")
Back to Journals » OncoTargets and Therapy » Volume 13
Up-Regulated CCDC34 Contributes to the Proliferation and Metastasis of Hepatocellular Carcinoma
Authors Lin Z , Qu S, Peng W, Yang P , Zhang R, Zhang P, Guo D , Du J , Wu W , Tao K, Wang J
Received 5 November 2019
Accepted for publication 18 December 2019
Published 7 January 2020 Volume 2020:13 Pages 51—60
DOI https://doi.org/10.2147/OTT.S237399
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Zhibin Lin,* Shibin Qu,* Wei Peng,* Peijun Yang, Ruohan Zhang, Pengcheng Zhang, Dongnan Guo, Jianbing Du, Wenlong Wu, Kaishan Tao, Jianlin Wang
Department of Hepatobiliary Surgery, Xijing Hospital, The Fourth Military Medical University, Xi’an, 710032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianlin Wang; Kaishan Tao
Department of Hepatobiliary Surgery, Xijing Hospital, The Fourth Military Medical University, 127 West Changle Street, Xi’an, Shaanxi 710032, People’s Republic of China
Tel +86 29 8477 5259
Fax +86 29 8477 1595
Email [email protected]; [email protected]
Background: Coiled-coil domain-containing protein 34 (CCDC34), which belongs to the CCDCs family, has been recently reported to be up-regulated in various kinds of tumors. However, its role in the development of hepatocellular carcinoma (HCC) still remains unclear.
Materials and methods: In this study, real-time polymerase chain reaction (RT-PCR) and Western blot analysis were performed to measure the mRNA and protein levels of CCDC34 in clinical samples. Kaplan-Meier method was used to analyze the relationship between CCDC34 and the prognosis in HCC patients. CCK-8 and colony formation assays were conducted to investigate CCDC34’s effect on the cell proliferation, and Transwell assays were used to detect CCDC34’s effect on the cell metastasis. Moreover, subcutaneous xenograft tumor model and lung metastasis model were applied to confirm the impact of CCDC34 on the HCC development. Lastly, RNA sequencing and Western blot analysis were performed to probe the underlying mechanism of CCDC34’s effect on HCC.
Results: CCDC34 was significantly induced in HCC tissues, and the overexpression of CCDC34 predicted the poor outcomes among HCC patients. It was verified by the in vitro and in vivo experiments that CCDC34-knockdown potently inhibited the proliferation and metastasis of HCC cells. Subsequent results indicated that CCDC34 inhibition can affect the activation of protein kinase B (PKB or AKT) as well as epithelial-mesenchymal transition (EMT) process.
Conclusion: CCDC34 is significantly associated with HCC. It will become a promising prognostic biomarker and therapeutic target against HCC.
Keywords: CCDC34, HCC, proliferation, EMT, PI3K/AKT, CCND1
Introduction
Hepatocellular carcinoma (HCC) is one of the most common tumors worldwide, and its mortality has surpassed that of lung cancer and gastric cancer, ranking the third among all tumors.1 The difficulty in the early diagnose and its rapid progress contribute to the poor overall prognosis of HCC patients. Though surgical resection, liver transplantation and radiofrequency ablation can improve the survival rate of patients, the 5-year recurrence rate is still as high as 80% to 90%.2,3 The occurrence and development of HCC are complex, multi-factor and multi-step processes, and the specific mechanism is unrevealed. Therefore, it is of great challenge to prevent and cure this disease. Moreover, it is clinically significant to probe the molecular mechanism of HCC and to find out some new potential targets for the diagnosis and treatment of HCC.
The coiled-coil domain (CCD), which consists of two to five α-helices twisting around one another, is widely expressed in various proteins. The spatial structure of CCD is highly flexible, allowing it to carry out a series of biological functions, such as regulating the cell movement, participating in the intercellular recognition system and being involved in the cellular signal transduction.4 Recently, abnormal activation of CCD-containing proteins (CCDC) has been observed in many tumors. For example, CCDC178, CCDC88A and CCDC8 are overexpressed in liver cancer, pancreatic cancer and lung cancer, respectively.5–7 CCDC34, also known as renal carcinoma antigen NY-REN-41, contains 373 amino acids, and is located on chromosomes 11p14.1.8 Previous studies have revealed the overexpression of CCDC34 in bladder, pancreatic, colon and esophageal cancers,8–11 but whether CCDC34 is involved in the occurrence and the development of HCC needs to be further explored.
In this study, the expression of CCDC34 was measured in the HCC tissues and the pare-cancer tissues. And the impact of CCDC34 on HCC cells was observed in both in vitro and in vivo experiments. Furthermore, bioinformatics and Western blot analysis were conducted to probe the underlying mechanism of CCDC34’s effect on HCC. In summary, this paper is the first one to demonstrate the role of CCDC34 in HCC, implicating that the regulation of CCDC34 can be chosen as a promising therapy against HCC.
Materials and Methods
Cell Lines and Tissue Samples
The HCC cell lines, MHCC97-H and SMMC-7721, were kindly provided by Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). All the cell lines were cultured in the dulbecco’s modified eagle medium (DMEM, Hyclone) supplemented with 10% fetal bovine serum (FBS), and then incubated in the humidified atmosphere containing 5% CO2 at 37 °C. 21 HCC samples and matched para-cancer tissues, since December 2017 to November 2018, were obtained from Xijing Hospital (Xi’an, China). The study was approved by the Ethics Committee of Xijing Hospital, and all patients were provided a signed written informed consent for the use of clinical specimens for the medical research.
