")
Back to Journals » OncoTargets and Therapy » Volume 10
Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy
Authors Robainas M, Otano R, Bueno S , Ait-Oudhia S
Received 16 January 2017
Accepted for publication 1 March 2017
Published 23 March 2017 Volume 2017:10 Pages 1803—1807
DOI https://doi.org/10.2147/OTT.S132508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Marianela Robainas, Rafael Otano, Stephen Bueno, Sihem Ait-Oudhia
Department of Pharmaceutics, Center for Pharmacometrics and Systems Pharmacology, College of Pharmacy, University of Florida, Orlando, FL, USA
Abstract: Autophagy is a vital, physiological catabolic process for cell survival by which cells clear damaged organelles and recycle nutrients when homeostasis is maintained. Cancer is a complex disease with uncontrolled growth of cancer cells. Recent studies have suggested the role of autophagy in cancer. A complex relationship exists between autophagy and cancer, since autophagy can contribute to the survival or the destruction of malignant cells depending on the stage of tumor development. In this review, we describe in detail the mechanism underlying autophagy in cancer cells and the intricate involvement of the programmed cell death-1 (PD1) receptor with its ligand (PD-L1). The overexpression of PD-L1 receptors on cancer cell membranes has been observed in several types of cancers. The interaction of PD-L1 on cancer cells with PD1 on the surface of T-cells causes cancer cells to escape from the immune system by preventing the activation of new cytotoxic T-cells in the lymph nodes and subsequent recruitment to the tumor. In addition to its immunopathogenicity, PD1 has been related to autophagy. Reduction of this receptor due to treatment increases autophagy, therefore promoting the recycling of nutrients and clearance of toxic species, consequently promoting cell survival. In addition, PD-L1/PD1 engagement can induce autophagy in nearby T-cells due to a decrease in the amino acids tryptophan and arginine and due to the deprivation of nutrients such as glucose followed by a reduction in glucose metabolism. Resistance to cancer therapies is attributed to various pathways in oncogenesis including, inhibition of tumor suppressors, alteration of the tumor metabolic environment, and upregulation of autophagy. Here we explore the interaction between the immunosuppressive PD-L1/PD1 engagement and autophagy mechanisms, and evaluate the impact of inhibition of these pathways in augmenting antitumor efficacy.
Keywords: immuno-oncology, cancer immunotherapy, autophagy, PD1, PD-L1
Introduction
Autophagy is a catabolic process by which cells recycle their own nutrients and remove damaged organelles and protein aggregates. The process begins with the formation of an autophagosome that engulfs fractions of the cytoplasm. The latter fuses with an endosome to form an amphisome, which carries the degraded contents to the lysosome.1 This is a physiological process that plays an important role in organogenesis, tissue homeostasis, metabolism, and immune responses, in addition to various diseases. While researchers continue looking at the role of autophagy in defeating cancer, the complexity of its underlying mechanisms remains a fundamental challenge. It was determined that autophagy plays a dual role, either by suppressing tumor cells or by promoting their survival. In the former case, tumorigenic capacity is reduced through induction of autophagy.1 An example of such autophagy-assisted tumorigenicity inhibition can be seen in beclin 1 (BECN1) protein, a mammalian homologue of yeast Atg6/Vps30 gene, where it is monoallelically deleted in several human cancers such as breast, ovarian, and prostate cancers.2 Liang et al3 observed autophagy induction with decrease in tumor cells in MCF7 breast cancer cells when BECN1 was introduced. Another common protein that has a tumor suppression