")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Understanding the Multiple Effects of PCBs on Lipid Metabolism
Authors Shan Q, Li H, Chen N, Qu F, Guo J
Received 24 June 2020
Accepted for publication 19 August 2020
Published 13 October 2020 Volume 2020:13 Pages 3691—3702
DOI https://doi.org/10.2147/DMSO.S264851
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Qiuli Shan,1,2,* Hongmei Li,1,* Ningning Chen,1,* Fan Qu,1 Jing Guo1
1College of Biological Science and Technology, University of Jinan, Jinan 250022, People’s Republic of China; 2State Key Laboratory of Environmental Chemistry and Eco-Toxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiuli Shan Email [email protected]
Abstract: Polychlorinated biphenyls (PCBs) are a typical class of environmental contaminants recently shown to be metabolism-disrupting chemicals. Lipids are a highly complex group of biomolecules that not only form the structural basis of biofilms but also act as signaling molecules and energy sources. Lipid metabolic disorders contribute to multiple diseases, including obesity, diabetes, fatty liver, and metabolic syndromes. Although previous literature has reported that PCBs can affect lipid metabolism, including lipid synthesis, uptake, and elimination, few systematic summaries of the detailed process of lipid metabolism caused by PCB exposure have been published. Lipid metabolic processes involve many molecules; however, the key factors that are sensitive to PCB exposure have not been fully clarified. Here, we summarize the recent developments in PCB research with a focus on biomarkers of lipid metabolic disorders related to environmental exposures.
Keywords: PCBs, metabolism-disrupting chemicals, hepatocytes, lipid metabolism
Introduction
Polychlorinated biphenyls (PCBs) are a group of chlorinated hydrocarbons that were widely produced and used in electrical equipment, building materials and other industrial applications in the 1930s–1970s.1 Although the production of PCBs was banned worldwide by the Stockholm Convention in 2001 because of concerns about their risks to human health and the environment, a total of 1.3 million tons of PCBs was manufactured during this time, including 209 different PCB congeners.2 PCBs are lipid-soluble and resistant to biodegradation, which enables them to persist in various environmental media, including air, soil, and water, and they are even biologically amplified through the food chain.3–5 In addition, unintentional releases of PCBs from old electrical equipment have recently increased in various countries, particularly in China.6 Therefore, environmental and health issues caused by PCBs are still a matter of concern.
Previous studies showed that PCBs have been classified as metabolism-disrupting chemicals (MDCs).7 The liver is known to be the main site of metabolism and is an important detoxifying organ in the body. Thus, hepatocytes are exposed to high concentrations of chemicals, and the subsequent occurrence of liver metabolism damage induced by PCBs has become a growing issue.8,9 For example, existing evidence from animal and human cell studies showed that PCB exposure could cause glucose and lipid metabolic disorders in the liver, thus initiating the onset of chronic systemic metabolic disorders, such as obesity, type 2 diabetes, fatty liver disease, cardiovascular disease and cancer.10–14 In fact, in addition to acting directly on the target organ, most PCBs that enter the body through breathing, drinking water and the diet can be stored in the liver and adipose tissue.15 When metabolic processes are disrupted in the body-especially, lipid metabolism-PCBs stored in adipose tissue are released into the blood circulation, potentially exposing the individual to various known adverse health effects for a second time.16,17 Therefore, it is important to understand the health risks of PCBs by assessing the effect of PCB exposure on lipid metabolism homeostasis.
In this paper, we summarize recent evidence that PCB exposure alters lipid metabolism and its underlying mechanisms in the livers of humans and rodents. We aim to elucidate the specific molecular mechanisms responsible for PCB-induced disruption of lipid metabolism and identify the related targets of PCB exposure. Our hope is that this paper will provide a therapeutic strategy in the treatment of PCB intoxication.
Mechanisms by Which PCBs Affect Hepatic Lipid Accumulation
Lipids are hydrophobic biomolecules that can be broadly divided by their chemical composition into three main categories: simple lipids (eg, triglycerides, TGs), compound lipids (eg, sphingolipids) and derived lipids (eg, cholesterol).18 However, most lipids are stored in adipose tissue in the form of the simple lipid TG. There are two main sources of lipids in the body: dietary intake and in vivo synthesis. Dietary lipids need to be broken down and resynthesized in the small intestine, while in vivo, hepatocytes have the strongest ability to synthesize and release lipids.19 Generally, lipids receive less attention in the life sciences than do other biomolecules, such as proteins and nucleic acids. However, lipids represent a complex group of biomolecules that play a vital role in a variety of biological processes, including cell composition, signal transduction, energy storage, and hormone production.20 Currently, the roles of lipids in different tissues have been extensively studied, and the results (as shown in Figure 1) show several adverse effects such as disordered energy homeostasis, including proinflammatory effects, proapoptotic effects, and dyslipidemia.21–26 Thus, lipid metabolism homeostasis is also vital to life.
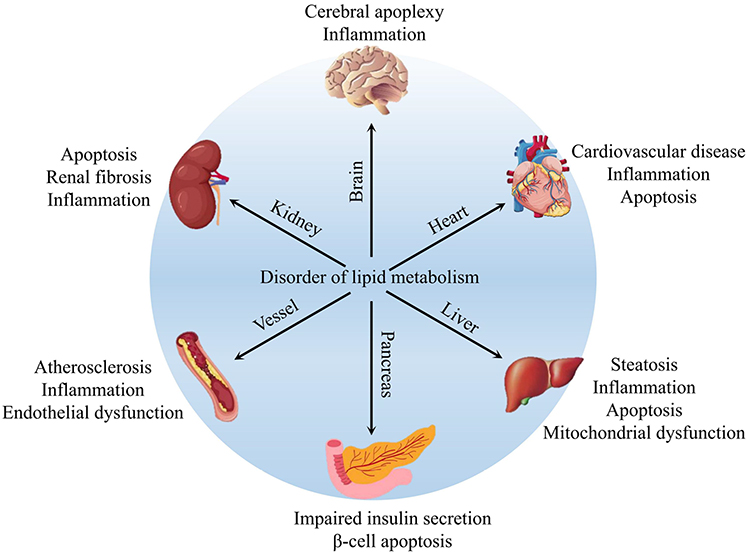 |
Figure 1 The relationship between lipid metabolic disorders in different organs and tissues. |
It is well recognized that disturbances in lipid metabolism homeostasis can be attributed to the imbalance of lipid acquisition and removal.27–29 The lipid acquisition process usually involves free FA uptake from blood by hepatocytes and de novo lipogenesis. The lipid removal process generally involves mitochondrial FAs oxidation and export as a component of VLDL particles. Therefore, PCB exposure-induced dysregulation of lipid metabolism homeostasis is also closely related to changes in these processes.