Lenti-Virus Transfection and Stable Cell Clone Establishment
The negative control lenti-virus, lenti-virus loading shRNA targeting genomic CCDC34 sequences (shCCDC34) and lenti-virus loading a plasmid carrying the CCDC34 gene (CCDC34) were purchased from GENECHEM (Shanghai, China). The sequence of shRNA is shown as TGAAGATGCCCATGATTCA. Cells were planted in 6-well plates and cultured overnight. The lenti-virus was infected into the HCC cell lines at 20 multiplicity of infection (MOI) with the transduction-enhancing solution. After 12 hrs, the medium was replaced with the complete medium.
RNA Isolation and Quantitative Polymerase Chain Reaction (q-PCR)
Total RNA was isolated from the HCC cell lines or frozen tissue samples using Trizol (Invitrogen) reagent. The total RNA was reverse-transcribed with PrimeScript™ Master Mix (Takara Biotechnology) at 37 °C for 15 min and 85 °C for 5 s, respectively. The mRNA levels were determined using SYBR Green PCR master mix (Takara Biotechnology) on a Bio-Rad IQ™5 detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) in the two-step reaction. β-actin was used as the quantitative control to normalize the mRNA expression levels of the target genes. Data were then collected and calculated using the 2−(ΔΔCt) method. The target specific primer sequences for the RT-PCR are shown below: CCDC34 (forward primer, ACAGAAACAGGTGCGCTTACC and reverse primer, CAGCCGGTCACG TTCTTCTTT); β-actin (forward primer, CATGTACGTTGCTATCCAGGC and reverse primer, CTCCTTAATGTCACGCACGAT).
Western Blot Analysis
Protein lysates were prepared with fresh lysis buffer (50 mM of Tris pH 7.4, 150 mM of NaCl, 1% TritonX-100, 1% sodium deoxycholate, 0.1% SDS, 2 mM of sodium pyrophosphate, and 1 mM EDTA; Beyotime, China), containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Total proteins were extracted and then separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and then transferred onto the PVDF membranes (Millipore, Billerica, MA, USA). After blocked for 60 mins at room temperature, those membranes were incubated with specific primary antibodies at 4 °C overnight. The antibodies are presented as following: CCDC34 (1:250; cat. no. ab122396; Abcam), AKT (1:1000; cat. no. 4060S; Cell Signaling Technology, Inc.), p-AKT (Ser473) (1:1000; cat. no. 9916T; Cell Signaling Technology, Inc.), β-actin (1:1000; cat. no. 3700; Cell Signaling Technology, Inc.), GAPDH (1:4000; cat. no. 5174; Cell Signaling Technology, Inc.), E-cadherin (1:1000; cat. no. 3195; Cell Signaling Technology, Inc.), N-cadherin (1:1000; cat. no. 13116; Cell Signaling Technology, Inc.), CCND1 (1:2000; cat. no. 60186-1-lg; Proteintech, Inc.), and P21 (1:500; cat. no. 10355-1-AP; Proteintech, Inc.).
CCK-8 and Colony Formation Assays
The transfected MHCC97-H and SMMC-7721 cells were seeded in 96-well plates with a density of 2000 cells/well, and cell proliferation was measured at the 1st, 2nd, 3rd, 4th and 5th day after seeding by using Cell Counting Kit-8 (CCK‑8; Beyotime, China) according to the manufacturer’s instructions. The experiments were performed with five replicates. Colony formation assays were carried out in 6-well culture plates with (1–2) × 103 cells/well. The cells were incubated for one to two weeks in the DMEM supplemented with 10% FBS. The colonies were then washed twice with phosphate‑buffered saline (PBS), fixed in 4% paraformaldehyde and stained with 1% crystal violet. The number of colonies was counted to evaluate the cell proliferation.
Migration and Invasion Assays
The HCC cell invasion was analyzed in a Matrigel-coated transwell cell culture chamber (EMD Millipore, Billerica, MA, USA). The MHCC97-H and SMMC-7721 cells (3–5 × 104 cells/well) were plated in the top chamber, and allowed to invade toward the lower chamber containing the complete medium. After incubated for 12 to 24 hrs, the cells were fixed with 4% paraformaldehyde and then stained with 1% crystal violet. The invaded cells were counted from five different fields at 200× magnification. The transwell cell culture chambers without Matrigel were used for the migration assays, and the process was conducted the same as the invasion assays.
Tumor Growth in Nude Mice
The MHCC97-H-shNC and MHCC97-H-shCCDC34 cells (2 × 106 cells per flank) were subcutaneously injected into both flanks of four-week-old male BALB/c nude mice, obtained from SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The mice were raised in a specific pathogen free (SPF) facility, and were given free access to water and rodent diet with a 12 hr light-dark cycle. After three weeks, the mice were sacrificed via CO2 inhalation according to the animal care guidelines. The tumors were harvested and weighed, and their volumes were calculated using the following formula: (length×width2)/2. To establish a lung metastasis model in the nude mice, the MHCC97-H-shNC and MHCC97-H-shCCDC34 cells (2 × 106 cells per mouse), suspended in PBS solution, were injected through the tail vein of the nude mice. After four weeks, the above process was repeated to harvest lung tissues and count the number of metastatic tumors. Thereafter, the obtained lung tissues were fixed with 4% paraformaldehyde for the hematoxylin-eosin staining. All experimental procedures on animals were conducted in accordance with the National Institutes of Health guidelines for the care and the use of laboratory animals, and approved by the Institutional Animal Care and Use Committee of The Fourth Military Medical University (Xi’an, Shaanxi, China).