effect on autophagy induction is endophilin B1 (BIF-1), which has been identified as a BECN1-interacting protein with tumor suppression properties.4 The role of these two genes in oncogenesis was confirmed clinically based on their downregulation observed in cancer patients and in BECN1−/- and Bif-1−/- knockout mice, which showed a higher frequency in tumor development and growth than their counterpart wild-type animals. In the latter case, the underlying molecular mechanism by which autophagy promotes tumor survival in already well-established tumors5 is still not fully understood. However, it is thought that this process is linked to the recycling of nutrients maintaining the cellular metabolism and enabling it to persist during cellular stress.1
In the past decade, a number of anticancer-targeted therapies have been discovered and proven effective in various hematological and solid malignancies.6 These therapies are tailored to interrupt specific pathways within the malignant cell and/or the tumor microenvironment, such as the hormonal axis, the receptor-mediated tyrosine kinase signaling pathway, and the immune system meshwork.3 The focus of this review is to examine the role of one of immune checkpoint pathways, the programmed cell death-1 receptor with its ligand (PD-L1)/PD1 axis, in inducing autophagy as the downstream pharmacological effect for killing cancer cells.7,8
PD-L1/PD1 interaction and its effects on autophagy
Cancer can escape the body’s immune surveillance. Numerous tumors and tumor-infiltrating immune cells express high levels of PD-L1. Under normal and steady-state conditions, the PD-L1 pathway can play an important role in maintaining immune homeostasis and protecting the immune system. In cancer, the PD-L1 pathway can protect tumors from cytotoxic T-cells by disrupting the cancer immunity cycle in two ways.9–11 The first is within the lymph nodes, where overexpression of PD-L1 on tumor-infiltrating immune cells can prevent the priming and activation of new cytotoxic T-cells in the lymph nodes and subsequent recruitment to the tumor.9–11 The second is within the tumor microenvironment, where upregulation of PD-L1 on dendritic cells results in deactivation of cytotoxic T-cells.10 In both cases, the interaction of PD-L1 with its cognate ligand PD1 on the surface of T-cells suppresses their function causing them to change phenotype, thus, developing a T-cell tolerance, inhibiting their proliferation, lowering their cytokine production, and hindering the recognition of tumor cells.
In addition to its immunopathogenesis role, the PD1/PD-L1 signaling pathway is recognized to play a key role in tumor intrinsic functions and survival.12 Autophagy is one example of these intrinsic functions affected by the PD-L1 ligand. The link between autophagy and the immune response is still not quite well understood. Recent findings from experiments with murine melanoma cells and human ovarian cancer cells indicated that cells that express high levels of the PD-L1 receptor are more sensitive to autophagy inhibitors, as compared to cells that weakly express PD-L1.11 This finding provides potential opportunities for the use of autophagy inhibitors in PD-L1-overexpressing cells as a new avenue in cancer medicine. Table 1 summarizes the autophagy inhibitors presently available and their mechanisms of action. However, in an animal model of ovarian cancer, this autophagy inhibitor-sensitization effect due to PD-L1 receptors was not observed. This suggests cell-specific effects of PD-L1 signaling and warrants further experiments to explore mechanisms of PD-L1 signaling and autophagy in different cell types. This, in turn, will assist in guiding whether autophagy inhibitors in combination with anti-PD-L1 therapy will augment antitumor response clinically.12 Figure 1 summarizes the mechanisms of autophagy induced by the blockade of the PD-L1/PD1 axis.