Effects of PCBs on de novo Lipogenesis
De novo lipogenesis refers to the incorporation of non-esterified FAs derived from glucose into TG synthesis within the liver.30 Although many cells and tissues still rely on lipid intake to meet survival requirements, numerous studies have demonstrated that de novo lipogenesis plays a substantial role in the pathogenesis of metabolic diseases-for example, 26% of TGs arise from de novo lipogenesis in Nonalcoholic fatty liver disease (NAFLD) patients.31 De novo lipogenesis is regulated by a variety of transcription factors, such as SREBP-1, ChREBP, LXRs, AhR and PXR, and ACC and FASN. However, available evidence suggests that PCB exposure only interferes with lipid metabolism by affecting the expression of some nuclear transcription factors and enzymes.
Nuclear Transcription Factors
The toxicity of PCBs often depends on their interaction with a variety of cellular receptors.32,33 PCBs have been classified as either dioxin-like or nondioxin-like based on whether they activate the AhR.34 Thus, AhR is a typical nuclear receptor of dioxin-like PCBs that has attracted widespread attention. Under normal physiological conditions, AhR is present in the cytoplasm in the form of nonactivated protein compounds. Upon binding to various exogenous agonists, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), dioxin-like PCBs, and benzo[a]pyrene (BaP), AhR is activated and then translocated into the nucleus, where it dimerizes with ARNT and binds to specific enhancer sequences adjacent to target promoters.35 It has been reported that the expressions of various AhR targets involved in detoxification metabolism and antioxidant defense, such as Phase I metabolic enzyme genes (Cyp1a1, Cyp1a2, Cyp1b1, and Cyp4 family), Phase II metabolic enzyme genes (GST), and antioxidant enzymes (SOD), are significantly induced by PCBs33,36,37 (Figure 2). Interestingly, even though the expression of AhR was reduced in mice exposed to Aroclor 1260 (PCB mixture, reduced 3.3-fold) and PCB 126 (reduced 3.2-fold), the transcriptional activity of AhR was not affected, as indicated by the increased expression of the AhR target gene Cyp1a2.32 Recent studies using AhR−/− mice have revealed that AhR deficiency can protect against high-fat diet (HFD)-induced obesity, hepatic steatosis, insulin resistance and inflammation.38 Additionally, Boverhof et al39 have shown that C57BL/6 mice treated with an AhR agonist (TCDD) for 7 days experienced lipid accumulation in their liver tissues. These results show that AhR is involved not only in the metabolism of xenobiotics but also in energy metabolism.
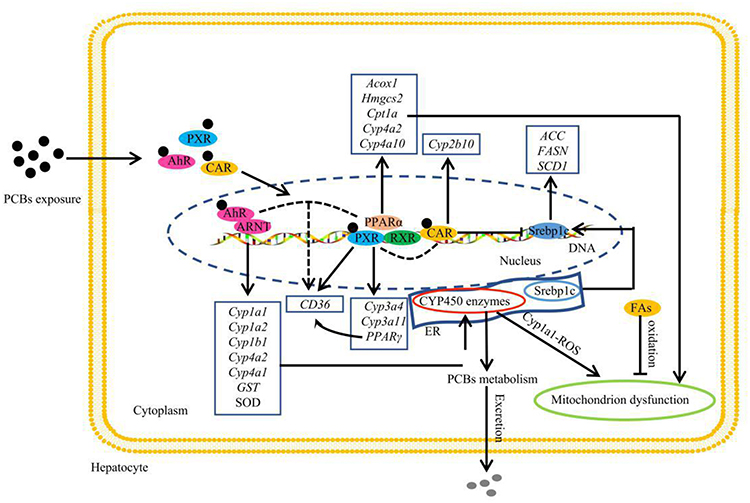 |
Figure 2 Effect of PCB156 exposure on nuclear receptors in hepatocytes. |
In fact, of all the target genes, Cyp1a1 is most strongly induced by AhR,40 and its coded enzymes are localized to the endoplasmic reticulum. Cyp1a1 can directly hydroxylate or oxidize PCBs that can then be secreted.41 On the other hand, PCBs have been shown to increase ROS primarily through a Cyp1a1 uncoupling-mediated mechanism.42 The increased ROS can damage biofilm structures and cause mitochondrial dysfunction, which causes the main FA oxidation pathways to be blocked in cells (Figure 2). In addition, Kawano et al43 previously reported that AhR activation significantly increased the levels of TGs and six long-chain monounsaturated FAs in the livers of mice by enhancing the expression of FAT/CD36, thus resulting in hepatic microvesicular steatosis. However, FAT/CD36 is not a direct target gene of AhR, and the AhR-induced activation of FAT requires other mediators, such as the reported PPARs.43 Interestingly, the upregulation of FAT/CD36 was also observed in the PCB-induced AhR and PPAR activation pathways.32,44 Together, these results further confirm that AhR plays an important role in PCB-induced lipid metabolism disorder.
PXR and CAR are two other xenobiotic receptors that regulate the expression of phase I and phase II metabolic enzyme systems.45 Non-dioxin-like PCBs may act as ligands of some nuclear receptors other than AhR, such as CAR and PXR.46 After PCB exposure, the expressions of the nuclear receptors PXR and CAR and their downstream genes Cyp3a4, Cyp3a11 (PXR target) and Cyp2b10 (a CAR target) were also obviously dysregulated in cells and animal models32,44,47,48 (Figure 2). Similar to the mechanism of action of AhR, ligand binding to PXR and CAR induces the translocation of these receptors to the nucleus. Subsequently, PXR and CAR enter the nucleus and form isodipolymers by binding with RXR, thus inducing the expression of Cyp3a or Cyp2b family members.49,50 In addition, PXR can dimerize with PPARα and induce the expression of Cyp3a subfamily members. Recently, PXR and CAR have also been considered as emerging new regulators of hepatic energy metabolism that connect sensing the chemical environment with metabolic health issues.51,52 Specifically, PXR activation mediates lipid accumulation, and its potential mechanisms most likely involve increased hepatic FA uptake by activating CD36. Moreover, in the same study, it was found that PXR also induced the expression of PPARγ, which is another positive regulator of CD36, suggesting that PXR can regulate CD36 directly or through the activation of PPARγ to increase the flow of FAs into hepatocytes53 (Figure 2). However, available evidence suggests that there is coordination between PXR and CD36 expression in PCB-induced steatosis, but the mechanism still requires further study.48
Unlike the activation of PXR, the activation of CAR may reduce lipid levels by interacting with Srebp154 (Figure 2). Although CAR has been proposed as a potential therapeutic target for lipid metabolic disease, some barriers exist for the clinical use of its agonists, and CAR also interacts with PPAR to regulate lipid and glucose homeostasis.50,51 Moreover, there is a complex relationship between CAR and PXR in PCB-induced metabolic disorders. For example, Aroclor 1260 could induce CAR and Cyp2b10 expression in wild-type mice, and CAR and Cyp2b10 expressions were significantly induced in PXR−/− mice.55 However, the presence of both PXR and CAR is required for Aroclor 1260 to induce NASH.55
PPARs are another crucial type of PCB-activated nuclear transcription factor superfamily and include PPARα, PPARβ/δ, and PPARγ. PPARs can form heterogenous dipolymers with retinol-type X receptors and then bind to DNA to regulate target gene transcription. A great deal of research has been done on the gene distribution and functional characteristics of this family.56–59 Therefore, a brief introduction is provided here. PPARα is most prominently expressed in hepatocytes, muscle and cardiomyocytes. PPARα is known to be the master regulator of β-oxidation, and it controls the expression of enzymes for FA oxidation in peroxisomes, mitochondria and the endoplasmic reticulum (microsomes). However, the exclusive effects of PCBs on PPARα-mediated FA oxidation are ambiguous. In male Sprague-Dawley (SD) rats, PCB 126 reduced the expressions of hepatic PPARα and its transcriptional targets, such as Acox1 and Hmgcs2.44 In the same study, other transcriptional targets of PPARα, such as Cpt1a and Cyp4a2, which are involved in mitochondrial and microsomal FA oxidation, were not decreased. Moreover, in C57BL/6 mice, PPARα and Cpt1a expressions were also significantly decreased in the HFD+PCB 153 group.13 But the expressions of PPARα, Cpt1α, and Cyp4a10 were significantly increased in PCB 126- or Aroclor 1260-exposed mice.32,44,55 Together, these results show that PPARα pathway disorders caused by PCB exposure play an important role in PCB-induced lipid metabolic disorders.