RNA Sequencing
The total RNA was isolated from BEL-7404-CCDC34-OE and BEL-7404-CCDC34-NC cells, and was analyzed by the RNA sequencing (BGI, Shenzhen, China) through the BGISEQ-500 platform. Genes that were significantly and differentially expressed were selected based on a fold change of >2.0 and a P-value of <0.05, and subsequently analyzed by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis and protein-protein interaction (PPI) network analysis.
Statistical Analysis
Each experiment was repeated at least three times. All data are presented as the mean ± SD. Student’s t-test was used to analyze the differences between groups with homogenous variance. Survival curves were calculated using the Kaplan-Meier method, and then compared by the Logrank test. SPSS 23.0 for Windows (IBM Corp., Armonk, NY, USA) was used to conduct the statistical analyses. P<0.05 indicated the statistical significance.
Results
Association Between CCDC34 and the Prognosis in HCC Patients
To investigate the underlying association between CCDC34 and HCC, the expression of CCDC34 was determined through consulting the Cancer Genome Atlas (TCGA) database. The results showed that CCDC34 was obviously overexpressed in the HCC tissues (Figure 1A). Furthermore, the Kaplan–Meier survival analysis demonstrated that the high CCDC34 expression was correlated with poor overall survival of HCC patients in TCGA database (Figure 1B). To confirm the results above, the q-PCR and Western blot assays were used to detect the CCDC34 expression in 21 pairs of HCC tissues and para-cancer tissues. The data consistently revealed that CCDC34 has significantly high level in HCC tissues (Figure 1C and D). Taken together, the above results indicated that CCDC34 is distinctly evoked in HCC tissues and predicts a poor prognosis in HCC patients.
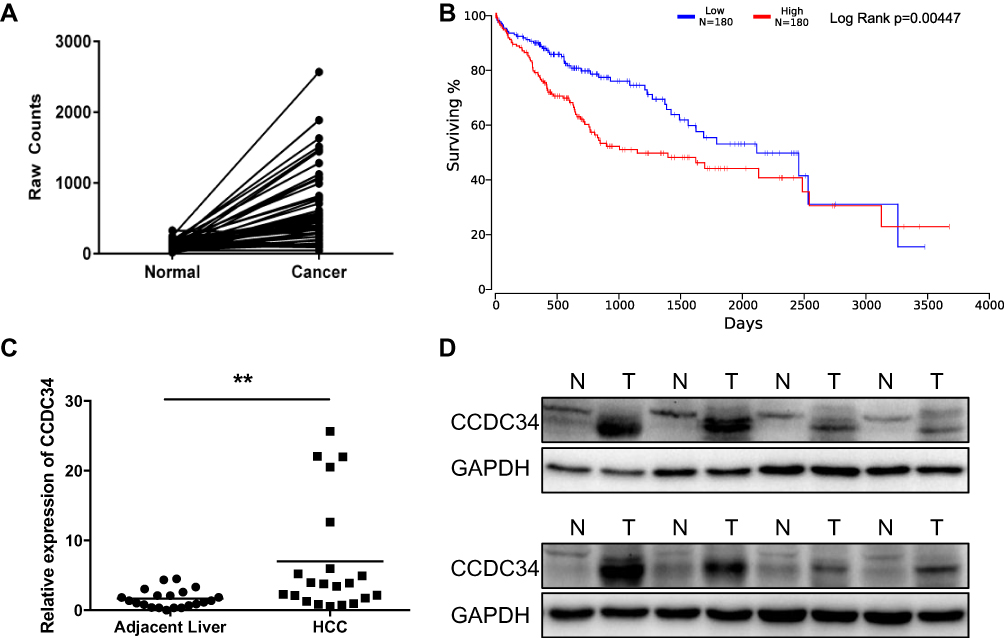 |
Figure 1 The overexpression of CCDC34 in HCC tissues. Notes: (A) The expression of CCDC34 in the HCC tissues and adjacent normal tissues from TCGA database (n=50). (B) The overall survival rate of patients with high or low CCDC34 expression in TCGA database (P=0.004). (C) The qRT-PCR assays for the CCDC34’s mRNA expression in the HCC tissues and adjacent normal tissues (n=21). **P<0.01. (D) Representative Western blot assays for the CCDC34 protein expression in the HCC tissues (T) and adjacent normal tissues (N).Abbreviations: HCC, hepatocellular carcinoma; TCGA, the Cancer Genome Atlas; qRT-PCR, quantitative real-time polymerase chain reaction. |
HCC Cell Proliferation and Clonogenicity in vitro Facilitated by CCDC34
Given that the expression of CCDC34 was highly induced in HCC tissues, the role of CCDC34 was investigated in the growth of HCC cells. The MHCC97-H and SMMC-7721 cells were both transfected with CCDC34-shRNA lentivirus (shCCDC34) and negative control lentivirus (NC), respectively. The knockdown (KD) efficiency was tested by q-PCR and Western blot analysis. The results showed that CCDC34 was markedly down-regulated in the cells transfected by the shCCDC34 (Figure 2A). Subsequently, the CCK-8 assays proved that the proliferation of both MHCC97-H and SMMC-7721 cells was inhibited after the CCDC34-KD (Figure 2B). Meanwhile, the colony formation assays illustrated that the CCDC34-KD significantly hampered the long-term proliferation of the two HCC cell lines (Figure 2C). Thereafter, CCDC34 was overexpressed in BEL-7404 and SMMC-7721 cell lines, and the overexpression efficiency was verified by q-PCR and Western blot methods (Figure 2D). Accordantly, the overexpressed CCDC34 facilitated the proliferation of HCC cells (Figure 2E and F).
