities from high-dose IL-2 can be severe and life-threatening when improperly managed. Toxicities mimic septic shock, with the appreciation of fevers, chills, hypotension, and tachycardia. In the above-mentioned pooled data from Atkins et al, 2.2% of patients died from treatment-related toxicity, with bacterial sepsis being the major cause of death. Use of prophylactic antibiotics has decreased the rate of bacterial sepsis and has made high-dose IL-2 therapy safer. Although toxicities can be severe, they can usually be anticipated, and vigilant monitoring and aggressive interventions allow for safe drug administration.
Onset of side effects usually occurs within 2 hours after the first dose of therapy, and these include hypotension and tachycardia. Nausea, vomiting, and diarrhea can be appreciated with subsequent doses. Intravascular volume depletion with overall volume overload from capillary leak syndrome is progressive throughout the course of therapy. Approximately 64% of patients experience hypotension; however, grade 4 hypotension is only seen in 1% of patients. Use of vasopressors is common, and the alpha agonist phenylephrine is often useful for pressure support in the setting of tachycardia. Progressive shortness of breath and pulmonary edema can also be seen, and patients often require supplemental oxygen during therapy. Discontinuation of therapy and diuresis is essential for significant pulmonary edema requiring more than 4 L of oxygen. Oliguria usually develops within 8 hours of initiating therapy and can often be managed by fluid boluses. If oliguria does not resolve with administration of 1–1.5 L of crystalloid fluids, use of low-dose dopamine (2 μg/kg/min) should be added to allow for urine output of at least 10–20 mL/hour. Most patients will require diuresis once blood pressure has stabilized while off vasopressors to mobilize fluid accumulated during therapy. Skin toxicities can also be seen, and usually include skin erythema and itching, which requires diligent use of emollient creams. Neuropsychiatric effects can also be appreciated, and vary from mild confusion to frank psychosis. Changes in mental status are short-lived, and symptoms improve with time following the last administration of high-dose IL-2.42,43
Aldesleukin combination regimens
Biochemotherapy (which combines chemotherapeutic agents, such as dacarbazine or temozolomide, with vinblastine and cisplatin, and the immune stimulators IL-2 and IFN-α) is used adjuvantly for high-risk stage III disease and for treatment in select patients with metastatic melanoma. A limitation is that IL-2 is administered as a 96-hour continuous infusion. This regimen has shown a relapse-free survival benefit over IFN-α in the adjuvant setting and has response rates of 45% in the metastatic setting.14,44,45 However, this regimen is not well tolerated and is associated with significant toxicity. Use of IL-2 in this regimen is hypothesized to lead to long-term survival in a subset of treated patients, with 5-year and 10-year survival rates of 17% and 15%, respectively, in patients with metastatic disease.46
In addition to biochemotherapy, IL-2 has been used in combination with other agents such as vaccines. Previous studies using the gp100 peptide vaccine were shown to increase circulating levels of antigen-specific T-cells with cytotoxic capabilities in vitro, thus leading to the hypothesis that concurrent use of vaccine with cytokine stimulation would be additive. A randomized Phase III clinical trial of 185 patients with metastatic melanoma was conducted using high-dose IL-2 alone or high-dose IL-2 with the gp100 HLA-A*0201-restricted peptide vaccine. Patients receiving the combination therapy had a statistically significant improvement in overall clinical response (16% versus 6%) which was associated with an improvement in overall survival (17.8 months versus 11.1 months). Toxicities were similar in both groups, but patients receiving the vaccine experienced more skin reactions at the injection site and a higher incidence of transient reversible sinus tachycardia and supraventricular tachycardia than patients who received high-dose IL-2 alone.47
Another treatment modality incorporating treatment with IL-2 involves the use of adoptive T-cell therapy (ACT). ACT using ex vivo-expanded T-cell clones or tumor-infiltrating lymphocytes (TILs) has been extensively studied in the treatment of metastatic melanoma. For TIL trials, T-cells are isolated from a tumor deposit and amplified in the laboratory setting to potentially billions of cells which are then infused back into the patient following administration of lymphodepleting chemotherapy. IL-2 is a critical component of T-cell expansion in vitro, and is also used via intravenous infusion following administration of TIL to potentiate the survival and persistence of the adoptively transferred T-cells. These regimens produce response rates of approximately 40%–50% and are associated with significant toxicities stemming from the use of lymphodepleting drugs, which make patients vulnerable to infection and bleeding complications as well as the toxicities of aldesleukin.48–50