PPARβ/δ is expressed ubiquitously and is similar to PPARα in promoting FA oxidation in metabolic tissues such as the skeletal muscle, liver and heart. Compared to what is known about PPARα and PPARγ, there is currently less research on the effects of PCBs on PPARβ/δ. Only two papers have reported a reduction in PPARβ/δ in Aroclor 1260-treated mice and in PCB 156-treated hepatocytes.60,61 In contrast, it has been extensively shown that exposure to PCBs, such as PCB 126 and PCB 77, can induce PPARγ expression in the livers of mice.11,62 PPARγ is expressed predominantly in adipose tissue and the immune system. PPARγ plays an important role in increasing insulin sensitivity, as well as in promoting FA uptake by adipocytes and adipocyte differentiation, and the net effect of these processes is an increase of TG storage in adipocytes. Importantly, studies have shown that PPARγ responds differently to PCBs in different liver pathological states. For example, in normal mice, increased PPARγ levels were observed in PCB 126-induced NAFLD,11 while PPARγ was downregulated in the PCB 126-induced liver injury group.63 In any case, PPARγ is likely to be one of the key factors in PCB-induced lipid disorders. The role of PPAR in PCB-induced lipid metabolism disorders is shown in Figure 2.
SREBPs and ChREBP are major nuclear transcription factors that regulate cellular lipid homeostasis. However, the current study focused on the effects of PCB exposure on SREBPs, and less information is known about ChREBP. SREBPs are composed of three major isoforms: Srebp1a, Srebp1c and Srebp2. The expression of Srebp1c, but not Srebp1a or Srebp2, is significantly induced by PCB exposure.9 Srebp1a and Srebp1c are isoforms of the same gene, Srebp1. Unactivated Srebp1c is usually bound to the membrane of the ER and is then cleaved to generate the mature active forms, which translocate to the nucleus to upregulate the expression of enzymes involved in the biosynthesis of FA and TG, such as ACC, FASN, and SCD164,65 (Figure 2). Aroclor 1260 is a mixture of PCBs that has demonstrated robust effects on the progression of steatohepatitis in models of obesity and NAFLD. C57BL/6 mice treated with Aroclor 1260 for 12 weeks had increased Srebp1c expression, especially compared to that produced with a HFD alone, and Aroclor 1260 promoted the development of steatohepatitis via the induction of Srebp1c mRNA in a dose-dependent manner.48 In addition, hepatic lipid deposition induced by PCB 1269 or PCB 15366 was also related to the induction of Srebp1c expression (Figure 2). The roles of all the nuclear transcription factors in PCB-induced lipid metabolism disorder are shown in Table 1.
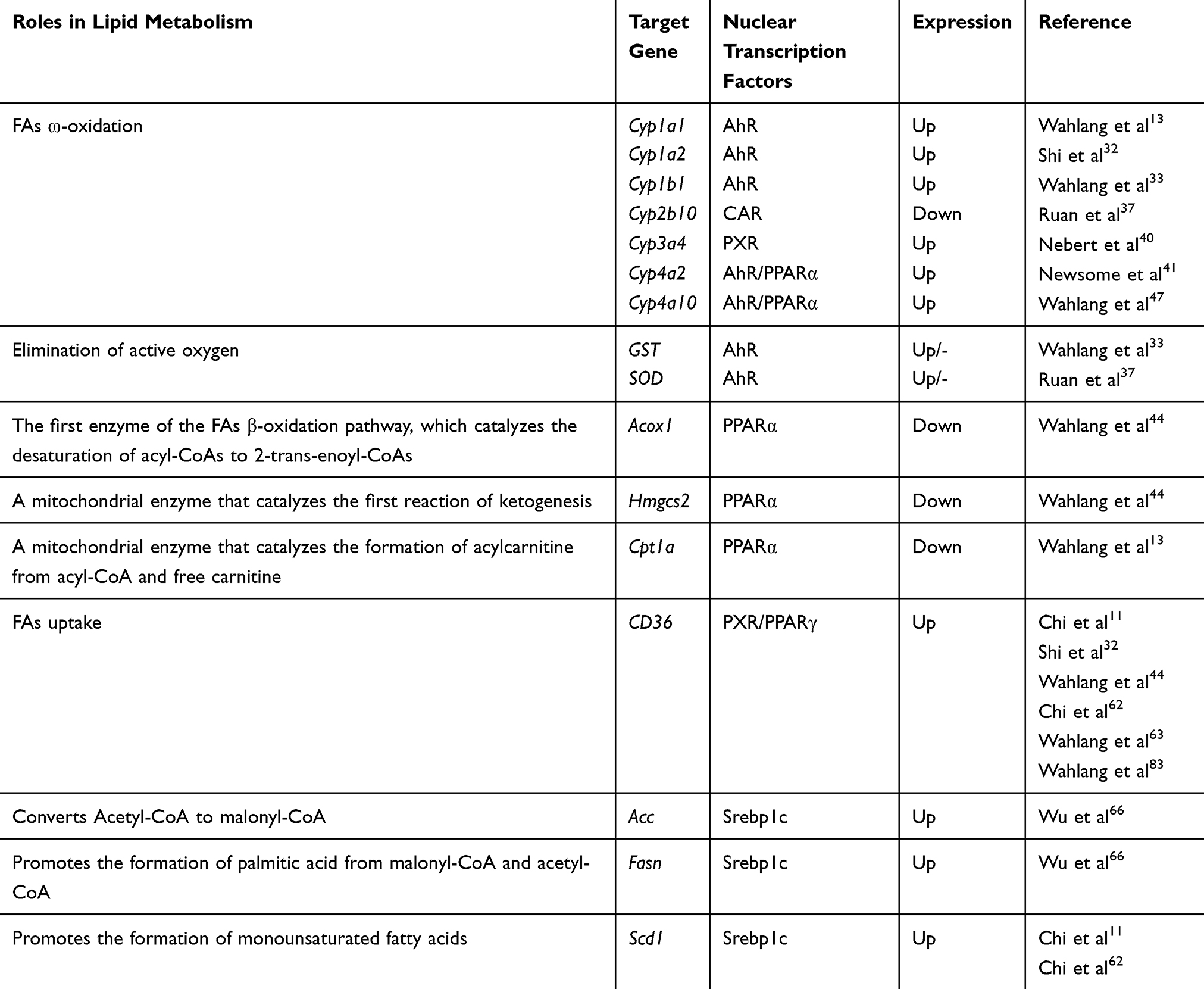 |
Table 1 The Roles of Nuclear Transcription Factors in PCB-Induced Lipid Metabolism Disorder |
Lipogenic Enzymes
