")
Back to Journals » Vascular Health and Risk Management » Volume 10
Unmet needs in the management of acute myocardial infarction: role of novel protease-activated receptor-1 antagonist vorapaxar
Authors Cho JR, Rollini F, Franchi F, Ferrante E, Angiolillo D
Received 14 January 2014
Accepted for publication 4 February 2014
Published 3 April 2014 Volume 2014:10 Pages 177—188
DOI https://doi.org/10.2147/VHRM.S36045
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jung Rae Cho, Fabiana Rollini, Francesco Franchi, Elisabetta Ferrante, Dominick J Angiolillo
University of Florida College of Medicine – Jacksonville, Jacksonville, FL, USA
Abstract: Platelet activation with subsequent aggregation is a complex process leading to thrombus formation, which remains a key component for atherothrombotic manifestations, in particular myocardial infarction. Therefore, antiplatelet therapies are pivotal for the treatment of these patients. Current oral antiplatelet therapies used for secondary prevention of ischemic recurrences include aspirin and adenosine diphosphate P2Y12 platelet-receptor antagonists. However, despite these therapies, patients who have experienced a myocardial infarction remain at risk for ischemic recurrences. Therefore, more aggressive secondary prevention measures have been an area of research, including identifying additional targets modulating platelet-activation and -aggregation processes. Among these, thrombin-mediated platelet activation via protease-activated receptors (PARs) has been subject to extensive clinical investigation. Several PAR-1 receptor antagonists have been developed. However, vorapaxar is the only one that has completed large-scale clinical investigation. The present manuscript will provide an overview on the role of thrombin-mediated signaling, the impact of PAR-1 blockade with vorapaxar on ischemic and bleeding outcomes, and the potential role for vorapaxar in clinical practice.
Keywords: platelet aggregation, antiplatelet agent, protease-activated receptor 1, vorapaxar
Introduction
Platelets have a crucial role in the pathogenesis of atherothrombotic disease manifestations, such as acute coronary syndrome (ACS), stroke, and peripheral artery disease (PAD).1,2 Multiple signaling pathways are implied in this process.1,2 Thromboxane (Tx) A2 and adenosine diphosphate (ADP) receptors have represented the main targets for current antiplatelet therapies used as the standard of care for patients with atherothrombotic disease manifestations.3 In particular, aspirin and clopidogrel are the most commonly used antiplatelet therapies among these patients. However, despite these therapies, rates of ischemic recurrences, especially in patients with ACS, remain high.4–6 More potent ADP P2Y12-inhibiting strategies, such as prasugrel and ticagrelor, have been shown to reduce ischemic event rates further compared with clopidogrel among ACS patients, albeit at the expense of an increased risk of bleeding.7,8 These observations may occur given the impact of P2Y12-mediated signaling on modulating hemostatic processes.9–11 Overall, these findings have led investigations in the field to assess alternative platelet signaling pathways to target, with the goal of optimizing clinical outcomes. Among these, thrombin-mediated platelet activation via protease-activated receptors (PARs) has been subject to extensive clinical investigation. In human platelets, PAR-1 has a key role in mediating platelet activation at low concentrations of thrombin.12 Several PAR-1 receptor antagonists have been developed.13 However, vorapaxar is the only one that has completed large-scale Phase III clinical investigation.14,15 The present manuscript provides an overview of the role of thrombin-mediated signaling, the impact of PAR-1 blockade with vorapaxar on ischemic and bleeding outcomes, and the potential role for vorapaxar in clinical practice.
Mechanism of thrombin-receptor antagonism for platelet inhibition
The role of PARs has been established in the field of vascular biology, atherothrombosis, and hemostasis as the receptor for thrombin, a potent agonist of platelet activation and aggregation.16,17 PAR is a G-protein-coupled receptor; it is constituted of a proteolytic enzyme that cleaves the extracellular loop of the receptor, and then the newly unmasked N-terminus binds to the proximally located transmembrane loop of the receptor itself.18 To date, four types of human PARs have been identified (PAR-1, -2, -3, and -4), and among these, only PAR-1 and PAR-4 are expressed on human platelets.19,20 PAR-1 has the principal role of mediating platelet activation at low concentrations of thrombin, while PAR-4 reacts at high concentrations.20–22 There are several signaling pathways for thrombin to activate PAR-1 (Figure 1). Once activated by thrombin, various phenotypic effects occur, which include Tx A2 production, ADP release, serotonin and adrenalin release, activation/mobilization of P-selectin and CD40 ligand, and finally platelet activation16–29 (Figure 1).
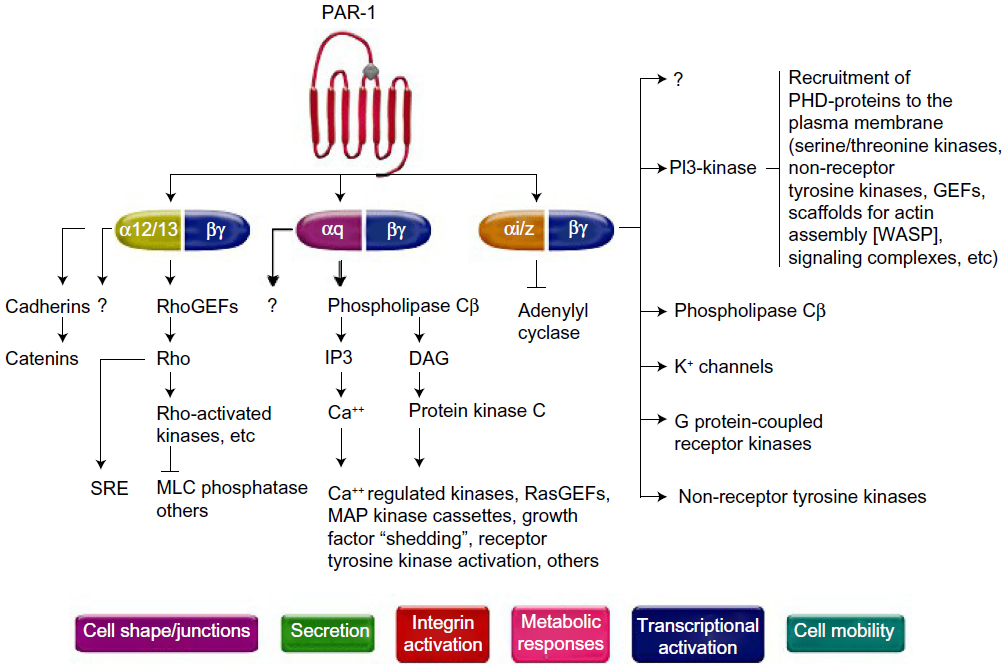 | Figure 1 Pathways of platelet protease-activated receptor (PAR)-1 activation. |
The key difference of PAR-1 in the development of pathologic atherothrombosis, compared to normal hemostasis, is that it lacks the ability to propagate the platelet-rich thrombus beyond the initial monolayer to become an occlusive clot, which is not found in aberrantly activated PAR-1.12 The prototype PAR-1 antagonist, FR 171113 was first tested in a guinea pig model.30 In this study, the use of FR 171113 did not inhibit ADP- or collagen-induced platelet aggregation, suggesting that PAR-1 antagonism does not affect other platelet signaling pathways. Preclinical studies with different molecules have been subsequently tested, and include SCH 530348 (vorapaxar; Merck, Whitehouse Station, NJ, USA), SCH 205831, SCH 602539, and E 5555 (atopaxar; Eisai, Tokyo, Japan). Among these, only vorapaxar and atopaxar have been tested in Phase II studies,13 and only vorapaxar has been tested in large-scale Phase III clinical investigations.14,15 The scope of this manuscript is to elaborate on vorapaxar. Details of other compounds go beyond the scope of this manuscript, and are described in more detail elsewhere.12,13