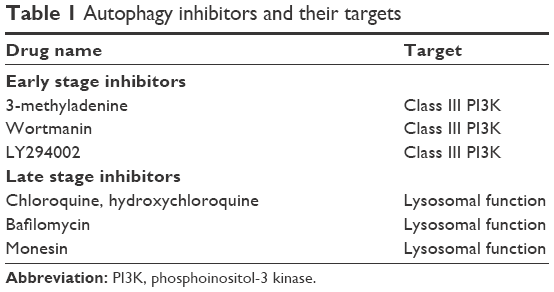 | Table 1 Autophagy inhibitors and their targets |
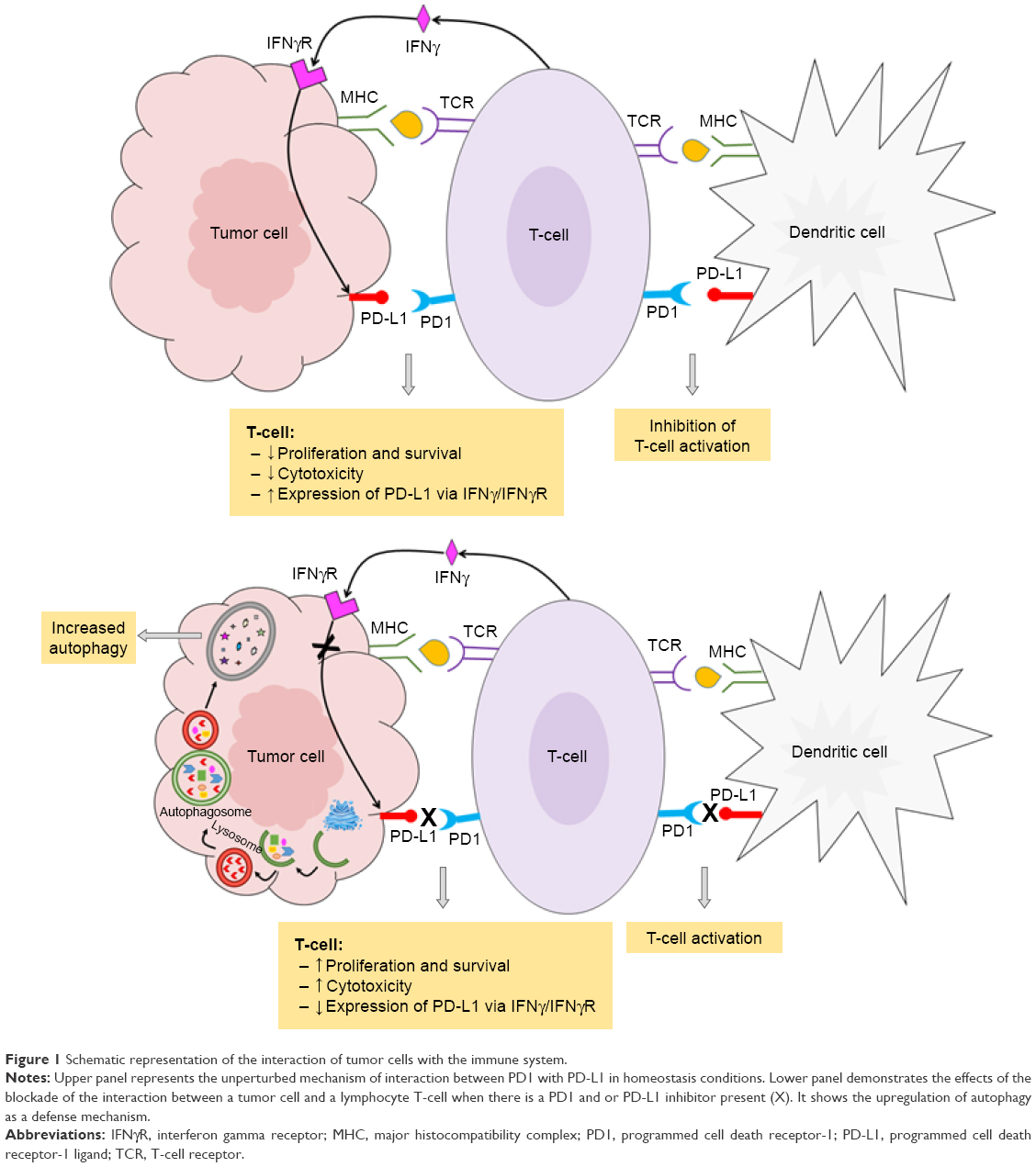 | Figure 1 Schematic representation of the interaction of tumor cells with the immune system. |
The effect of autophagy is seen not only in tumor cells but also in tumor-free cells interacting with it. For example, Parry et al13 demonstrated that PD1 engagement in T-cells inhibited glucose uptake and glycolytic rate through blockade of CD28-mediated nutrient metabolism. Depletion of glucose induces autophagy due to nutrient starvation via the AMP-activated protein kinase (AMPK) and mammalian target of rapamycin complex-1 (mTORC1) pathway.13,14 It was also shown that when tumors expressing indoleamine 2,3-dioxygenase (IDO) engage with T-cells, they cause immunosuppression, due to the generation of metabolites of tryptophan, 3-hydroxyantranylic acid (HAA), and quinolinic acid (QUIN). Tryptophan metabolite-mediated T-cell death is caused due to induction of apoptosis, however, it has also been hypothesized that depletion of tryptophan itself, an essential amino acid, may lead to autophagy in T-cells.15,16
Although autophagy inhibitors have been proven efficacious on their own, recent investigations have focused on their combinations with other chemotherapeutics in search of synergistic interactions with the prospect of cancer eradication.17 Given the dual role of autophagy in cancer, studies that hypothesize the rationale of using autophagy inhibitors are only useful if they target the prosurvival role of autophagy in tumor cells. For instance, Rao et al18 concluded from their studies that autophagy inhibits tumorigenesis at the beginning of tumor progression and reverses its role to facilitate tumor survival and progression with increasing immune suppression through PD-L1 with Atg5-regulated autophagy in KRASG12D-driven lung cancer. Table 2 summarizes some combination therapies and Table 3 summarizes the current US Food and Drug Administration (FDA)-approved PD1 and PD-L1 inhibitors.