Enzymes are one of the most critical factors that can be directly involved in lipid metabolism. Thus, changes in lipogenic enzyme activity have long been considered one of the key mechanisms involved in lipid metabolism disorders caused by PCBs. In de novo lipogenesis, pyruvate generated by glycolysis first enters mitochondria and participates in the TAC, thus producing the FA synthesis precursor acetyl-CoA. Acetyl-CoA is then catalyzed to malonyl-CoA under Acc, initiating FA synthesis (Figure 3). Finally, FAs that have been synthesized from acetyl-CoA or taken up from the blood are involved in the biosynthesis of TGs by sn-glycerol-3-phosphate (G3P) or monoacylglycerol (MAG). Notably, the G3P pathway is dominant in the liver, whereas the MAG pathway is essential for the biosynthesis of TGs in the small intestine. In this paper, we only focused on the G3P pathway of TG synthesis.
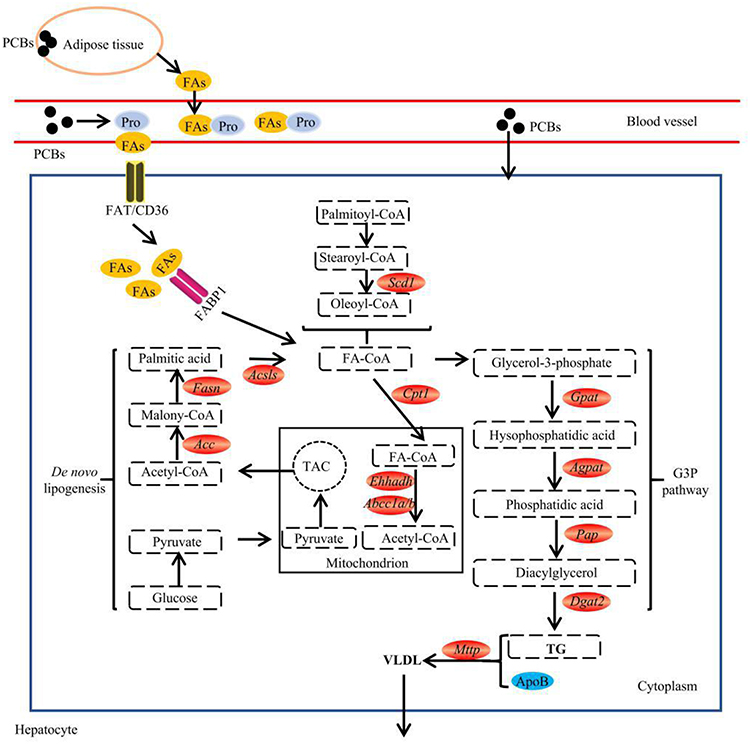 |
Figure 3 Effect of PCB156 exposure on lipid metabolism-related enzymes. |
Acc is a key rate-controlling enzyme in de novo FA lipogenesis, and there are two isoforms of Acc in rodents and humans: Acc1 and Acc2. Acc1 is highly expressed in hepatocytes and adipocytes, whereas Acc2 is mainly associated with the heart and skeletal muscle. It has been reported that inhibition of both Acc1 and Acc2 reduces hepatic TGs and insulin resistance in rat models.67 In addition to Acc, Fasn catalyzes the last step in the FA biosynthetic pathway, in which malonyl-CoA is converted to palmitic acid. Once fatty acyl-CoA synthetase-mediated activation occurs, palmitic acids are either incorporated into TAG or broken down through a series of mitochondrial β-oxidation processes into acetyl-CoA. After PCB exposure, the mRNA levels of Acc and Fasn are significantly induced at the same time in AML-12 hepatocytes and 3T3-L1 preadipocytes of mice.66 In addition, PCB exposure affects the structure of Scd1. Previous studies have shown that Scd1 is a microsomal enzyme that can catalyze the synthesis of monounsaturated long-chain FAs from saturated FAs.68 The formation of unsaturated fatty acids is a key factor involved in important physiological metabolic processes for TG synthesis. In PCB-induced steatosis, the expression of Scd1 is always significantly induced11,62 (Figure 3).
In the G3P pathway, G3P is converted to DAG under the action of GPAT, AGPAT, and PAP (Figure 3). Recent studies have shown that PCB exposure can interfere with the gene expression of the above mentioned enzymes by affecting the expression of the core rhythm genes in the liver.69 The final step in TG synthesis requires Dgat, which is the rate-limiting enzyme in TG synthesis. Dgat exists in two isoforms: Dgat1 and Dgat2. Experimental evidence suggests that Dgat1 knockout mice have 50% less TG in their tissues, and Dgat2 knockout mice die shortly after birth because of severe lipopenia.68,70 However, it has been recently reported that Dgat inhibition increased the hepatic free FA levels and exacerbated hepatic oxidative damage and hepatocyte death, thereby promoting the progression from steatosis to fibrosis.71 Interestingly, PCB 126 exposure could induce Dgat1 and Dgat2 expression in the livers of mice fed a control diet.11 However, in the MCD diet-induced liver injury, PCB 126 significantly reduced Dgat2 expression in the livers of mice.63 Thus, the effect of PCBs on Dgat may be associated with the liver condition (Figure 3).