Pharmacokinetics and pharmacodynamics of vorapaxar
Vorapaxar (formerly known as SCH 530348) is a synthetic tricyclic 3-phenylpyridine analog of himbacine, a natural product that has been modified as a crystalline salt (Figure 2).12,31,32 It is a potent antagonist of the PAR-1 receptor, blocking thrombin-mediated platelet activation without interfering with thrombin-mediated cleavage of fibrinogen. In particular, vorapaxar is a nonprotein small molecule with high affinity, and is an orally active, competitive inhibitor of PAR-1.12,31,32 After oral administration, this drug is absorbed rapidly with high bioavailability. In pharmacokinetic studies, vorapaxar showed a dissociation half-life as long as 20 hours, thereby providing consistent antiplatelet effects. The route of elimination is mainly feces and secondarily by urine (less than 5%).17 Vorapaxar is metabolized by cytochrome P450 (CYP) 3A4; therefore, coadministration of drugs interfering with CYP 3A4 activity (such as ketoconazole or rifampin) could potentially affect its antiplatelet effects.33
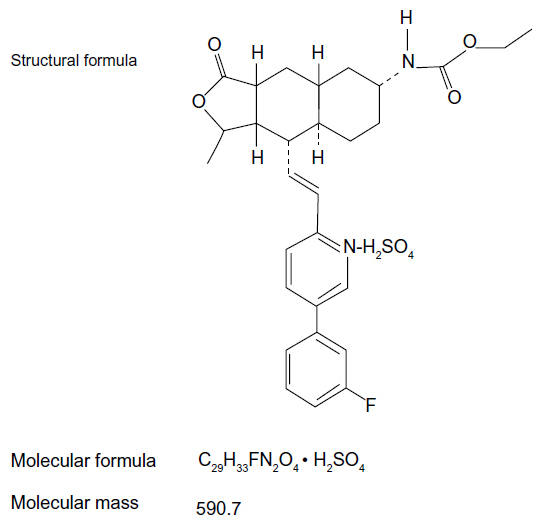 | Figure 2 Chemical structure of vorapaxar. |
In a Phase I study with 50 healthy volunteers, a single oral dose of vorapaxar (5–40 mg) was able to produce >90% mean inhibition of thrombin receptor-agonist peptide (TRAP)-induced platelet aggregation in all subjects for >72 hours, with low interindividual variability in plasma concentrations.34 Another Phase I study showed that a single 40 mg dose of vorapaxar was safe and well tolerated in healthy Caucasian and Japanese subjects, exhibiting >80% inhibition of TRAP-induced platelet aggregation in both populations.35,36 These findings encouraged further investigation of vorapaxar in clinical settings, as follows.
Clinical trial data
Phase II
Thrombin Receptor Antagonist Percutaneous Coronary Intervention (TRA-PCI) was a Phase II, randomized, double-blind, placebo-controlled trial with vorapaxar in patients undergoing non-urgent PCI or coronary angiography with planned PCI (n=1,030).37 In this trial, different loading doses of vorapaxar (10, 20, and 40 mg) were compared with matched placebo (3:1 ratio) on top of standard antiplatelet-treatment regimens, which included many patients on standard dual-antiplatelet therapy (DAPT) with aspirin and clopidogrel. The vorapaxar group, who subsequently underwent PCI (primary PCI cohort), continued taking a maintenance dose (0.5, 1, or 2.5 mg per day), and the placebo group continued placebo for 60 days. Bleeding events according to the Thrombosis In Myocardial Infarction (TIMI) scale represented the primary end point. The study showed no increased bleeding with vorapaxar (as a whole and according to each dosing regimen) compared with placebo. The secondary ischemic end point (composite of death, myocardial infarction [MI], and stroke) among PCI-treated patients (primary PCI cohort) was nonsignificantly lower with vorapaxar at each dosing regimen compared with placebo (placebo 9%, 10 mg 8%, 20 mg 5% and 40 mg 5%; odds ratio 0.67, 95% confidence interval [CI] 0.33–1.34). Overall, these findings suggest vorapaxar to have a favorable safety profile, which then led to Phase III clinical investigation.
Phase III
Two large-scale Phase III clinical studies have been conducted14,15: the TRACER (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome) and the TRA 2°P – TIMI 50 (Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events - Thrombolysis in Myocardial Infarction 50) trials. The key findings of these trials as well as the most recent updates in the literature derived from these pivotal investigations on vorapaxar are summarized in the following sections.
TRACER
The TRACER trial compared vorapaxar, at a loading dose of 40 mg and a daily maintenance dose of 2.5 mg thereafter, with placebo in 12,944 patients with ACS without ST-segment elevation (non-STE ACS) on top of standard treatment regimens, including DAPT with aspirin and clopidogrel.14 The primary composite end point comprised death from cardiovascular causes, MI, stroke, recurrent ischemia with hospitalization, or urgent coronary revascularization. The primary safety end points were a composite of moderate or severe bleeding according to the Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO) classification and clinically significant bleeding according to the TIMI classification, defined as TIMI major or minor bleeding or bleeding that required unplanned medical or surgical treatment or laboratory evaluation.14 The primary composite ischemic end point was nonsignificantly lower in patients randomized to vorapaxar compared with placebo (18.5% versus 19.9%, hazard ratio [HR] 0.92, 95% CI 0.85–1.01; P=0.07) at 2-year follow-up. A key secondary ischemic end point represented by the composite of death from cardiovascular causes, MI, or stroke occurred significantly less frequently with vorapaxar (14.7% versus 16.4%, HR 0.89, 95% CI 0.81–0.98; P=0.02) (Table 1). However, this occurred at the expense of increased bleeding (GUSTO moderate-to-severe bleeding) with vorapaxar compared with placebo (7.2% versus 5.2%, HR 1.35, 95% CI 1.16–1.58; P<0.001). Importantly, the rate of intracranial bleeding was over threefold higher with vorapaxar (1.1% versus 0.2%, HR 3.39, 95% CI 1.78–6.45; P<0.001) (Table 1). In January 2011, due to these safety concerns, the Data and Safety Monitoring Board (DSMB) decided to terminate the study prematurely.38 Overall, the results from this trial led to the conclusion that antiplatelet therapy with vorapaxar on top of standard antiplatelet-treatment regimens, including mostly patients on DAPT, provides only marginal ischemic beneficial effects, and is hampered by significantly increased bleeding rates, including intracranial hemorrhage, in patients with non-STE ACS.14 However, important predefined subgroup and post hoc analysis have been reported, providing important insights on the impact of vorapaxar on clinical outcomes.