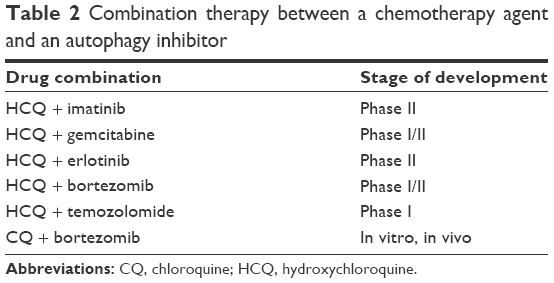 | Table 2 Combination therapy between a chemotherapy agent and an autophagy inhibitor |
 | Table 3 Summary of biologic (monoclonal antibodies) inhibitors of the PD-L1/PD1 axis |
Additionally, Yang et al19 demonstrated that CYT387, a Tank binding kinase-1 (TBK1) and Janus kinase (JAK) inhibitor, not only inhibited autophagy but also suppressed inflammation and feedback cytokines induced by KRAS-TBK1 signaling, pancreatic dysplasia, and PD-L1 expression, suggesting a potentially beneficial role of such type of autophagy inhibitors and their possible synergy with anti-PD-L1 therapy. In a preclinical study conducted on mice xenografted with human melanoma cancer, it was shown that the combination of anthracycline with autophagy inhibitors resulted in an enhanced antitumor T-cell response.20 The same study also demonstrated that autophagy inhibition did not have any major negative effect on the immune system. These results provide a novel opportunity in cancer therapy for prolonged and more efficacious combinatorial effects of anticancer immunotherapies such as with drugs targeting the PD-L1/PD1 axis with autophagy inhibitors.20
In contrast, however, a study by Peng et al indicated that the loss of phosphatase and tensin homologue (PTEN) in tumors was associated with reduced autophagy and subsequent reduction in T-cell infiltration and reduced T-cell-mediated tumor cell death. Their findings suggested that blockade of the PI3K-Akt pathway, more specifically by PI3Kβ inhibitors, would compensate the immunosuppressive effects of loss of PTEN-mediated autophagy reduction in tumor cells.21
Thus, although autophagy inhibitors provide opportunities for enhancing antitumor activity when used in combination with anti-PD-L1 agents, autophagy inhibition is only one component of a complex network of pathways that affect immune system-mediated tumor cell death, and further experiments are necessitated to discern the various scenarios in which autophagy inhibitors may prove to be beneficial in cancer therapy.