Effects of PCB Exposure on the Uptake of Hepatic Nonesterified FAs
FAs are a group of molecules consisting of long hydrocarbon chains of different lengths (carbon atoms) and saturation levels (double bond numbers). Within the cell, FAs are one of the most important raw materials for lipid synthesis.19,72 Generally, there are two sources of FAs: glycolysis and plasma. However, FAs derived from glycolysis were introduced in the lipogenic enzyme section, and the primary discussion here will be FAs derived from plasma. Donnelly et al31 demonstrated that approximately 60% of hepatic TGs in human subjects are derived from nonesterified fatty acids in the plasma. Importantly, PCBs have been reported to accelerate steatosis or worsen diet-induced steatosis by promoting hepatic FA uptake from plasma.11,32,62 In fact, increased plasma FAs are due to increased FA release from adipose tissue. Moreover, the increased fat mass directly contributes to increased FA release from adipose tissue.73 FAs are poorly soluble in blood and are mainly present bound to proteins such as albumin.74 Therefore, FAs need to be separated from the albumin to which they are bound before entering the cell. Subsequently, FAs diffuse to the membrane surface, either binding with receptors on the membrane surface or inserting directly into the lipid bilayer.75 It is now clear that the main transporters involved in FA transmembrane translocation are caveolins, FATPs, FAT/CD36 and FABPs.28,59 Interestingly, available experimental evidence shows that PCB exposure increases FA intake mostly through FAT/CD36 and FABPs33 (Figure 3).
FAT/CD36
FAT/CD36 is an 80 kD membrane protein with heavy glycosylation. FAT/CD36 can be expressed in a wide range of cells, such as microvascular endothelial cells, immune cells, adipocytes and myocytes.76 In 1993, Abumrad et al77 first found that FAT/CD36 promotes cell intake of FAs. This role was then well demonstrated in rodents and humans.78,79 FAT/CD36 can accelerate FA dissociation from albumin and catalyze the integration of protonated FAs into the outer phospholipid bilayer of the plasma membrane, promoting the intake of FAs in cells.59 Therefore, multiple findings suggest that increased FAT/CD36 activity may be critical for the development of steatosis.80–82 Interestingly, mice that were administered the PCB mixture Aroclor 1260 had increased CD36 expression.32,44,48,55,83 Moreover, treatment with individual congeners (eg, PCB 126, PCB 77 or PCB 156) has also been found to increase CD36 expression in mice or cells.11,32,44,62,63,83
FABPs
In parallel with the increase in CD36, PCB exposure also increases the gene expression of FABPs.32 FABPs are another highly expressed protein superfamily that facilitates the transport of FAs and other lipid mediators across cellular membranes. In fact, FABPs are mainly located in the cytoplasm, where they may facilitate the transport of FAs from the cytoplasm to their nuclear receptors, thus controlling the transcription of downstream target genes.59 FABPs include nine different members (FABP1, 2, 3, 5, 6, 7, 8 and 9), and PCB exposure mainly increases the expression of FABP1,32,83 which is also known as L-FABP and is primarily expressed in the liver, small intestine, pancreas, and kidney. Exposure to both Aroclor 1260 and PCB 126 increases the expression of FABP1 in the livers of mice.32 Furthermore, 1 week after an acute injection of PCB 126 in rats, the hepatic gene and protein levels of FABP1 were higher than those in the corn oil group.84 A summary of the effects of PCBs on fatty acid intake is shown in Figure 3.
FA Oxidative Pathways
FA oxidation, especially mitochondrial FA β-oxidation, is a crucial pathway of aerobic ATP production in mammalian organisms. PPARα is known as the master regulator of β-oxidation and controls the expression of enzymes for FA oxidation in mitochondria. In the de novo lipogenesis section, we summarized the effects of PCB exposure on PPARα. In this section, we mainly analyze the effect of PCB exposure on the expression of key factors that are directly involved in the β-oxidation process. FAs that enter cells are first converted into acyl-CoA under the action of long-chain ACSLs and then transported to mitochondria by CPT1 for oxidation. CPT1 has been confirmed as a rate-limiting enzyme in the oxidation of FAs. Available literature has shown that PCB exposure reduces FA oxidation mainly by decreasing CPT1 expression.13,44,63,85 After acyl-CoA enters the mitochondrial matrix, an oxidation cycle that includes oxidation, hydration, dehydrogenation, and thiolytic cleavage occurs with the assistance of acyl-CoA oxidases, hydratase, 3-hydroxyacyl-CoA dehydrogenase, and peroxisomes harboring the D-bifunctional enzymes L-bifunctional enzyme (Ehhadh) and 3-ketoacyl CoA thiolase A/B (Acaa1a/b). Emerging transcriptomics evidence supports that PCBs could lead to the dysregulation of Ehhadh and Acaa1a/b expressions. These results showed that PCB exposure-induced diseases may be closely related to abnormal lipid oxidative processes (Figure 3).
In addition, some special oxidation pathways especially, ω-oxidation are carried out in the endoplasmic reticulum in hepatocytes and require cytochrome P450 enzymes. Interestingly, cytochrome P450 enzymes are mostly downstream effectors for AhR, PXR or CAR, which are widely involved in the metabolism of xenobiotics, including PCBs. Thus, PCB exposure is also closely related to FA ω-oxidation through effects on the expression of CYP450 enzymes.32
TG Secretion
Hepatocytes can synthesize lipids using glucose and then form VLDL particles to transport lipids to peripheral tissues, including skeletal muscle, cardiac muscle and adipose tissue. In hepatocytes, each VLDL particle is composed of apolipoprotein B 100 (apoB 100), which is assembled with TG and cholesteryl esters. These constituents are then delivered to peripheral tissues when VLDL is converted by lipoprotein lipase to higher-density and smaller-sized atherogenic particles, including LDL and IDL. There is growing evidence that PCBs block hepatic VLDL secretion, and the corresponding mechanism may be related to the dysregulated expression of MTTP.9 The key event responsible for hepatic VLDL metabolism is the lipidation of apoB 100, and this process is known to be regulated by Mttp, which is an ER resident protein that has both apoB 100 binding and lipid transfer domains.59 A small quantity of TG is first transferred to apoB 100 in the ER by MTTP. The apoB then continues to bind with larger droplets of TG to form VLDL particles. Therefore, a decrease in Mttp expression hampers VLDL assembly, preventing liver lipids from being secreted. Furthermore, the results of experimental studies using animal or cell models support the hypothesis that reduced Mttp activity is sufficient to induce hepatic steatosis. In particular, a significant decrease in Mttp was also observed in PCB-induced hepatic lipid accumulation in rats,9,63,84 further indicating that PCBs causes hepatic lipid metabolic dysfunction (Figure 3).