It is not known how much of the clinical response to TIL therapy is due to high-dose IL-2, and it is hypothesized that much lower doses of IL-2 would be sufficient to support the transferred T-cells. In an initial Phase I study using non-myeloablative lymphodepleting chemotherapy in combination with adoptively transferred antigen-specific T-cells, three different dosing regimens of IL-2 were utilized, including standard high-dose (720,000 IU/kg intravenously three times a day for a maximum of 12 doses), low-dose (72,000 IU/kg intravenously three times a day for a maximum of 15 doses), or no IL-2. Although there were no objective clinical responses seen in this very early ACT trial, transient tumor reductions were seen in patients who received low-dose IL-2 therapy, thereby showing the feasibility of using lower doses of IL-2 in conjunction with ACT.51
A novel Phase I study investigated adoptively transferred CD8+ T-cell clones targeting tumor-associated antigens MART-1, MelanA, and gp100 in patients with metastatic melanoma. In this trial, four infusions of autologous T-cell clones were administered with or without IL-2 for the first infusion, and then graduated levels of low-dose IL-2 at 0.25, 0.50, and 1×106 units/m2 daily for the second, third, and fourth infusions, respectively. This study demonstrated the ability of the T-cell to track into the tumor, to persist in response to low-dose IL-2, and to produce minor responses or stable disease lasting up to 21 months in eight of ten treated patients. The median survival of transfused T-cells was significantly improved by administration of IL-2 compared with no administration of IL-2; however, the persistence of T-cells was not increased over what was seen with administration of the lowest dose of IL-2. The T-cell infusions and low-dose IL-2 were well tolerated with no observed grade 3 or 4 toxicity.52
A small pilot study in Denmark tested the feasibility of substituting high-dose IL-2 with low-dose subcutaneous IL-2 in the setting of TIL therapy for patients with previously treated metastatic melanoma. In this study, standard fludarabine and cyclophosphamide non-myeloablative lymphodepleting chemotherapy and autologous TIL were infused as per established protocols.48,49 Instead of using high-dose IL-2, low-dose subcutaneous IL-2 (2 million IU subcutaneously daily for 14 days) was administered, starting on the evening of TIL infusion. Six patients were treated with this regimen and partial responses were seen in two patients, two patients progressed, and two patients maintained stable disease. The median time to progression for the six patients was 8.2 months and overall survival was 12 months, with two having ongoing complete responses. The toxicity associated with this regimen was largely attributable to the lymphodepleting chemotherapy, including complications from pancytopenia. Low-dose IL-2 was well tolerated, and all planned injections were administered without any dose reductions required. Side effects associated with the injections included fever, chills, nausea, and fatigue, although none of these exceeded grade 2 toxicity. Although the response rates and duration of response were low compared with results seen in TIL trials using high-dose IL-2, it should be noted that the average amount of infused T-cells was only 26 billion per patient.53 It is established that higher absolute number of infused T-cells correlates with improved clinical outcomes, so it is feasible that the relatively poor outcomes in this study correlated more with T-cell expansion than with low-dose IL-2.49
Ongoing clinical trials
There are numerous ongoing clinical trials using IL-2 in varying doses in combination with additional agents, as described in Tables 1 and 2. While most trials are using aldesleukin in combination, there is an ongoing prospective registry study documenting the efficacy and toxicity of aldesleukin as a single agent for metastatic renal cell cancer and melanoma (NCT01415167). Additionally, the IL-2 SELECT trial is a prospective tissue and blood collection study for identification of biomarkers predictive of response or treatment failure (NCT01288963) which may help aldesleukin to remain as a viable option for treatment of melanoma patients, even with the recent developments in both targeted and immunotherapies.