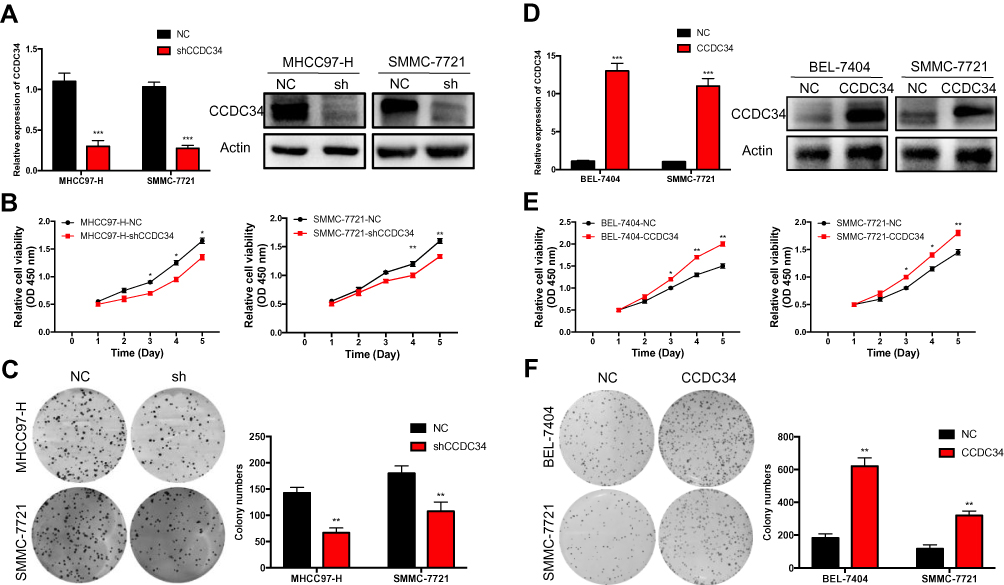 |
Figure 2 HCC cell proliferation and clonogenicity in vitro facilitated by CCDC34. Notes: (A) The knockdown efficiency of shCCDC34 in the MHCC97-H and SMMC-7721 cell lines measured by the qRT-PCR and Western blot. (B and C) The proliferation ability of the MHCC97-H and SMMC-7721 cells transfected with shCCDC34 or NC lentivirus were detected by the CCK-8 assays (B) and the colony formation assays (C). The “sh” represents the shCCDC34 group and NC stands for the negative control group. (D) The overexpression efficiency of CCDC34 in the BEL-7404 and SMMC-7721 cell lines were measured by the qRT-PCR and Western blot (E and F). The proliferation ability of the BEL-7404 and SMMC-7721 cells transfected with the CCDC34-OE or NC lentivirus were detected by the CCK-8 assays (E) and the colony formation assays (F). The CCDC34 represents the CCDC34-OE group and NC stands for the negative control group. *P<0.05; **P<0.01; ***P<0.001.Abbreviations: HCC, hepatocellular carcinoma; qRT-PCR, quantitative real-time polymerase chain reaction; NC, negative control; CCK-8, Cell Counting Kit-8; OE, overexpression. |
HCC Cell Migration and Invasion in vitro Regulated by CCDC34
The transwell assays were conducted to explore whether CCDC34 could promote the migration and invasion of HCC cells. The results showed that both the migration and invasion of MHCC97-H and SMMC-7721 cells were significantly inhibited after the CCDC34-KD (Figure 3A and B), while both were enhanced by the overexpression of CCDC34 (Figure 3C and D). It suggested that CCDC34 exerts an important role in the metastasis of HCC cells.
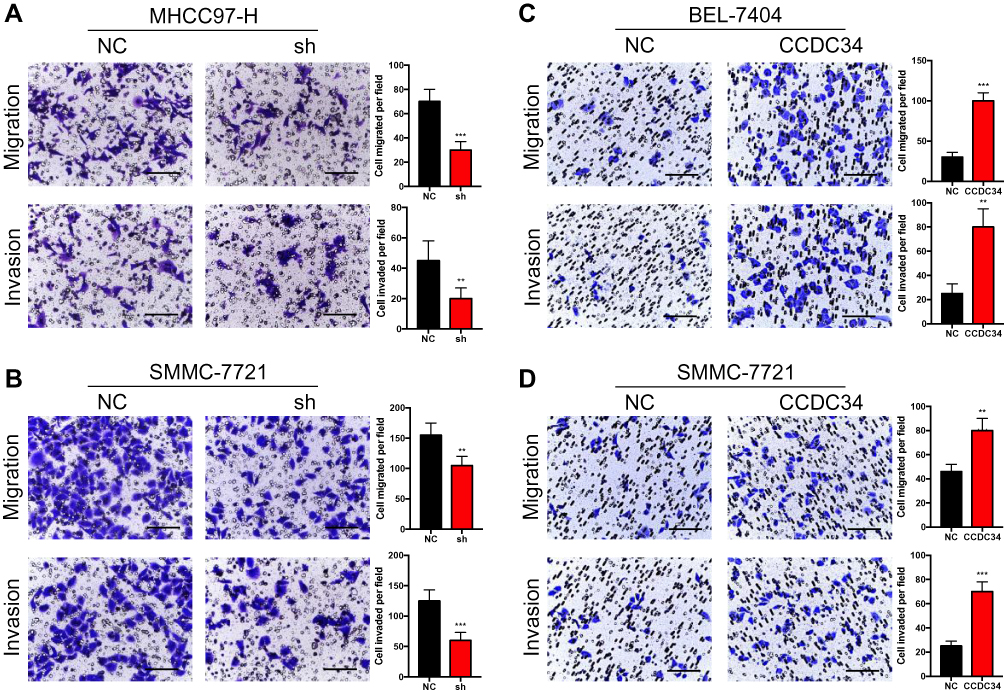 |
Figure 3 HCC cell migration and invasion in vitro regulated by CCDC34. Notes: (A and B) The migration and invasion ability of the MHCC97-H (A) and SMMC-7721 (B) cells transfected with the shCCDC34 or NC lentivirus were measured by the transwell assays. (C and D). The migration and invasion ability of the BEL-7404 (C) and SMMC-7721 (D) cells transfected with the CCDC34-OE or NC lentivirus were measured by the transwell assays. Scale bar = 100 μm. **P<0.01; ***P<0.001.Abbreviations: HCC, hepatocellular carcinoma; NC, negative control; OE, overexpression. |
HCC Cell Growth and Metastasis in vivo Suppressed by CCDC34-KD