Conclusions
During the past two decades, numerous studies have suggested that PCB exposure is significantly associated with disturbances in lipid metabolism. However, the pathogenesis of hepatic lipid metabolism disorders induced by PCBs is very complex and only partially understood. Based on existing evidence, the present review focused on the specific mechanisms by which PCB exposure is involved in lipid metabolism. Overall, the key events responsible for PCB-induced lipid accumulation are (i) increased lipid influx due to the upregulated expression of CD36 and Fabp1; (ii) increased lipogenesis due to the activated expression of AhR, CAR, PXR, Srebp1c, Acc, Fasn and Scd1; (iii) decreased FA oxidation due to the downregulated expression of PPARα; and (iv) decreased lipid efflux due to the reduced expression of Mttp. These key factors and mechanisms are summarized in Table 2. Although further research is needed, the elucidation of each of these steps will offer novel therapeutic options in the field of PCB-induced metabolic diseases in the coming years.
 |
Table 2 The Mechanisms of PCBs in Lipid Metabolism |
Abbreviations
Acaa1a/b, 3-ketoacyl CoA thiolase A/B; ACC, acetyl CoA carboxylase; Acox1, acyl-CoA oxidase 1; ACSLs, acyl-CoA synthetases; AGPAT, acylglycerol-3-phosphate acyltransferase; AhR, aryl hydrocarbon receptor; ARNT, AhR nuclear translocator; CAR, Constitutive androstane receptor; ChREBP, carbohydrate-responsive element binding protein; Cpt1a, carnitine palmitoyltransferase 1A; CYP450, cytochrome P450; DAG, diacylglycerol; Ehhadh, L-bifunctional enzyme; ER, endoplasmic reticulum; FATPs, FA transport proteins; Fas, fatty acids; FASN, FA synthase; FAT/CD36, fatty acid translocase; FMO3, flavin-containing dimethylaniline monoxygenase 3; G3P, glycerol-3-phosphate; Gpat, glycerol-3-phosphate acyltransferase; GST, glutathione S transferase; Hmgcs2, 3-hydroxy-3-methylglutaryl-CoA synthase 2; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LXRs, liver X receptors; MCD, methionine-choline deficient; MTTP, microsomal triglyceride transfer protein; NAFLD, nonalcoholic fatty liver disease; NASH, non-alcoholic-steatohepatitis; PAP, phosphatidic acid phosphatase; PPARs, peroxisome proliferator-activated receptors; PXR, pregnane X receptor; ROS, reactive oxygen species; RXR, retinoid X receptor; SCD1, stearoyl CoA desaturase-1; SOD, superoxide dismutase; TAC, tricarboxylic acid cycle; SREBP-1, sterol regulatory element binding protein-1 SREBP-1; VLDL, very low-density lipoprotein.
Funding
This work was funded under the National Natural Science Foundation of China (NSFC) [grant numbers 21507040] and the Natural Science Foundation of Shandong Province [grant numbers BS2015SW023].
Disclosure
The authors have no potential conflicts of interest to disclose.
References
1. Desforges JP, Hall A. Predicting global killer whale population collapse from PCB pollution. Science. 2018;361(6409):1373–1376.
2. Raffetti E, Donato F, Speziani F, Scarcella C, Gaia A, Magoni M. Polychlorinated biphenyls (PCBs) exposure and cardiovascular, endocrine and metabolic diseases: a population-based cohort study in a North Italian highly polluted area. Environ Int. 2018;120:215–222. doi:10.1016/j.envint.2018.08.022
3. Gao Q, Ben Y, Dong Z, Hu J. Age-dependent human elimination half-lives of dioxin-like polychlorinated biphenyls derived from biomonitoring data in the general population. Chemosphere. 2019;222:541–548.
4. Wu WL, Deng XL, Zhou SJ, et al. Levels, congener profiles, and dietary intake assessment of polychlorinated dibenzo-p-dioxins/dibenzofurans and dioxin-like polychlorinated biphenyls in beef, freshwater fish, and pork marketed in Guangdong Province, China. Sci Total Environ. 2018;615:412–421. doi:10.1016/j.scitotenv.2017.09.273
5. Yang L, Jin F, Liu G, et al. Levels and characteristics of polychlorinated biphenyls in surface sediments of the Chaobai river, a source of drinking water for Beijing, China. Ecotoxicol Environ Saf. 2019;109922.
6. Cui S, Fu Q, Ma WL, Song WW, Liu LY, Li YF. A preliminary compilation and evaluation of a comprehensive emission inventory for polychlorinated biphenyls in China. Sci Total Environ. 2015;533:247–255. doi:10.1016/j.scitotenv.2015.06.144
7. Heindel JJ, Blumberg B, Cave M, et al. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol. 2017;68:3–33. doi:10.1016/j.reprotox.2016.10.001
8. Armstrong LE, Guo GL. Understanding environmental contaminants’ direct effects on non-alcoholic fatty liver disease progression. Curr Environ Health Rep. 2019;6(3):95–104. doi:10.1007/s40572-019-00231-x
9. Boucher MP, Lefebvre C, Chapados NA. The effects of PCB126 on intra-hepatic mechanisms associated with non alcoholic fatty liver disease. J Diabetes Metab Disord. 2015;14(1):88. doi:10.1186/s40200-015-0218-2
10. Arsenescu V, Arsenescu R, Parulkar M, et al. Polychlorinated biphenyl 77 augments angiotensin II-induced atherosclerosis and abdominal aortic aneurysms in male apolipoprotein E deficient mice. Toxicol Appl Pharmacol. 2011;257(1):148–154. doi:10.1016/j.taap.2011.08.028
11. Chi Y, Lin Y, Lu Y, Huang Q, Ye G, Dong S. Gut microbiota dysbiosis correlates with a low-dose PCB126-induced dyslipidemia and non-alcoholic fatty liver disease. Sci Total Environ. 2019;653:274–282. doi:10.1016/j.scitotenv.2018.10.387
12. Loiola RA, Dos Anjos FM, Shimada AL, et al. Long-term in vivo polychlorinated biphenyl 126 exposure induces oxidative stress and alters proteomic profile on islets of Langerhans. Sci Rep. 2016;6(1):27882. doi:10.1038/srep27882
13. Wahlang B, Falkner KC, Gregory B, et al. Polychlorinated biphenyl 153 is a diet-dependent obesogen that worsens nonalcoholic fatty liver disease in male C57BL6/J mice. J Nutr Biochem. 2013;24(9):1587–1595. doi:10.1016/j.jnutbio.2013.01.009
14. Whysner J, Wang CX. Hepatocellular iron accumulation and increased cell proliferation in polychlorinated biphenyl-exposed sprague-dawley rats and the development of hepatocarcinogenesis. Toxicol Sci. 2001;62(1):36–45. doi:10.1093/toxsci/62.1.36
15. La Merrill M, Emond C, Kim MJ, et al. Toxicological function of adipose tissue: focus on persistent organic pollutants. Environ Health Perspect. 2013;121(2):162–169. doi:10.1289/ehp.1205485
16. Chevrier J, Dewailly E, Ayotte P, Mauriege P, Despres JP, Tremblay A. Body weight loss increases plasma and adipose tissue concentrations of potentially toxic pollutants in obese individuals. Int J Obes Relat Metab Disord. 2000;24(10):1272–1278. doi:10.1038/sj.ijo.0801380
17. Kim MJ, Marchand P, Henegar C, et al. Fate and complex pathogenic effects of dioxins and polychlorinated biphenyls in obese subjects before and after drastic weight loss. Environ Health Perspect. 2011;119(3):377–383. doi:10.1289/ehp.1002848
18. Bumpus TW, Baskin JM. Greasing the wheels of lipid biology with chemical tools. Trends Biochem Sci. 2018;43(12):970–983. doi:10.1016/j.tibs.2018.09.011
19. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2019.