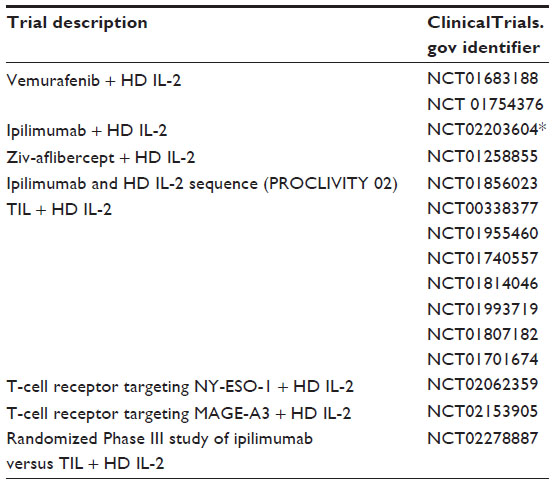 | Table 1 Ongoing clinical trials using high-dose interleukin-2 |
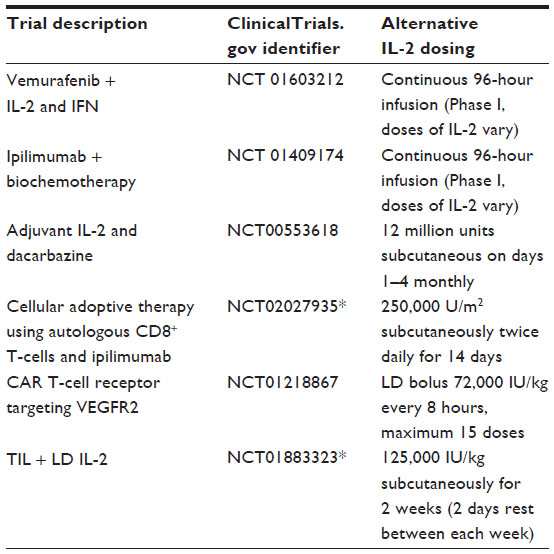 | Table 2 Ongoing or planned clinical trials using alternative interleukin-2 dosing strategies |
Patient considerations: quality of life, acceptability, and adherence
The toxicity of high-dose IL-2 is severe but predictable. Administration of this therapy should be restricted to treatment centers with high volume and clinical expertise. Patient eligibility for aldesleukin therapy should be restricted to those with excellent performance status (Eastern Cooperative Oncology Group 0–1) and excellent lung and cardiac function as determined by pretreatment pulmonary function tests and cardiac stress tests, respectively. Treated patients require cardiac monitoring and vigilant monitoring of clinical status in the inpatient setting. Vital signs, urine output, and mental status must be assessed prior to each dose. Adherence to the regimen is generally good, given that it usually requires week-long inpatient hospital stays. Quality of life is reasonable for patients, because the toxicities are experienced during the inpatient stay, during which maximal medical management is available. Most toxicities resolve within 72 hours of the last dose, and patients are usually able to be discharged from hospital within 72 hours of completing their last dose.
A significant benefit of this form of therapy is the short duration of the regimen and the potential for long-term disease control or complete responses in a small subset of patients. This is in contrast with targeted therapy, where response rates are high but durability of responses is quite limited. Ipilimumab, a checkpoint inhibitor, also entails a short course of therapy (four doses over 12 weeks), but side effects may be long-lasting in comparison with the toxicities observed with IL-2. Other checkpoint inhibitors currently approved for the treatment of melanoma, ie, pembrolizumab and nivolumab, must be given continuously every 3 weeks and 2 weeks, respectively.
Conclusion: place in therapy
Aldesleukin as a single agent remains a viable treatment approach for medically fit patients with metastatic melanoma. However, its use is now much more limited in light of the expertise required for its administration and the availability of other regimens for therapy. The 2013 Society for Immunotherapy of Cancer consensus statement on tumor immunotherapy for the treatment of cutaneous melanoma recommends aldesleukin as first-line therapy for patients with good performance status regardless of BRAF mutational status.54 However, recent progress in the development of targeted therapies and checkpoint blockade for metastatic melanoma makes it prudent to re-evaluate the role of this therapy within the changing landscape of treatment for melanoma. Checkpoint blockade can be administered in the outpatient setting with significantly less toxicity (especially in the case of PD-1-directed therapies), and preliminary evidence suggests that these agents can also produce durable responses. Additionally, there is no clear biomarker of response to aldesleukin, making it difficult to predict the group of patients who will respond favorably to this form of therapy. Identification of biomarkers is essential, and collection of critical baseline tumor and blood samples for the currently accruing IL-2 SELECT trial is taking place. Until there is a better understanding of which group of patients will derive the most benefit from aldesleukin, we believe that only young, medically fit patients with excellent performance status and limited disease burden (lymph node, soft tissue, limited lung metastases) should receive this therapy in the first-line setting, regardless of BRAF mutational status (Figure 3). Due to the safety and ease of administration, it can be argued that checkpoint blockade should be offered in the first-line setting for this group of patients as well, and we have noted that more providers at our institution favor first-line administration of checkpoint blockade over aldesleukin.