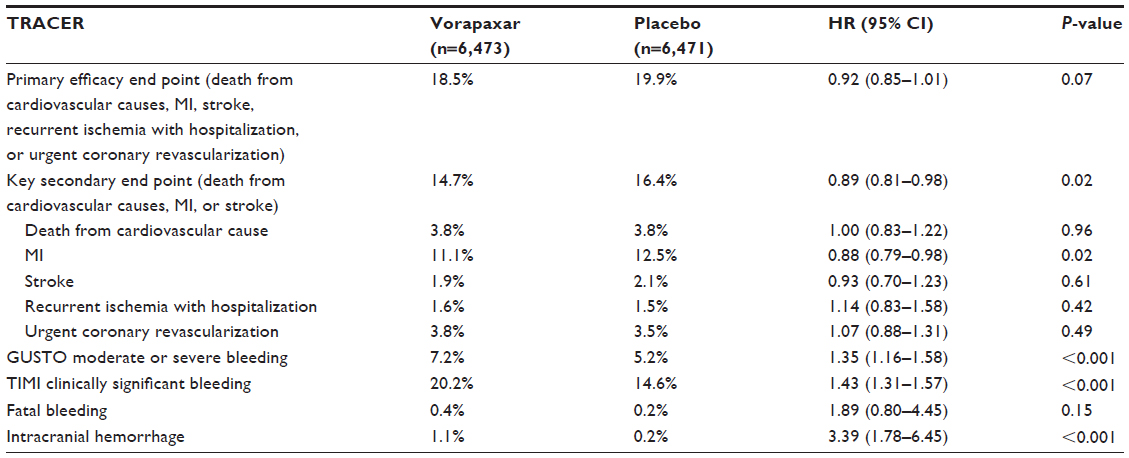 | Table 1 Efficacy and safety end points in the TRACER (at 2 years) trial |
MI analysis
A key driver of the reduction of ischemic events with vorapaxar was from MI rates. A blinded, independent central end-point adjudication committee prospectively defined and classified MI according to the universal MI definition.39,40 During follow-up, 1,580 MIs occurred in 1,319 patients. The majority (n=1,025, 64.9%) were type 1 (spontaneous) MI, followed by type 4a (periprocedural, n=352, 22.3%). Vorapaxar reduced the hazard of first MI of any type by 12% (HR 0.88, 95% CI 0.79–0.98; P=0.021) and the hazard of total MIs by 14% (HR 0.86, 95% CI 0.77–0.97; P=0.014), and these effects were sustained over time.39 Of note, vorapaxar reduced type 1 MI by 17% (HR 0.83, 95% CI 0.73–0.95; P=0.007), whereas there was no significant effect on type 4a MI (HR 0.90, 95% CI 0.73–1.12; P=0.35) compared with placebo, suggesting that vorapaxar is beneficial for the prevention of clinically relevant MI. Although exploratory in nature, this post hoc analysis suggests the potential role of vorapaxar in the management of patients with ACS to prevent future MI events.
CABG analysis
The TRACER trial was selectively performed in non-STE ACS patients. Therefore, the management of these patients could vary and include patients undergoing PCI (placebo 57.4% versus vorapaxar 58.1%), medical management (placebo 32.2% versus vorapaxar 32.0%), and coronary artery bypass graft (CABG; placebo 10.4% versus vorapaxar 9.9%).14 Most recently, an analysis of the subgroup of patients undergoing CABG (n=1,312, 10.1% of total TRACER population) during the index hospitalization was reported.41 This analysis showed that vorapaxar was associated with a 45% relative risk reduction in the primary end point compared with placebo (HR 0.55, 95% CI 0.36–0.83; P=0.005), with a positive interaction among these subjects (P=0.012). Notably, CABG-related TIMI major bleeding was numerically higher with vorapaxar, but not significantly different between vorapaxar and placebo (9.7% versus 7.3%, HR 1.36, 95% CI 0.92–2.02; P=0.12), without excess in fatal bleeding (0% versus 0.3%) or need for reoperation (4.7% versus 4.6%).41 These findings suggest that CABG patients in whom thrombin generation is known to be accelerated may represent a potential target for the use of vorapaxar, thereby increasing the risk of thrombotic complications such as graft occlusion or native coronary thrombosis.42–44 However, there are several considerations to be made of this analysis.45 The lack of differences in bleeding outcomes may be simply attributed to the small sample size of the CABG cohort (10.8% of TRACER population), making it insufficiently powered to draw any definitive conclusions. Clopidogrel was also held prior to CABG, with only 39% receiving the drug within 5 days of surgery. Upon discharge, clopidogrel use in this CABG cohort was only 18%, compared with 84% in the non-CABG cohort. Therefore, the benefits observed with vorapaxar may be attributed to the fact that the control arm was not on optimal secondary prevention therapy. Whether CABG patients represent a niche specific for vorapaxar requires further investigation.
PCI analysis
The safety and efficacy of vorapaxar were also assessed in a subgroup of TRACER patients who underwent PCI as a primary treatment strategy, in which data were analyzed according to stent type.46 Among 12,944 TRACER patients, 7,479 (58%) underwent PCI, with either bare-metal stent (BMS) or drug-eluting stent (DES) during index hospitalization. The type of stent (BMS or DES) used in the vorapaxar and placebo groups was well balanced. In line with current clinical practice, the duration of thienopyridine therapy was longer among patients treated with DES than those with BMS (median 221 versus 133 days). At 1 year, the primary end point occurred in 10.4% of patients with vorapaxar versus 10.6% of those with placebo (HR 0.96, 95% CI 0.83–1.10; P-interaction for primary end point =0.605). The key secondary end point (cardiovascular death, MI, or stroke) occurred in 6.8% with vorapaxar and 7.3% with placebo (HR 0.89, 95% CI 0.75–1.05; P-interaction for key secondary end point =0.72). The rate of Academic Research Consortium-defined definite or probable stent thrombosis was no different between vorapaxar and placebo (1.7% versus 1.5%, HR 1.12, 95% CI 0.78–1.62; P=0.54).14 There was a trend for interaction on cardiovascular death, MI, and stroke between stent type, with patients receiving BMS showing a relatively greater effect from vorapaxar (HR 0.70 versus 0.96; P-interaction =0.069).46 GUSTO moderate/severe bleeding was increased overall with vorapaxar, with no statistical interaction (P-interaction =0.316), although the relative increase appeared less with BMS (HR 1.27, 95% CI 0.85–1.90) than with DES (HR 1.70; 95% CI 1.14-2.53), likely attributed to the different duration in clopidogrel use.