Conclusion
Cancer cells have several mechanisms to escape the immune system, proliferate, and survive. Among which, the most commonly observed are the activation of oncogenic pathways, the inhibition of tumor suppressors, and the alteration of tumor metabolic environment (ie, oxygen, pH, and nutrients) due to rapid cancer cell division. Therefore, tumors tend to adapt through different survival mechanisms to compensate for the deprivation of resources. One of these mechanisms is autophagy, which although beneficial to normal cells, in cancer conditions, it helps malignant cells to adjust and adapt to an unfavorable environment, and hence, develop and continue growing. Additionally, autophagy has an immunomodulatory effect. Blocking the PD-L1/PD1 axis via antibodies such as anti-PD1 or anti-PD-L1 triggers autophagy in tumor cells, which allows the recycling of nutrients and signals by the cells in the vicinity and the release of cytokines and extracellular vesicles that signal the ongoing damage in the malignant cell.11 Due to the versatile ways in which autophagy promotes tumor cell survival and decreases the effectiveness of chemotherapy by enhancing cancer cell metabolism and inhibiting its cell death pathway, there have been suggestions about using immune checkpoint inhibitors in combination with autophagy inhibitors. A hypothetical way in which this could result in a synergistic interaction is by combining a biologic such as anti-PD-L1 receptor or anti-PD1 causing an increase in autophagy with an autophagy inhibitor. However, more studies are necessary to guide this synergistic interaction particularly in the onset of tumor progression.
In summary, autophagy is a physiological phenomenon that researchers are presently extensively studying in the field of anticancer therapy due to the different mechanisms by which it promotes tumor survival and ability to decrease anticancer therapy effectiveness. A deeper understanding of how autophagy is regulated at the molecular level (ie, genetic, epigenetic, protein, metabolic, and posttranslational) and how autophagy can regulate itself plays a role in the immune system, and be regulated by drugs, extracellular components, and metabolites, may point to new therapeutic targets that can directly or indirectly modulate autophagy.
Acknowledgment
SAO would like to thank Tanaya R Vaidya and Yi Ting (Kayla) Lien for their help.
Disclosure
The authors report no conflicts of interest in this work.
References
Lorin S, Hamai A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Semin Cancer Biol. 2013;23(5):361–379. | ||
Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–734. | ||
Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. | ||
Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochim Biophys Acta. 2009;1793(9):1516–1523. | ||
Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–666. | ||
Brown C. Targeted therapy: an elusive cancer target. Nature. 2016;537(7620):S106–S108. | ||
Blank C, Brown I, Peterson AC, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64(3):1140–1145. | ||
Ishida M, Iwai Y, Tanaka Y, et al. Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol Lett. 2002;84(1):57–62. | ||
Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy – inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012;18(24):6580–6587. | ||
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Clark CA, Gupta HB, Sareddy G, et al. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 2016;76(23):6964–6974. | ||
Parry RV. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. | ||
Moruno F, Pérez-Jiménez E, Knecht E. Regulation of autophagy by glucose in mammalian cells. Cells. 2012;1(3):372–395. | ||
Lu B, Finn OJ. T-cell death and cancer immune tolerance. Cell Death Differ. 2008;15(1):70–79. | ||
Fallarino F, Grohmann U, Vacca C, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9(10):1069–1077. | ||
Viry E, Paggetti J, Baginska J, et al. Autophagy: an adaptive metabolic response to stress shaping the antitumor immunity. Biochem Pharmacol. 2014;92(1):31–42. | ||
Rao S, Tortola L, Perlot T, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. | ||
Yang S, Imamura Y, Jenkins RW, et al. Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol Res. 2016;4(6):520–530. | ||
Starobinets H, Ye J, Broz M, et al. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest. 2016;126(12):4417–4429. | ||
Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.