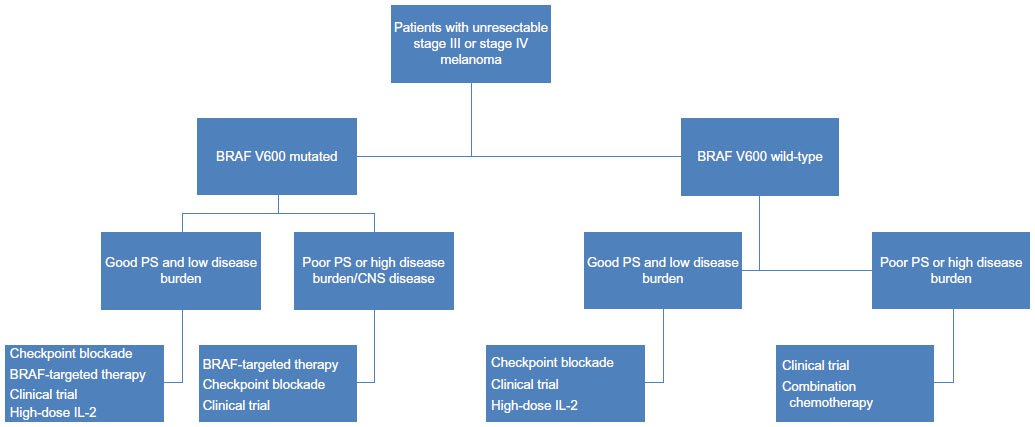 | Figure 3 First-line therapy options for patients with metastatic melanoma. The treatment algorithm is stratified by BRAF mutation analysis result, patient performance status, and disease burden. |
Exploration of alternative dosing strategies and aldesleukin-based combinations should be the priority in moving forward, as IL-2 remains an important agent to consider, particularly in combination with other strategies. Alternate dosing strategies are being utilized in ACT trials, and may provide a better understanding of the actual dose required for successful T-cell persistence. Additional research is clearly needed to identify those patients likely to respond to IL-2, and insights should be gained from ongoing clinical trials. However, added translational research will help to further define the role of IL-2, either as monotherapy in select patients or in combination with other strategies, and such studies are currently underway. Overall, this research will add to the body of knowledge to help improve treatment options for patients with melanoma.
Acknowledgments
JAW acknowledges grants from the National Institutes of Health (1K08CA160692-01A1, U54CA163125-01) and the generous philanthropic support of several families whose lives have been affected by melanoma.
Disclosure
JAW has received honoraria for participation on the speakers’ bureau of Dava Oncology and is an advisory board member for GlaxoSmithKline, Roche/Genentech, and Amgen. The other authors report no conflicts of interest in this work.