Analysis according to ischemic and bleeding risk
A recent post hoc analysis evaluated the risk–benefit profile of vorapaxar by categorizing patients according to their risk of ischemic events and bleeding complications.47 In particular, patients were categorized into four groups: low bleeding/low ischemia, low bleeding/high ischemia, high bleeding/low ischemia, and high bleeding/high ischemia. The risk of bleeding and ischemia were derived post hoc from the TRACER population. The net benefit, defined as the difference in ischemic and bleeding event rates, was assessed. In this study, a large proportion of patients were categorized as low bleeding/low ischemia risk (63%), in whom there was a neutral effect with vorapaxar (net benefit −0.1%). Regardless of ischemic risk (high or low), patients at high bleeding risk (cumulatively representing 11% of the total population) had more harm than benefit (net benefit ranged from −2.93% to −3.83%). Ultimately, among patients categorized as low bleeding/high ischemia risk, representing 26% of the TRACER population, there was a net benefit (+2.76%) with vorapaxar. The results of this analysis, although post hoc in nature, do emphasize the potential for harm of vorapaxar among patients at high risk of bleeding, irrespective of their ischemic risk, but also suggest a potential role for vorapaxar in patients at low risk of bleeding and high risk of ischemic events. However, the risk score used in this analysis needs to be externally validated, and dedicated prospective studies need to be performed to support these post hoc findings.
Impact of thienopyridines
Another analysis that was performed to elucidate factors affecting the risk of bleeding in patients treated with vorapaxar was an assessment of patients on DAPT. In the TRACER trial 11,307 (87%) patients were on DAPT with aspirin and a thienopyridine, mostly represented by clopidogrel (91.8%), whereas 1,637 (13%) patients were not on a thienopyridine. Patients without thienopyridine therapy had a lower risk of bleeding, as assessed by all bleeding scales: GUSTO moderate/severe (HR thienopyridine, 1.45 versus no thienopyridine, 0.95; P-interaction =0.044), TIMI significant (HR 1.46 versus 1.24; P-interaction =0.25), GUSTO severe (HR 1.71 versus 1.43; P-interaction =0.60), and TIMI major (HR 1.62 versus 1.15; P-interaction =0.25).48 The high rate of thienopyridine use is in line with the fact that the TRACER trial enrolled patients with ACS, many undergoing PCI, in whom the use of DAPT forms part of the standard of care. It is important to note that the design of the TRACER trial was to evaluate the impact of more comprehensive platelet blockade by adding vorapaxar to standard antiplatelet-treatment regimens, including DAPT. Therefore, based on this analysis, we cannot infer a potential for the use of vorapaxar without a thienopyridine, because this would require a dedicated trial.
Impact of aspirin dosing
Aspirin-dosing regimens have been a subject of interest in recent years. In particular, studies have shown that high-dose aspirin does confer added benefit, but can increase the risk of bleeding when used in combination with clopidogrel.49,50 Although aspirin dosing has not been shown to impact outcomes in patients treated with prasugrel,51 reduced ischemic benefits have been shown in ticagrelor-treated patients when high-dose (≥300 mg) aspirin was used.52 This has indeed raised interest in understanding the impact of aspirin dosing on clinical outcomes in patients treated with vorapaxar in the TRACER trial.53 Overall, 96% (n=12,515) of the entire TRACER population was on aspirin at randomization at the following dosing regimens: low dose (≤100 mg, n=7,523, 60%), medium dose (100–300 mg, n=1,049, 8.4%), and high dose (≥300 mg, n=3,943, 31%). Compared with low-dose aspirin, high-dose aspirin was associated with a trend to a higher hazard of bleeding (adjusted HR for GUSTO severe bleeding: 1.88 [high dose] versus 1.63 [low dose]; P-interaction =0.954) and less efficacy (adjusted HR for primary efficacy end-point event: 1.05 [high dose] versus 0.90 [low dose]; P-interaction =0.140) with vorapaxar compared with placebo, although this did not reach statistical significance.53
Pharmacodynamic substudy
A pharmacodynamic substudy enrolling a total of 249 patients in TRACER was conducted.54 Pharmacodynamic assessments were conducted at baseline and 1 month after study enrollment in patients on aspirin and clopidogrel, and included light-transmission aggregometry following stimuli with a variety of agonists (20 μM ADP, 15 μM TRAP, and combination of 2.5 μg/mL collagen-related peptide, 5 μM ADP, and 15 μM TRAP (CAT)) and vasodilator-stimulated phosphoprotein (VASP).54 TRAP and CAT-induced platelet aggregation were significantly inhibited with vorapaxar compared with placebo, while nonsignificant reductions in platelet aggregation were observed following ADP stimuli. The platelet-reactivity index determined by VASP, which is regarded as most specific to P2Y12-receptor function, was significantly lower in both treatment arms at 1 month compared with baseline, although numerically lower in the vorapaxar group. These results suggest that PAR-1 receptor inhibition mediated by vorapaxar may have some synergistic effects on P2Y12 receptor-mediated signaling. This supports prior studies suggesting an interplay between these two pathways,55 which can lead to enhanced P2Y12-inhibitory effects in patients concomitantly treated with vorapaxar and clopidogrel.54
TRA 2°P – TIMI 50
The TRA 2°P – TIMI 50 trial was a secondary prevention study that enrolled 26,449 patients with atherothrombotic disease.15 Eligible patients had to have a history of atherosclerosis, which was defined as a spontaneous MI or ischemic stroke within the previous 2 weeks to 12 months or PAD associated with a history of intermittent claudication in conjunction with either ankle-brachial index of less than 0.85 or previous revascularization for limb ischemia. Patients were randomized to receive vorapaxar (2.5 mg daily dose) or matching placebo on top of standard-of-care therapy, which could include aspirin and/or clopidogrel, with an expected follow-up period up to 30 months.15 The primary end point was determined as the composite of death from cardiovascular causes, MI, or stroke. After 3 years, the primary end point was achieved, with lower event rate with vorapaxar compared with placebo (9.3% versus 10.5%, HR 0.87, 95% CI 0.80–0.94; P<0.001). In addition, a key secondary end point represented by the composite of cardiovascular death, MI, stroke or recurrent ischemia leading to revascularization also occurred less frequently with vorapaxar (11.2% versus 12.4%, HR 0.88, 95% CI 0.82–0.95; P=0.001). The ischemic benefit observed with vorapaxar was driven by a reduction in MI rates (5.2% versus 6.1%, HR 0.83, 95% CI 0.74–0.93; P=0.001) (Table 2). There was a trend towards a positive interaction (P=0.058) among patients who had a prior MI (0.80 [0.72–0.89]) as a qualifying event to enter the study compared with patients with a prior stroke (1.03 [0.85–1.26]) or established PAD (0.94 [0.78–1.14]). The details of these subgroups are described below. However, at a median of 24 months of follow-up after completion of enrollment, the DSMB of TRA 2°P – TIMI 50 reported an excess of intracranial bleeding in patients with a history of stroke in the vorapaxar group, and recommended discontinuation of the drug in all patients with previous stroke, including those with a new stroke during the trial. The DSMB also recommended continuation of the trial in patients without a history of stroke.15 After 3 years, there was increased bleeding (GUSTO moderate and severe) with vorapaxar compared with placebo (4.2% versus 2.5%, HR 1.66, 95% CI 1.43–1.93; P=0.001). The rate of intracranial bleeding was almost twofold higher with vorapaxar (1.0% versus 0.5%, HR 1.94, 95% CI 1.39–2.70; P<0.001) (Table 2). Similarly to the TRACER trial, there were important analyses deriving from the TRA 2°P trial that are useful for understanding the potential role of vorapaxar as an adjunct antiplatelet therapy for secondary prevention of ischemic events.