Tumorigenesis assays in nude mice were used to confirm the effect of CCDC34-KD on the proliferation of HCC cells. MHCC97-H cells transfected with either NC or shCCDC34 virus were subcutaneously planted into the nude mice to establish a xenograft tumor model. Mice were sacrificed on the 24th day after injection, and their tumors were dissected and weighed. Compared with the control group, the MHCC97-H cells treated with shCCDC34 exhibited weakened proliferation ability in vivo, which manifested as the decreased tumor weight and volume (Figure 4A and B). To simulate the process of the HCC metastasis in vivo, the MHCC97-H-NC or MHCC97-H-shCCDC34 cells were injected into the nude mice via their tail veins. After 28 days, the mice were sacrificed, and the H&E staining on the lung tissues was performed to indicate the lung metastasis nodules. The data showed that metastasis nodules in the shCCDC34 group were greatly reduced in both quantity and volume compared to those in the NC group (Figure 4C and D). Taken together, the results revealed that the MHCC97-H cells transfected with shCCDC34 possessed lower tumorigenic and metastatic properties in vivo than the control cells.
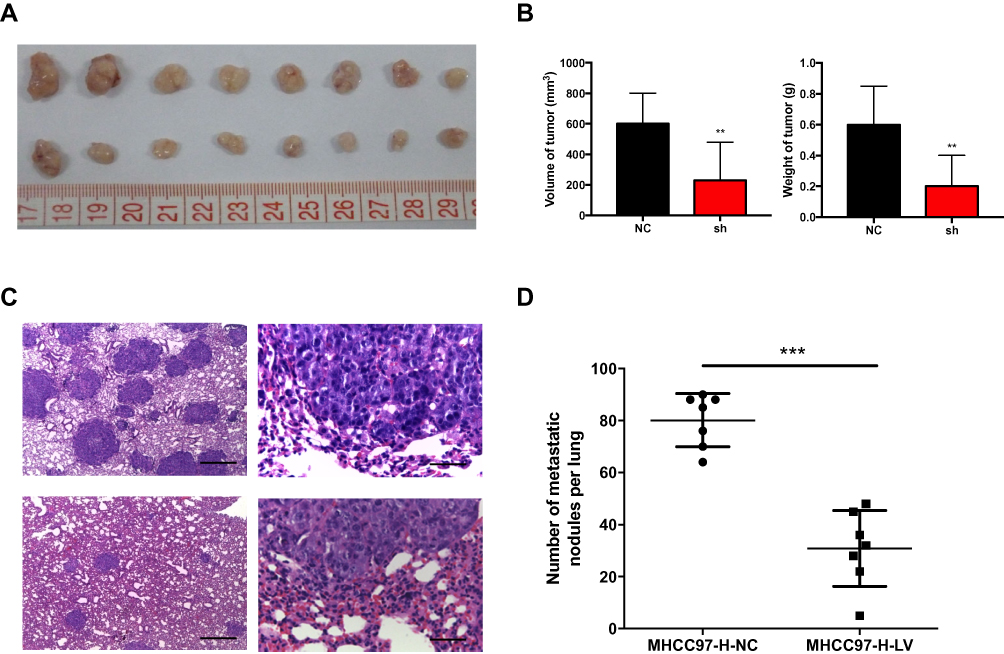 |
Figure 4 HCC cell growth and metastasis in vivo suppressed by CCDC34-KD.Notes: (A) The MHCC97-H cells transfected with shCCDC34 or NC lentivirus were subcutaneously injected into the nude mice (n=8). The top tumors were produced by MHCC97-H-NC cells, while the bottom by MHCC97-H-shCCDC34 cells. (B) The volume and weight of tumors harvested from the nude mice were measured. (C) Representative H&E staining in lung sections obtained from the nude mice injected with the MHCC97-H-shCCDC34 or NC cells through the tail veins (n=7). Scale bar = 500 μm (left) and 50 μm (right). The top tumors were produced by MHCC97-H-NC cells, while the bottom by MHCC97-H-shCCDC34 cells. (D) The number of metastatic tumors per lung was counted. **P<0.01; ***P<0.001.Abbreviations: KD, knockdown; HCC, hepatocellular carcinoma; NC, negative control. |
The Activation of AKT Pathways and EMT Process Hampered by Silencing of CCDC34
To identify genes modified by the overexpression of CCDC34 in BEL-7404 cell line, RNA sequencing was conducted, showing that when compared to the NC group, 148 genes were up-regulated and 101 genes were down-regulated in the BEL-7404-CCDC34 group (Figure 5A). The KEGG pathway enrichment analysis of differentially expressed genes (DEGs) revealed that the PI3K/AKT signaling pathway, cell cycle, and another HCC-associated signaling pathways possessed the majority of the DEGs (Figure 5B). To find the candidate downstream substrates of CCDC34, the DEGs were extracted from the five HCC-related modules (red) of Figure 5B, and presented in a heatmap (Figure 5C). Subsequently, protein-protein interaction (PPI) network analysis was used to display the interaction relationship