20. Tumanov S, Kamphorst JJ. Recent advances in expanding the coverage of the lipidome. Curr Opin Biotechnol. 2017;43:127–133. doi:10.1016/j.copbio.2016.11.008
21. Wang SP, Yang H, Wu JW, Gauthier N, Fukao T, Mitchell GA. Metabolism as a tool for understanding human brain evolution: lipid energy metabolism as an example. J Hum Evol. 2014;77:41–49. doi:10.1016/j.jhevol.2014.06.013
22. Palomer X, Barroso E, Zarei M, Botteri G, Vazquez-Carrera M. PPARbeta/delta and lipid metabolism in the heart. Biochim Biophys Acta. 2016;1861(10):1569–1578. doi:10.1016/j.bbalip.2016.01.019
23. Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol. 2017;8(1):1–8.
24. Sunami Y, Rebelo A, Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers. 2017;10(1):3. doi:10.3390/cancers10010003
25. Zhao L, Varghese Z, Moorhead JF, Chen Y, Ruan XZ. CD36 and lipid metabolism in the evolution of atherosclerosis. Br Med Bull. 2018;126(1):101–112. doi:10.1093/bmb/ldy006
26. Gai Z, Wang T, Visentin M, Kullak-Ublick GA, Fu X, Wang Z. Lipid accumulation and chronic kidney disease. Nutrients. 2019;11(4):722. doi:10.3390/nu11040722
27. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. 2019;76(1):99–128.
28. Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. doi:10.1007/s00535-013-0758-5
29. Knebel B, Fahlbusch P, Dille M, et al. Fatty liver due to increased de novo lipogenesis: alterations in the hepatic peroxisomal proteome. Front Cell Dev Biol. 2019;7:248. doi:10.3389/fcell.2019.00248
30. Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism. 2014;63(7):895–902. doi:10.1016/j.metabol.2014.04.003
31. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi:10.1172/JCI23621
32. Shi H, Jan J, Hardesty JE, et al. Polychlorinated biphenyl exposures differentially regulate hepatic metabolism and pancreatic function: implications for nonalcoholic steatohepatitis and diabetes. Toxicol Appl Pharmacol. 2019;363:22–33.
33. Wahlang B, Hardesty JE, Jin J, Falkner KC, Cave MC. Polychlorinated biphenyls and nonalcoholic fatty liver disease. Curr Opin Toxicol. 2019;14:21–28. doi:10.1016/j.cotox.2019.06.001
34. Safe S, Bandiera S, Sawyer T, et al. PCBs: structure-function relationships and mechanism of action. Environ Health Perspect. 1985;60:47–56.
35. Larigot L, Juricek L, Dairou J, Coumoul X. AhR signaling pathways and regulatory functions. Biochim Open. 2018;7:1–9.
36. Petriello MC, Hoffman JB, Sunkara M, et al. Dioxin-like pollutants increase hepatic flavin containing monooxygenase (FMO3) expression to promote synthesis of the pro-atherogenic nutrient biomarker trimethylamine N-oxide from dietary precursors. J Nutr Biochem. 2016;33:145–153. doi:10.1016/j.jnutbio.2016.03.016
37. Ruan J, Guo J, Huang Y, Mao Y, Yang Z, Zuo Z. Adolescent exposure to environmental level of PCBs (aroclor 1254) induces non-alcoholic fatty liver disease in male mice. Environ Res. 2019;108909.
38. Xu CX, Wang C, Zhang ZM, et al. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int J Obes (Lond). 2015;39(8):1300–1309. doi:10.1038/ijo.2015.63
39. Boverhof DR, Burgoon LD, Tashiro C, et al. Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in sprague dawley rats and C57BL/6 mice. Toxicol Sci. 2006;94(2):398–416. doi:10.1093/toxsci/kfl100
40. Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59(1):65–85. doi:10.1016/S0006-2952(99)00310-X
41. Newsome BJ, Petriello MC, Han SG, et al. Green tea diet decreases PCB 126-induced oxidative stress in mice by up-regulating antioxidant enzymes. J Nutr Biochem. 2014;25(2):126–135. doi:10.1016/j.jnutbio.2013.10.003
42. Lim EJ, Majkova Z, Xu S, et al. Coplanar polychlorinated biphenyl-induced CYP1A1 is regulated through caveolae signaling in vascular endothelial cells. Chem Biol Interact. 2008;176(2–3):71–78. doi:10.1016/j.cbi.2008.08.007
43. Kawano Y, Nishiumi S, Tanaka S, et al. Activation of the aryl hydrocarbon receptor induces hepatic steatosis via the upregulation of fatty acid transport. Arch Biochem Biophys. 2010;504(2):221–227. doi:10.1016/j.abb.2010.09.001
44. Wahlang B, Perkins JT, Petriello MC, Hoffman JB, Stromberg AJ, Hennig B. A compromised liver alters polychlorinated biphenyl-mediated toxicity. Toxicology. 2017;380:11–22. doi:10.1016/j.tox.2017.02.001
45. Tolson AH, Wang H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev. 2010;62(13):1238–1249. doi:10.1016/j.addr.2010.08.006
46. Al-Salman F, Plant N. Non-coplanar polychlorinated biphenyls (PCBs) are direct agonists for the human pregnane-X receptor and constitutive androstane receptor, and activate target gene expression in a tissue-specific manner. Toxicol Appl Pharmacol. 2012;263(1):7–13. doi:10.1016/j.taap.2012.05.016
47. Wahlang B, Falkner KC, Clair HB, et al. Human receptor activation by aroclor 1260, a polychlorinated biphenyl mixture. Toxicol Sci. 2014;140(2):283–297. doi:10.1093/toxsci/kfu083
48. Wahlang B, Song M, Beier JI, et al. Evaluation of aroclor 1260 exposure in a mouse model of diet-induced obesity and non-alcoholic fatty liver disease. Toxicol Appl Pharmacol. 2014;279(3):380–390. doi:10.1016/j.taap.2014.06.019
49. Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23(5):687–702.