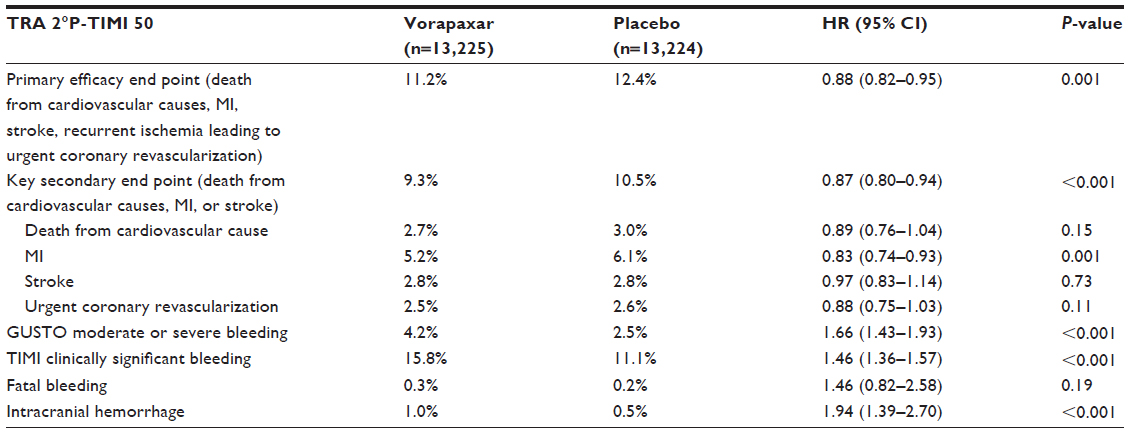 | Table 2 Efficacy and safety end points in the TRA 2°P – TIMI 50 (at 3 years) trial |
Analysis in patients with prior stroke
A total of 4,883 patients enrolled in TRA 2°P had a history of prior ischemic stroke.56 In these patients, vorapaxar was associated with an increased risk of intracranial hemorrhage compared with placebo (2.5% versus 1.0%, HR 2.52, 95% CI 1.46–4.36; P<0.001), as well as GUSTO moderate and severe bleeding (4.2% versus 2.4%, HR 1.93, 95% CI 1.33–2.79; P<0.001). This occurred without any significant improvement in the primary ischemic end point (13.0% versus 11.7%, P=0.75), including ischemic stroke (8.6% versus 7.1%, HR 0.99, 95% CI 0.78–1.25; P=0.90).56 Overall, these data indicate the potential for harm with vorapaxar in patients with prior stroke, discouraging the potential role of this agent in this setting.56
Analysis in patients with prior MI
Patients with a previous MI represented the largest subgroup of subjects enrolled in the trial (n=17,779 patients, 67% of the entire TRA 2°P – TIMI 50 population), and drove most of the benefit with vorapaxar compared with patients with prior stroke or PAD (P-interaction =0.058). In this cohort of patients, the benefit of vorapaxar on reducing the primary ischemic end point was more profound compared with placebo (8.1% versus 9.7%, HR 0.80, 95% CI 0.72–0.89; P<0.0001) (Figure 3).57 Furthermore, this benefit was consistent regardless of qualifying MI from the time of randomization: <3 months (HR 0.82, 95% CI 0.70–0.95; P=0.011), 3–6 months (HR 0.79, 95% CI 0.65–0.97; P=0.023), and >6 months (HR 0.78, 95% CI 0.62–0.97; P=0.026) (Figure 4).57 This was also confirmed in a landmark analysis assessing patients before (3.2% versus 4.0%, HR 0.79, 95% CI 0.67–0.92; P=0.003) and after 1 year (5.5% versus 6.5%, HR 0.82, 95% CI 0.71–0.94; P=0.004) from randomization. These findings were consistent in the all key subgroups, including qualifying MI (non-ST-elevation MI or ST-elevation MI, with or without thienopyridine use, with or without prior stent).
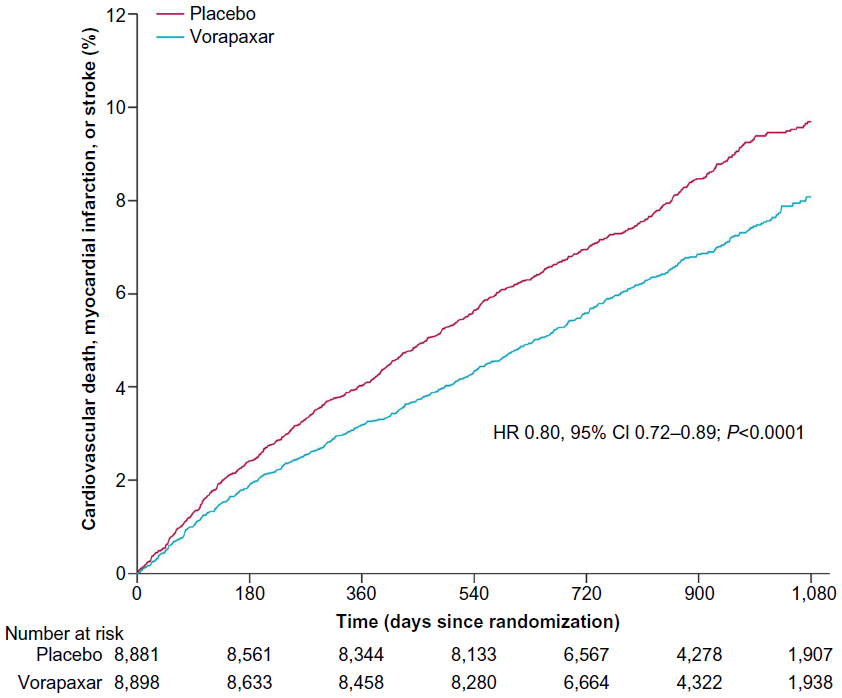 | Figure 3 Kaplan–Meier curve of estimated occurrence of cardiovascular death, myocardial infarction, or stroke in TRA 2°P – TIMI 50 prior myocardial infarction cohort. |
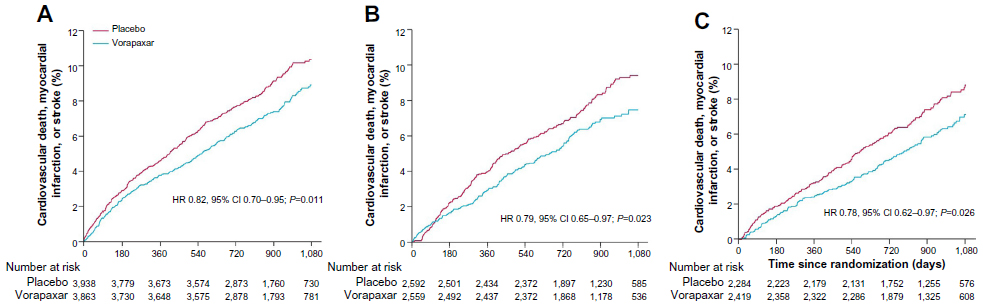 | Figure 4 Kaplan–Meier estimates of cardiovascular death, myocardial infarction, or stroke according to time from qualifying myocardial infarction to randomization: <3 months (A), 3–6 months (B), and >6 months (C) in the TRA 2°P–TIMI 50 prior myocardial infarction cohort. |
However, GUSTO moderate or severe bleeding was more common in the vorapaxar group than the placebo group (3.4% versus 2.1%, HR 1.61, 95% CI 1.31–1.97; P<0.0001). Similarly, TIMI non-CABG major (2.2% versus 1.6%, HR 1.29, 95% CI 1.02–1.64; P=0.033) and TIMI clinically significant bleeding (15.1% versus 10.4%, HR 1.49, 95% CI 1.36–1.63; P<0.0001) were also increased with vorapaxar.57 However, there was only a trend for increased intracranial bleeding with the vorapaxar group, which was infrequent overall (0.6% versus 0.4%, HR 1.54, 95% CI 0.96–2.48; P=0.076), and there were no differences in fatal bleeding (0.2% versus 0.1%, HR 1.56, 95% CI 0.67–3.60; P=0.30). The net clinical outcome (presented as a composite of primary efficacy outcome and GUSTO moderate or severe bleeding) was in favor of vorapaxar compared with placebo (12.5% versus 13.4%, HR 0.91, 95% CI 0.84–0.99; P=0.038).57