between the DGEs mentioned above (Figure 5D). The results revealed that CCND1 with the largest number of neighboring nodes, was the hub node of this network. Meanwhile, other important factors in the cell cycle, CCNB1 and CCND3, also possessed several neighboring nodes (Figure 5D). Then, the Western blot assays were performed in order to verify the result of RNA sequencing. The results indicated that the expression of phosphorylation-AKT, together with its downstream effectors CCND1 and P21,12 changed dramatically after the CCDC34-KD in both MHCC97-H and SMMC-7721 cell lines (Figure 5E). Subsequently, the Western blot was used to determine whether the CCDC34’s effect on HCC metastasis is associated with the EMT process, which is characterized by the inactivation of E-cadherin and ZO-1 or the activation of N-cadherin and Vimentin.13 As expected, the knockdown of CCDC34 significantly reduced the expression of N-cadherin and enhanced the expression of E-cadherin in the two types of HCC cell lines (Figure 5E).
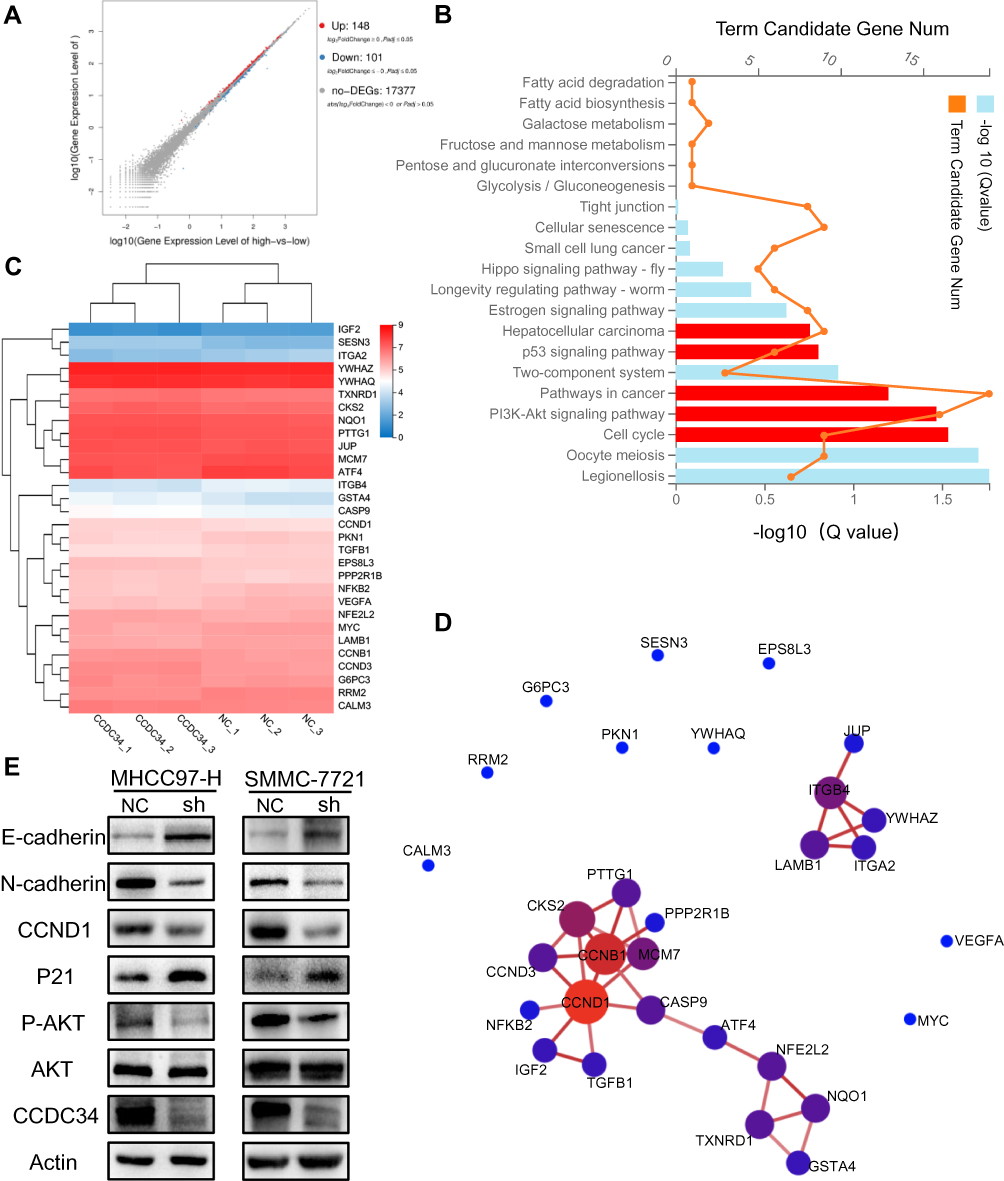 |
Figure 5 The activation of AKT pathways and EMT process hampered by silencing of CCDC34. Notes: (A) A scatter plot shows the number of the DEGs analyzed by the RNA sequencing in the BEL-7404-CCDC34-OE cells (fold change >2.0 and P-value <0.05). (B) The KEGG pathway enrichment analysis of all the DEGs. (C) A heatmap consisting of the DEGs extracted from the five HCC-related modules (red) of Figure 5B. (D) Protein-protein interaction (PPI) network analysis for the DEGs extracted from the five HCC-related modules (red) of Figure 5B. The size of the nodes indicates the number of neighboring nodes directly connected to the node. (E) Representative Western blot assays for the protein level of CCDC34, AKT, p-AKT, P21, CCND1, N-cadherin and E-cadherin.Abbreviations: EMT, epithelial-mesenchymal transition; DEGs, differentially expressed genes; OE, overexpression; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction. |
Discussion
HCC, the fourth cause of most common cancer-related death worldwide, has a low patient survival rate.2 Despite some potent treatments for HCC at early stage, the effective therapies for treating advanced liver cancer is rather limited, which results from the lack of efficient therapeutic targets for HCC. Therefore, researchers all over the world have devoted to investigating the molecular mechanisms underlying the development of HCC. Up to now, several signaling pathways involved in the development of HCC have been identified.14–16 However, a thorough understanding of HCC regulation network is badly desired.