References
Carbone PP, Costello W. Eastern Cooperative Oncology Group studies with DTIC (NSC-45388). Cancer Treat Rep. 1976;60(2):193–198. | |
Patel PM, Suciu S, Mortier L, et al. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer. 2011;47(10):1476–1483. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | |
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | |
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | |
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010; 468(7326):968–972. | |
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | |
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | |
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–4861. | |
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–20416. | |
Whittaker SR, Theurillat JP, Van Allen E, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3(3):350–362. | |
Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. | |
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | |
Flaherty LE, Othus M, Atkins MB, et al. Southwest Oncology Group S0008: a phase III trial of high-dose interferon Alfa-2b versus cisplatin, vinblastine, and dacarbazine, plus interleukin-2 and interferon in patients with high-risk melanoma – an Intergroup Study of cancer and leukemia Group B, Children’s Oncology Group, Eastern Cooperative Oncology Group, and Southwest Oncology Group. J Clin Oncol. 2014; 32(33):3771–3778. | |
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | |
Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006;18(2):206–213. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363(8):711–723. | |
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011; 364(26):2517–2526. | |
Schadendorf D, Hodi FS, Robert C. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. Paper presented at the European Cancer Congress (ECCO-ESMO-ESTRO), September 27 to October 1, 2013, Amsterdam, The Netherlands. | |
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | |
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. | |
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. | |
Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. | |
Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol. 1993;11:245–268. | |
Gaffen SL. Signaling domains of the interleukin 2 receptor. Cytokine. 2001;14(2):63–77. | |
Sugamura K, Takeshita T, Asao H, et al. The IL-2/IL-2 receptor system: involvement of a novel receptor subunit, gamma chain, in growth signal transduction. Tohoku J Exp Med. 1992;168(2):231–237. | |
Lord JD, McIntosh BC, Greenberg PD, Nelson BH. The IL-2 receptor promotes proliferation, bcl-2 and bcl-x induction, but not cell viability through the adapter molecule Shc. J Immunol. 1998;161(9):4627–4633. | |
Friedmann MC, Migone TS, Russell SM, Leonard WJ. Different interleukin 2 receptor beta-chain tyrosines couple to at least two signaling pathways and synergistically mediate interleukin 2-induced proliferation. Proc Natl Acad Sci USA. 1996;93(5):2077–2082. | |
Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev. 2011;241(1):63–76. | |
Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30(2):204–217. | |
Martins GA, Cimmino L, Liao J, Magnusdottir E, Calame K. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J Exp Med. 2008;205(9):1959–1965. | |
Weiss GR, Grosh WW, Chianese-Bullock KA, et al. Molecular insights on the peripheral and intratumoral effects of systemic high-dose rIL-2 (aldesleukin) administration for the treatment of metastatic melanoma. Clin Cancer Res. 2011;17(23):7440–7450. | |
Janas ML, Groves P, Kienzle N, Kelso A. IL-2 regulates perforin and granzyme gene expression in CD8+ T cells independently of its effects on survival and proliferation. J Immunol. 2005;175(12):8003–8010. | |
Tsung K, Meko JB, Peplinski GR, Tsung YL, Norton JA. IL-12 induces T helper 1-directed antitumor response. J Immunol. 1997;158(7):3359–3365. | |
Panelli MC, Wang E, Phan G, et al. Gene-expression profiling of the response of peripheral blood mononuclear cells and melanoma metastases to systemic IL-2 administration. Genome Biol. 2002;3(7):RESEARCH0035. | |
de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34(9):2480–2488. | |
Sadlack B, Lohler J, Schorle H, et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995; 25(11):3053–3059. | |
Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. | |
Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8(5):615–623. | |
Konrad MW, Hemstreet G, Hersh EM, et al. Pharmacokinetics of recombinant interleukin 2 in humans. Cancer Res. 1990;50(7):2009–2017. | |
Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–2116. | |
Schwartzentruber DJ. Guidelines for the safe administration of high-dose interleukin-2. J Immunother. 2001;24(4):287–293. | |
Sasse AD, Sasse EC, Clark LG, Ulloa L, Clark OA. Chemoimmunotherapy versus chemotherapy for metastatic malignant melanoma. Cochrane Database Syst Rev. 2007;1:CD005413. | |
Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2,621 patients. J Clin Oncol. 2007;25(34):5426–5434. | |
Bedikian AY, Johnson MM, Warneke CL, et al. Systemic therapy for unresectable metastatic melanoma: impact of biochemotherapy on long-term survival. J Immunotoxicol. 2008;5(2):201–207. | |
Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. | |
Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. | |
Radvanyi LG, Bernatchez C, Zhang M, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18(24):6758–6770. | |
Besser MJ, Shapira-Frommer R, Itzhaki O, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. 2013;19(17):4792–4800. | |
Dudley ME, Wunderlich JR, Yang JC, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25(3):243–251. | |
Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99(25):16168–16173. | |
Ellebaek E, Iversen TZ, Junker N, et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose interleukin-2 in metastatic melanoma patients. J Transl Med. 2012;10:169. | |
Kaufman HL, Kirkwood JM, Hodi FS, et al. The society for immunotherapy of cancer consensus statement on tumor immunotherapy for the treatment of cutaneous melanoma. Nat Rev Clin Oncol. 2013;10(10):588–598. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.