A further analysis of the MI cohort assessed outcomes excluding patients at high risk of bleeding. In particular, patients with prior transient ischemic attack/stroke, <60 kg, and >75 years of age (factors that in another pivotal trial of prasugrel in ACS patients were shown to be associated with an increased risk of bleeding6) were identified. In the TRA 2°P trial, these subjects also had high rates of bleeding overall, and rates were further increased among patients treated with vorapaxar compared with placebo (6.9% versus 3.9%). In an analysis excluding such patients (n=14,909), known as a low-bleeding-risk cohort, the primary combined ischemic end point was significantly reduced with vorapaxar compared with placebo (6.8% versus 8.6%, HR 0.75, 95% CI 0.66–0.85; P<0.0001), and there was also a significant reduction in cardiovascular death (1.5% versus 2.0%, HR 0.73, 95% CI 0.56–0.95; P=0.02). GUSTO moderate or severe bleeding was lower than in the overall trial, although still higher with vorapaxar than placebo (2.7% versus 1.8%).59 Overall, this analysis conducted among patients who were randomized in the TRA 2°P trial indicates that among appropriately selected patients, prolonged treatment with vorapaxar when added to aspirin with or without a thienopyridine may be beneficial for long-term secondary prevention in patients with prior MI.
Diabetic subgroup analysis
Another subgroup analysis evaluated patients with prior MI according to the presence or absence of diabetes mellitus (DM).58 In this analysis, patients with DM (n=3,936) had a higher risk of both ischemic and bleeding events compared with non-DM (n=13,843) subjects, consistent with prior investigations. Vorapaxar showed consistent effects on reducing ischemic event rates, irrespective of DM status (P-interaction =0.51). In particular, vorapaxar reduced adverse events compared with placebo patients with DM (12.6% versus 15.7%, HR 0.77; P=0.004) and without DM (6.8% versus 7.9%, HR 0.83; P=0.005). The benefit of vorapaxar in patients with DM was driven by a reduction in MI (8.6% versus 11.4%, HR 0.73, 95% CI 0.59–0.91; P=0.01, P-interaction =0.38), and severe recurrent ischemia (3.9% versus 5.6%; HR 0.63, 95% CI 0.46–0.86; P=0.004, P-interaction =0.02). GUSTO moderate/severe bleeding was increased in DM patients (4.7% versus 2.8%, HR 1.59, 95% CI 1.09–2.32; P=0.02) and non-DM (3.0% versus 1.9%, 95% CI 1.27–2.06, HR 1.62; P<0.001) patients treated with vorapaxar than placebo, which was not different between DM and non-DM patients (HR 1.59 versus 1.62, P-interaction =0.95).58
PAD subgroup analysis
The PAD cohort of the TRA 2°P trial represented 14.3% of the study population (n=3,787). Vorapaxar failed to reduce the primary efficacy end point compared with placebo (11.3% versus 11.9%, HR 0.94, 95% CI 0.78–1.14; P=0.53).59 However, vorapaxar did reduce the rate of hospitalization for acute limb ischemia (2.3% versus 3.9%, HR 0.58, 95% CI 0.39–0.86; P=0.006) and peripheral artery revascularization (18.4% versus 22.2%, HR 0.84, 95% CI 0.73–0.97; P=0.017). This occurred at the expense of more frequent GUSTO moderate and severe bleeding (7.4% versus 4.5%, HR 1.62, 95% CI 1.21–2.18; P=0.001).59 Given that PAR-1 receptors are also present on a number of cells (including endothelial cells) other than platelets, it could be hypothesized that these study findings may be attributed to an effect of vorapaxar on vascular remodeling and vascular perfusion.59 However, these findings warrant further investigation.
Impact of thienopyridines
In contrast to TRACER, in the TRA 2°P study comparing patients with and without thienopyridine (n=15,356 [58%] versus n=11,093 [42%]), there were no differences in bleeding complications among patients treated with vorapaxar.60 However, this may be attributed to differences in baseline characteristics. In fact, in TRACER patients presented with an ACS leading to early use of DAPT, which occurred in approximately nearly 90% of patients.
Impact of aspirin dosing
In the substudy of TRA 2°P – TIMI 50 by Scirica et al, including patients with prior MI, they divided the group according to each different dose of aspirin (<100 mg, 100–162 mg, >162 mg). A total of 6,988 patients (40%) received <100 mg aspirin daily, 7,704 patients (44%) 100–162 mg, and 2,755 patents (16%) >162 mg. Of note, the relative risk of bleeding was not higher with vorapaxar at the higher aspirin doses.61 Apparently, the different criteria of aspirin doses in both the TRACER and TRA 2°P – TIMI 50 studies influenced the bleeding outcome. As we mentioned before, low-dose aspirin would be an appropriate option with the combined use of vorapaxar.