Current studies have shown that CCDC34 is up-regulated in various kinds of tumors and predicts poor survivals in cancer patients,8–11 indicating that CCDC34, a novel oncogene, may be an effective therapeutic target against HCC. Similarly, the experiments conducted in this paper verified that the level of CCDC34 was highly induced in HCC tissues, and the higher expression of CCDC34 was associated with poorer survival among HCC patients. Moreover, the results, which revealed that both the knockdown and overexpression of CCDC34 have distinct impacts on the proliferation and migration of HCC, is consistent with that of other studies. The bioinformatics and Western blot analysis for the underlying mechanism suggested that CCDC34’s impact on HCC may be related to PI3K/AKT signaling pathway and EMT process.
The activation of the PI3K/AKT pathway has been reported to be essential for the malignant transformation of hepatocytes, and is closely related to the proliferation and migration of HCC cells.17–19 Meanwhile, it has been reported that CCDC34 silencing can suppress the cell proliferation as well as the activation of AKT in bladder cancer.8 In the present research, the results of the RNA sequencing showed that the overexpression of CCDC34 led to an extensive change in DEGs related to PI3K/AKT pathway. And the Western blot analysis presented that silencing of CCDC34 hampered the phosphorylation of AKT. Therefore, it can be deduced that CCDC34 has a tight relationship with the PI3K/AKT signaling pathway. Moreover, the PPI network analysis showed that one of the downstream substrates of the PI3K/AKT pathway, CCND1,20,21 was located in the central position of the network. At present, CCND1 has been identified as an oncogene.22,23 The overexpression of CCND1 can result in a faster cell cycle progression and a quicker exit, which leads to a chromosome missegregation, causing the chromosomal instability and eventually inducing the hepatocarcinogenesis.24 The CCND1-CDK4 complex can drive the cell cycle progression from G1 into S phase by sequestering the P21.25 And the Western blot results revealed that the CCDC34-KD led to a lower level of CCND1 and a higher level of P21. Hence, it is likely that CCDC34 accelerates the progress of the cell cycle by up-regulating the expression of CCND1 and eventually results in the occurrence of HCC. Taken together, there seems to be a CCDC34/AKT/CCND1 axis in the development of HCC. However, this regulatory axis needs to be strictly validated in further investigation.
The EMT refers to a process, in which epithelial cells lose their cell to cell adhesion and become more liable to proliferate and migrate. During the EMT, cells acquire characteristics of mesenchymal stem cells, such as transcriptional inactivation of E-cadherin and ZO-1 or the activation of N-cadherin and Vimentin.26 Since the present results of both in vitro and in vivo assays revealed that the CCDC34-KD can affect the tumor metastasis. And the Western blot analysis revealed that the expression of N-cadherin and E-cadherin changed accordingly after CCDC34-KD. Therefore, it is possible that CCDC34 prompts HCC metastasis through regulating the EMT process. However more studies should be conducted to thoroughly investigate the underlying mechanism. Besides, it has been reported that PI3K/AKT axis exerts a positive effect on EMT.27,28 Therefore, it can be supposed that the CCDC34 promotes the EMT process through activating PI3K/AKT axis, which still needs to be strictly verified.