50. Xu P, Zhai Y, Wang J. The role of PPAR and its cross-talk with CAR and LXR in obesity and atherosclerosis. Int J Mol Sci. 2018;19(4):1260. doi:10.3390/ijms19041260
51. Xiao L, Xie X, Zhai Y. Functional crosstalk of CAR–LXR and ROR–LXR in drug metabolism and lipid metabolism. Adv Drug Deliv Rev. 2010;62(13):1316–1321. doi:10.1016/j.addr.2010.07.006
52. Zhou J, Febbraio M, Wada T, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134(2):556–567. doi:10.1053/j.gastro.2007.11.037
53. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241–252. doi:10.1016/S0092-8674(00)81575-5
54. Roth A, Looser R, Kaufmann M, et al. Regulatory cross-talk between drug metabolism and lipid homeostasis: constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Mol Pharmacol. 2008;73(4):1282–1289. doi:10.1124/mol.107.041012
55. Wahlang B, Prough RA, Falkner KC, et al. Polychlorinated biphenyl-xenobiotic nuclear receptor interactions regulate energy metabolism, behavior, and inflammation in non-alcoholic-steatohepatitis. Toxicol Sci. 2016;149(2):396–410. doi:10.1093/toxsci/kfv250
56. Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137(1):354–366. doi:10.1210/endo.137.1.8536636
57. Drew PD, Xu J, Storer PD, Chavis JA, Racke MK. Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int. 2006;49(2):183–189. doi:10.1016/j.neuint.2006.04.003
58. Liss KHH, Finck BN. PPARs and nonalcoholic fatty liver disease. Biochimie. 2017;136:65–74. doi:10.1016/j.biochi.2016.11.009
59. Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog Lipid Res. 2009;48(1):1–26. doi:10.1016/j.plipres.2008.08.001
60. Hardesty JE, Wahlang B, Falkner KC. Proteomic analysis reveals novel mechanisms by which polychlorinated biphenyls compromise the liver promoting diet-induced steatohepatitis. J Proteome Res. 2019;18(4):1582–1594. doi:10.1021/acs.jproteome.8b00886
61. Chen N, Shan Q, Qi Y, Liu W, Tan X, Gu J. Transcriptome analysis in normal human liver cells exposed to 2, 3, 3ʹ, 4, 4ʹ, 5 - hexachlorobiphenyl (PCB 156). Chemosphere. 2019;239:124747. doi:10.1016/j.chemosphere.2019.124747
62. Chi Y, Wang H, Lin Y, et al. Gut microbiota characterization and lipid metabolism disorder found in PCB77-treated female mice. Toxicology. 2019;420:11–20. doi:10.1016/j.tox.2019.03.011
63. Wahlang B, Barney J, Thompson B, et al. Editor’s highlight: PCB126 exposure increases risk for peripheral vascular diseases in a liver injury mouse model. Toxicol Sci. 2017;160(2):256–267.
64. Ruiz R, Jideonwo V, Ahn M, et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J Biol Chem. 2014;289(9):5510–5517. doi:10.1074/jbc.M113.541110
65. Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99(5):846–854. doi:10.1172/JCI119248
66. Wu H, Yu W, Meng F, et al. Polychlorinated biphenyls-153 induces metabolic dysfunction through activation of ROS/NF-kappaB signaling via downregulation of HNF1b. Redox Biol. 2017;12:300–310. doi:10.1016/j.redox.2017.02.026
67. Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116(3):817–824. doi:10.1172/JCI27300
68. Dobrzyn P, Dobrzyn A, Miyazaki M, et al. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci U S A. 2004;101(17):6409–6414. doi:10.1073/pnas.0401627101
69. Shen X, Chen Y, Zhang J, et al. Low-dose PCB126 compromises circadian rhythms associated with disordered glucose and lipid metabolism in mice. Environ Int. 2019;128:146–157. doi:10.1016/j.envint.2019.04.058
70. Stone SJ, Myers HM, Watkins SM, et al. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J Biol Chem. 2004;279(12):11767–11776. doi:10.1074/jbc.M311000200
71. Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45(6):1366–1374. doi:10.1002/hep.21655
72. Juarez-Hernandez E, Chavez-Tapia NC, Uribe M, Barbero-Becerra VJ. Role of bioactive fatty acids in nonalcoholic fatty liver disease. Nutr J. 2016;15(1):72. doi:10.1186/s12937-016-0191-8
73. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51(2):679–689. doi:10.1002/hep.23280
74. van der Vusse GJ. Albumin as fatty acid transporter. Drug Metab Pharmacokinet. 2009;24(4):300–307. doi:10.2133/dmpk.24.300
75. Glatz JF, Luiken JJ, van Nieuwenhoven FA, Van der Vusse GJ. Molecular mechanism of cellular uptake and intracellular translocation of fatty acids. Prostaglandins Leukot Essent Fatty Acids. 1997;57(1):3–9. doi:10.1016/S0952-3278(97)90485-3
76. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. 2014;34(1):281–303. doi:10.1146/annurev-nutr-071812-161220
77. Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268(24):17665–17668.
78. Coburn CT, Knapp FF
79. Hirano K, Kuwasako T, Nakagawa-Toyama Y, Janabi M, Yamashita S, Matsuzawa Y. Pathophysiology of human genetic CD36 deficiency. Trends Cardiovasc Med. 2003;13(4):136–141. doi:10.1016/S1050-1738(03)00026-4
80. Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology. 2016;157(2):570–585. doi:10.1210/en.2015-1866
81. Zhao L, Zhang C, Luo X, et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J Hepatol. 2018;69(3):705–717. doi:10.1016/j.jhep.2018.04.006
82. Ding D, Ye G, Lin Y, et al. MicroRNA-26a-CD36 signaling pathway: pivotal role in lipid accumulation in hepatocytes induced by PM2.5 liposoluble extracts. Environ Pollut. 2019;248:269–278. doi:10.1016/j.envpol.2019.01.112
83. Wahlang B, Jin J, Hardesty JE, et al. Identifying sex differences arising from polychlorinated biphenyl exposures in toxicant-associated liver disease. Food Chem Toxicol. 2019;129:64–76.
84. Chapados NA, Boucher MP. Liver metabolic disruption induced after a single exposure to PCB126 in rats. Environ Sci Pollut Res Int. 2017;24(2):1854–1861. doi:10.1007/s11356-016-7939-8
85. Gadupudi GS, Elser BA, Sandgruber FA, Li X, Gibson-Corley KN, Robertson LW. PCB126 inhibits the activation of AMPK-CREB signal transduction required for energy sensing in liver. Toxicol Sci. 2018;163(2):440–453. doi:10.1093/toxsci/kfy041
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.