Conclusion and future directions
Patients with MI remain at risk for ischemic recurrences, underscoring the need for aggressive secondary preventive measures. Intensifying antithrombotic treatment regimens by means of using more potent P2Y12-receptor antagonists,6,7 adding an anticoagulant agent to standard DAPT,62–64 or adding a third antiplatelet agent14,15,65 have all represented treatment strategies tested in clinical trials. While these strategies aim to reduce ischemic recurrences, they all come at the risk of increased bleeding complications. Vorapaxar has been tested as an add-on strategy for patients on standard antiplatelet-treatment regimens, mostly represented by aspirin and clopidogrel. The clinical trial data clearly indicate a potential for harm in certain subgroups, such as patients with a prior cerebrovascular event, but also indicate subjects, such as those with prior MI, in whom there may be adjunctive benefit. Whether findings from key subgroups of patients identified from large clinical trial data can be translated into clinical practice remains unknown at this time, and will require experience from real-world clinical practice. Most recently, vorapaxar has been recommended by the US Food and Drug Administration Drug Advisory Committee for clinical use in the management of a select group of patients with a history of MI, excluding those with a history of stroke, transient ischemic attack, intracranial hemorrhage, or active pathologic bleeding.66 In this context, vorapaxar will represent an agent to add to the armamentarium of agents used to reduce the long-term risk of MI patients. Indeed, further studies are warranted, although the current evidence suggests vorapaxar is most likely to have a niche use after careful patient selection, in light of the availability of other novel antiplatelet therapies that have also been shown to reduce the risk of recurrent MI.67
Disclosure
DJA received payment as an individual for: consulting fees or honoraria from Bristol-Myers Squibb, Sanofi-Aventis, Eli Lilly, Daiichi Sankyo, The Medicines Company, AstraZeneca, Merck, Evolva, Abbott Vascular, and PLx Pharma; participation in review activities from Johnson and Johnson, St Jude, and Sunovion; institutional payments for grants from Bristol-Myers Squibb, Sanofi-Aventis, GlaxoSmithKline, Otsuka, Eli Lilly, Daiichi Sankyo, The Medicines Company, AstraZeneca, Evolva, and Gilead; and has other financial relationships with Esther and King Biomedical Research Grant. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
Davì G, Patrono C. Platelet activation and atherothrombosis. N Eng J Med. 2007;357(24):2482–2494. | |
Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. 2010;74(4):597–607. | |
Ueno M, Kodali M, Tello-Montoliu A, Angiolillo DJ. Role of platelets and antiplatelet therapy in cardiovascular disease. J Atheroscler Thromb. 2011;18(6):431–442. | |
Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs. 2012;72(16):2087–2116. | |
Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, et al. Variability in individual responsiveness to clopidogrel: clinical implications, management, and future perspectives. J Am Coll Cardiol. 2007;49(14):1505–1516. | |
Tantry US, Bonello L, Aradi D, et al. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol. 2013;62(24):2261–2273. | |
Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–2015. | |
Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–1057. | |
Leon C, Alex M, Klocke A, et al. Platelet ADP receptors contribute to the initiation of intravascular coagulation. Blood. 2004;103(2):594–600. | |
van der Meijden PE, Feijge MA, Giesen PL, Huijberts M, van Raak LP, Heemskerk JW. Platelet P2Y12 receptors enhance signalling towards procoagulant activity and thrombin generation. A study with healthy subjects and patients at thrombotic risk. Thromb Haemost. 2005;93(6):1128–1136. | |
Angiolillo DJ, Capranzano P, Desai B, et al. Impact of P2Y(12) inhibitory effects induced by clopidogrel on platelet procoagulant activity in type 2 diabetes mellitus patients. Thromb Res. 2009;124(3):318–322. | |
Angiolillo DJ, Capodanno D, Goto S. Platelet thrombin receptor antagonism and atherothrombosis. Eur Heart J. 2010;31(1):17–28. | |
Capodanno D, Bhatt DL, Goto S, et al. Safety and efficacy of protease-activated receptor-1 antagonists in patients with coronary artery disease: a meta-analysis of randomized clinical trials. J Thromb Haemost. 2012;10(10):2006–2015. | |
Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012; 366(1):20–23. | |
Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404–1413. | |
Martorell L, Martínez-González J, Rodríguez C, Gentile M, Calvayrac O, Badimon L. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb Haemost. 2008;99(2):305–315. | |
Ueno M, Ferreiro JL, Angiolillo DJ. Mechanism of action and clinical development of platelet thrombin receptor antagonists. Expert Rev Cardiovasc Ther. 2010;8(8):1191–1200. | |
Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3(8):1800–1814. | |
Kahn ML, Zheng YW, Huang W, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394(6694):690–694. | |
Vu T-KH, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64(6):1057–1068. | |
Kahn ML, Nakanish-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103(6):879–887. | |
Xu WF, Anderson H, Whitemore TE, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95(12):6642–6646. | |
Henriksen RA, Hanks VK. PAR-4 agonist AYPGKF stimulates thromboxane production by human platelets. Arterioscler Thromb Vasc Biol. 2002;22(5):861–866. | |
Covic L, Singh C, Smith H, Kuliopulos A. Role of PAR4 thrombin receptor in stabilizing platelet-platelet aggregates as revealed by a patient with Hermansky-Pudlak syndrome. Thromb Haemost. 2002;87(4):722–727. | |
Lova P, Campus F, Lombardi R, et al. Contribution of protease-activated receptors 1 and 4 and glycoprotein Ib-IX-V in the G(i)-independent activation of platelet Rap1B by thrombin. J Biol Chem. 2004;279(24):25299–25306. | |
Smith CC, Wilson AP, Prichard BN, Betteridge DJ. Stimulus-induced release of endogenous catecholamines from human washed platelets. Clin Sci (Lond). 1986;70(5):495–500. | |
Henn V, Slupsky JR, Grafe M, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391(6667):591–594. | |
Stenberg PE, McEver RP, Shuman MA, Jacques YV, Bainton DF. A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J Cell Biol. 1985;101(3):880–886. | |
Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389(6647):183–186. | |
Kato Y, Kita Y, Hirasawa-Taniyama Y, et al. Inhibition of arterial thrombosis by a protease-activated receptor 1 antagonist, FR171113, in the guinea pig. Eur J Pharmacol. 2003;473(2–3):163–169. | |
Chackalamannil S, Davies RJ, Asberom T, Doller D, Leone D. A highly efficient total synthesis of (+)-himbacine. J Am Chem Soc. 1996;118(40):9812–9813. | |