Conclusion
This study highlighted that CCDC34 was up-regulated in HCC tissues, and the CCDC34 overexpression predicted poor prognosis in HCC patients. Furthermore, CCDC34 played a crucial role in the proliferation and metastasis of HCC both in vitro and in vivo. And the underlying mechanism should be associated with activated PI3K/AKT signal, disturbed cell cycle and EMT process. In one word, CCDC34 can serve as a prognostic biomarker and an effective therapeutic target for HCC treatment.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 81670593), the National High-Tech Research and Development Program of China (grant no. 2016YFC0905902) and the Basic Research Plan of Natural Science of Shannxi Province (grant no. S2016YFJM0747).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. doi:10.1038/s41575-019-0186-y
3. Xia F, Wu LL, Lau WY, et al. Adjuvant sorafenib after heptectomy for barcelona clinic liver cancer-stage C hepatocellular carcinoma patients. World J Gastroenterol. 2016;22(23):5384–5392. doi:10.3748/wjg.v22.i23.5384
4. Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11(2):82–88. doi:10.1016/S0962-8924(00)01898-5
5. Hu X, Zhao Y, Wei L, et al. CCDC178 promotes hepatocellular carcinoma metastasis through modulation of anoikis. Oncogene. 2017;36(28):4047–4059. doi:10.1038/onc.2017.10
6. Tanouchi A, Taniuchi K, Furihata M, et al. CCDC88A, a prognostic factor for human pancreatic cancers, promotes the motility and invasiveness of pancreatic cancer cells. J Exp Clin Cancer Res. 2016;35(1):190. doi:10.1186/s13046-016-0466-0
7. Jiang GY, Zhang XP, Zhang Y, et al. Coiled-coil domain-containing protein 8 inhibits the invasiveness and migration of non-small cell lung cancer cells. Hum Pathol. 2016;56:64–73. doi:10.1016/j.humpath.2016.06.001
8. Gong Y, Qiu W, Ning X, et al. CCDC34 is up-regulated in bladder cancer and regulates bladder cancer cell proliferation, apoptosis and migration. Oncotarget. 2015;6(28):25856–25867. doi:10.18632/oncotarget.v6i28
9. Qi W, Shao F. Q H. expression of coiled-coil domain containing 34 (CCDC34) and its prognostic significance in pancreatic adenocarcinoma. Med Sci Monit. 2017;23:6012–6018. doi:10.12659/MSM.907951
10. Geng W, Liang W, Fan Y, Ye Z, Zhang L. Overexpression of CCDC34 in colorectal cancer and its involvement in tumor growth, apoptosis and invasion. Mol Med Rep. 2018;17(1):465–473. doi:10.3892/mmr.2017.7860
11. Hu DD, Li PC, He YF, Jia W, Hu B. Overexpression of coiled-coil domain-containing protein 34 (CCDC34) and its correlation with angiogenesis in esophageal squamous cell carcinoma. Med Sci Monit. 2018;24:698–705. doi:10.12659/MSM.908335
12. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi:10.1016/j.cell.2017.04.001
13. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166(1):21–45. doi:10.1016/j.cell.2016.06.028
14. Xu Z, Xu M, Liu P, et al. The mTORC2-Akt1 cascade is crucial for c-Myc to promote hepatocarcinogenesis in mice and humans. Hepatology. 2019;70:1600–1613. doi:10.1002/hep.v70.5
15. Liu JJ, Li Y, Chen WS, et al. Shp2 deletion in hepatocytes suppresses hepatocarcinogenesis driven by oncogenic beta-catenin, PIK3CA and MET. J Hepatol. 2018;69(1):79–88. doi:10.1016/j.jhep.2018.02.014
16. Bouattour M, Raymond E, Qin S, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology. 2018;67(3):1132–1149. doi:10.1002/hep.v67.3
17. Yang YF, Zhang MF, Tian QH, et al. SPAG5 interacts with CEP55 and exerts oncogenic activities via PI3K/AKT pathway in hepatocellular carcinoma. Mol Cancer. 2018;17(1):117. doi:10.1186/s12943-018-0872-3
18. Klingenberg M, Gross M, Goyal A, et al. The long noncoding RNA cancer susceptibility 9 and RNA binding protein heterogeneous nuclear ribonucleoprotein L form a complex and coregulate genes linked to AKT signaling. Hepatology. 2018;68(5):1817–1832. doi:10.1002/hep.v68.5
19. Chen Z, Gao W, Pu L, et al. PRDM8 exhibits antitumor activities toward hepatocellular carcinoma by targeting NAP1L1. Hepatology. 2018;68:994–1009. doi:10.1002/hep.29890
20. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203. doi:10.1038/nrm3290
21. Pauta M, Rotllan N, Fernandez-Hernando A, et al. Akt-mediated foxo1 inhibition is required for liver regeneration. Hepatology. 2016;63(5):1660–1674. doi:10.1002/hep.v63.5
22. Wu SY, Lan SH, Liu HS. Degradative autophagy selectively regulates CCND1 (cyclin D1) and MIR224, two oncogenic factors involved in hepatocellular carcinoma tumorigenesis. Autophagy. 2019;15(4):729–730. doi:10.1080/15548627.2019.1569918
23. Chen RW, Bemis LT, Amato CM, et al. Truncation in CCND1 mRNA alters miR-16-1 regulation in mantle cell lymphoma. Blood. 2008;112(3):822–829. doi:10.1182/blood-2008-03-142182
24. Chan TH, Chen L, Liu M, et al. Translationally controlled tumor protein induces mitotic defects and chromosome missegregation in hepatocellular carcinoma development. Hepatology. 2012;55(2):491–505. doi:10.1002/hep.24709
25. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93–115. doi:10.1038/nrc.2016.138
26. Pradella D, Naro C, Sette C, Ghigna C. EMT and stemness: flexible processes tuned by alternative splicing in development and cancer progression. Mol Cancer. 2017;16(1):8. doi:10.1186/s12943-016-0579-2
27. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3ʹ kinase/AKT pathways. Oncogene. 2005;24(50):7443–7454. doi:10.1038/sj.onc.1209091
28. Rafael D, Doktorovova S, Florindo HF, et al. EMT blockage strategies: targeting Akt dependent mechanisms for breast cancer metastatic behaviour modulation. Curr Gene Ther. 2015;15(3):300–312. doi:10.2174/1566523215666150126123642
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.