Doller D, Chakalamannil S, Czarniecki M, McQuade R, Ruperto V. Design, synthesis, and structure-activity relationship studies of himbacine derived muscarinic receptor antagonists. Bioorg Med Chem Lett. 1999;9(6):901–906. | |
Xia Y, Chackalamannil S, Greenlee WJ, et al. Discovery of a vorapaxar analog with increased aqueous solubility. Bioorg Med Chem Lett. 2010;20(22):6676–6679. | |
Kosoglou T, Reyderman L, Tiessen R, et al. TRAP-induced platelet aggregation following single and multiple rising doses of SCH 530348, a novel thrombin receptor antagonist, in healthy volunteers. Pharmacol Ther. 2009;85(Suppl 1):S21. | |
Kosoglou T, Reyderman L, Tiessen RG, et al. Pharmacodynamics and pharmacokinetics of the novel PAR-1 antagonist vorapaxar (formerly SCH 530348) in healthy subjects. Eur J Clin Pharmacol. 2012;68(3):249–258. | |
Kosoglou T, Reyderman L, Kasserra C, et al. No differences in the pharmacodynamics and pharmacokinetics of the thrombin receptor antagonist vorapaxar between healthy Japanese and Caucasian subjects. Eur J Clin Pharmacol. 2012;68(3):291–300. | |
Becker RC, Moliterno DJ, Jennings LK, et al. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomized, double-blind, placebo-controlled phase II study. Lancet. 2009;373(9667):919–928. | |
Business Wire. Merck says additional information provided to investigators in clinical study of vorapaxar. 2011. Available from: http://www.businesswire.com/news/home/20110119006900/en/Merck-Additional-Information-Investigators-Clinical-Study-Vorapaxar. Accessed January 3, 2014. | |
Leonardi S, Tricoci P, White HD, et al. Effect of vorapaxar on myocardial infarction in the thrombin receptor antagonist for clinical event reduction in acute coronary syndrome (TRACER) trial. Eur Heart J. 2013;34(23):1723–1731. | |
Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60(16):1581–1598. | |
Whellan DJ, Tricoci P, Chen E, et al. Vorapaxar in acute coronary syndrome patients undergoing coronary artery bypass graft surgery: subgroup analysis from the TRACER trial. J Am Coll Cardiol. Epub November 6, 2013. | |
Boyle EM Jr, Lille ST, Allaire E, Clowes AW, Verrier ED. Endothelial cell injury in cardiovascular surgery: atherosclerosis. Ann Thorac Surg. 1997;63(3):885–894. | |
Reilly CF, McFall RC. Platelet-derived growth factor and transforming growth factor beta regulate plasminogen activator inhibitor-1 synthesis in vascular smooth muscle cells. J Biol Chem. 1991;266(15):9419–9427. | |
Verrier ED, Boyle EM Jr. Endothelial cell injury in cardiovascular surgery. Ann Thorac Surg. 1996;62(3):915–922. | |
Ben-Yehuda O. Vorapaxar in patients undergoing CABG: insights from a subgroup analysis. J Am Coll Cardiol. Epub November 6, 2013. | |
Valgimigli M, Tricoci P, Huang Z, et al. Vorapaxar, a platelet thrombin-receptor antagonist, in patients with non-ST-segment elevation acute coronary syndrome undergoing percutaneous coronary intervention: results from the TRACER trial. Circulation. 2012;126: A18805. | |
Tricoci P, Huang Z, van de Werf F, et al. Net clinical benefit of vorapaxar in NSTE ACS: role of ischemic and bleeding risk stratification. Circulation. 2012;126:A19049. | |
Tricoci P, Huang Z, van de Werf F, et al. Vorapaxar with and without thienopyridine use in acute coronary syndromes: results from the TRACER trial. Eur Heart J. 2012;33:495–496. | |
Peters RJ, Mehta SR, Fox KA, et al. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) study. Circulation. 2013;108(14):1682–1697. | |
CURRENT-OASIS 7 Investigators, Mehta SR, Bassand JP, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med. 2010;363(10):930–942. | |
Kohli P, Udell JA, Murphy SA, et al. Discharge aspirin dose and clinical outcomes in patients with acute coronary syndromes treated with prasugrel versus clopidogrel: an analysis from the TRITON-TIMI 38 study (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis In Myocardial Infarction 38). J Am Coll Cardiol. 2014;63(3):225–232. | |
Mahaffey KW, Wojdyla DM, Carroll K, et al. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation. 2011;124(5):544–554. | |
Mahaffey KW, Huang Z, Wallentin L, et al. Association of aspirin dose and vorapaxar safety and efficacy in patients with non-ST-segment elevation acute coronary syndrome (from the TRACER Trial). Am J Cardiol. Epub December 25, 2013. | |
Storey RF, Kotha J, Smyth S, et al. Effects of vorapaxar on platelet reactivity and biomarker expression in non-ST-elevation acute coronary syndromes. The TRACER Pharmacodynamic Substudy. Thromb Haemost. 2014;111(5). | |
Dorsam RT, Tuluc M, Kunapuli SP. Role of protease-activated and ADP receptor subtypes in thrombin generation on human platelets. J Thromb Haemost. 2004;2(5):804–812. | |
Morrow DA, Alberts MJ, Mohr JP, et al. Efficacy and safety of vorapaxar in patients with prior ischemic stroke. Stroke. 2013;44(3):691–698. | |
Scirica BM, Bonaca MP, Braunwald E, et al. Vorapaxar for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2°P-TIMI 50 trial. Lancet. 2012;380(9850):1317–1324. | |
Scirica BM, Bonaca MP, Morais J, et al. Vorapaxar in patients with diabetes and prior MI: findings from the TRA 2P-TIMI 50 trial. Circulation. 2013;128:A10398. | |
Bonaca MP, Scirica BM, Creager MA, et al. Vorapaxar in patients with peripheral artery disease: results from TRA2°P-TIMI 50. Circulation. 2013;127(14):1522–1529. | |
Bonaca MP, Scirica BM, Braunwald E, et al. Efficacy of vorapaxar is not modified by thienopyridine therapy: results from TRA 2P-TIMI 50 trial. Circulation. 2012;126:A18595. | |
Scirica BM, Bonaca MP, Braunwald E, et al. Vorapaxar for secondary prevention after myocardial infarction according to aspirin dose – insights from the TRA 2P-TIMI 50 trial. Circulation. 2012;126:A14508. | |
Alexander JH, Lopes RD, James S, et al. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365(8):699–708. | |
Oldgren J, Budaj A, Granger CB, et al. Dabigatran vs. placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J. 2011;32(22):2781–2789. | |
Mega JL, Braunwald E, Wiviott SD, et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366(1):9–19. | |
Friedland SN, Eisenberg MJ, Shimony A. Meta-analysis of randomized controlled trials on effect of cilostazol on restenosis rates and outcomes after percutaneous coronary intervention. Am J Cardiol. 2012;109(10):1397–1404. | |
US Food and Drug Administration. Vorapaxar Advisory Committee briefing document. 2014. Available from: http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/cardiovascularandrenaldrugsadvisorycommittee/ucm381330.pdf. Accessed January 31, 2014. | |
Ferreiro JL, Angiolillo DJ. New directions in antiplatelet therapy. Circ Cardiovasc Interv. 2012;5(3):433–445. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.