")
Back to Journals » Cancer Management and Research » Volume 13
Understanding and Monitoring Chronic Myeloid Leukemia Blast Crisis: How to Better Manage Patients
Authors Wang L, Li L, Chen R, Huang X, Ye X
Received 5 April 2021
Accepted for publication 13 June 2021
Published 23 June 2021 Volume 2021:13 Pages 4987—5000
DOI https://doi.org/10.2147/CMAR.S314343
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Lulu Wang,1,2,* Li Li,1,* Rongrong Chen,1,2 Xianbo Huang,1 Xiujin Ye1
1Department of Hematology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 2Program in Clinical Medicine, School of Medicine of Zhejiang University, Hangzhou, Zhejiang Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiujin Ye; Xianbo Huang
Department of Hematology, the First Affiliated Hospital, College of Medicine, Zhejiang University, 79# Qingchun Road, Hangzhou, 310003, People’s Republic of China
Tel + 86-571-87236702
; +86-571-87235875
Fax + 86-571-87235560
Email [email protected]; [email protected]
Abstract: Chronic myeloid leukemia (CML) is triggered primarily by the t(9;22) (q34.13; q11.23) translocation. This reciprocal chromosomal translocation leads to the formation of the BCR-ABL fusion gene. Patients in the chronic phase (CP) experience a good curative effect with tyrosine kinase inhibitors. However, cases are treatment refractory, with a dismal prognosis, when the disease has progressed to the accelerated phase (AP) or blast phase (BP). Until now, few reports have provided a comprehensive description of the mechanisms involved at different molecular levels. Indeed, the underlying pathogenesis of CML evolution comprises genetic aberrations, chromosomal translocations (except for the Philadelphia chromosome), telomere biology, and epigenetic anomalies. Herein, we provide knowledge of the biology responsible for blast transformation of CML at several levels, such as genetics, telomere biology, and epigenetic anomalies. Because of the limited treatment options available and poor outcomes, only the therapeutic response is monitored regularly, which involves BCR-ABL transcript level assessment and immunologic surveillance, with the optimal treatment strategy for patients in CP adapted to evaluate disease recurrence or progression. Overall, selecting optimal treatment endpoints to predict survival and successful TFR improves the quality of life of patients. Thus, identifying risk factors and developing risk-adapted therapeutic options may contribute to a better outcome for advanced-phase patients.
Keywords: chronic myeloid leukemia, blast crisis, mechanism, monitor, management
Introduction
Chronic myeloid leukemia is a myeloproliferative neoplasm marked by excessive malignant accumulation of myeloid cells in peripheral blood and the bone marrow (BM). The disease is caused by the BCR-ABL fusion gene generated from the t(9;22)(q34;q11) reciprocal translocation, encoding the BCR-ABL oncoprotein, which constitutively activates ABL kinase and drives hematopoietic cell proliferation and leukemic transformation.1–6 CML is a natural triphasic course disease starting with the indolent CP, which is characterized by a remarkable increase in myeloid precursors and mature cells and lasts approximately 3–5 years.7 Without therapeutic intervention, after a median interval of 3–18 months, the disease spontaneously progresses to AP and eventually to highly aggressive BP, characterized by a rapid expansion of primitive cells in the bone marrow that spill into circulation,8,9 similar to acute leukemia.
With the development of tyrosine kinase inhibitor (TKI) imatinib and subsequent advent of second-generation drugs such as nilotinib, dasatinib, and bosutinib, which target the BCR-ABL fusion protein, treatment outcomes for chronic CML have improved significantly. The subsequent introduction of ponatinib, which is effective in patients with genetic abnormalities that make them resistant to TKIs, has further improved treatment outcomes, and the overall survival rate is up to 90%.6−10−12
However, a proportion of chronic patients fail to respond to tyrosine kinase inhibitors and progress to blast crisis (BC), generally regarded as a heterogeneous disease at the molecular level,9 and conventional chemotherapy has been much less effective, followed by a response rate of only 30% and a substantially shortened time before the development of resistance.13 The median overall survival (OS) and failure-free survival (FFS) of BP-CML are 12 and 5 months, respectively.14
The underlying biological basis that drives the transformation to BC remains unclear. According to a deep-sequencing study of BP-CML, genetic changes occur in almost 76.9% of cases. Gene defects and additional chromosomal abnormalities are found at a higher rate in BP-CML patients than in CP patients. However, no single mutation has been proven to play a definitive role in blast crisis.7,15 BP-CML may partly come from a critical number of accumulated mutations. Additionally, telomere biology and epigenetic anomalies are frequently present in the advanced phase but rarely in the CP.16 Disease progression may well be propelled by the “right” combination of molecular-level changes.
Considering the poor prognosis of BP patients, along with the current research being constantly updated, we present an overview of the current relevant research findings from multiple dimensions, such as the molecular mechanism of blast crisis, treatment response assessment, and risk factor identification, to establish a thorough understanding of this disease. Treatment response assessments, such as regular BCR-ABL monitoring and immunologic surveillance, and timely intervention help prevent relapse or progression after treatment. Optimal endpoint evaluation reveals how long and deep the treatment response has predictive value in improving the long-term survival and successful TFR rate. Identifying risk factors may be beneficial for developing risk-adapted therapeutic options, focusing more on and preparing for upfront HSCT.
Evidence That Drives CML Progression is Increasingly Explored at Different Molecular Levels
The pathobiology of CML-BP involves multiple intricate pathways but is not fully understood.17 Higher expression and activation of BCR-ABL (P=0.0079 at the protein level, P=0.015 at the RNA level) were found.3,18 On the one hand, the cytoplasmic location advantage of the BCR-ABL oncoprotein allows it to access many cell substrates that are not accessible by major nuclear ABL proteins, which regulate the activation of survival and proliferation pathways,19 such as RAS/STAT5/PI-3K.20–23 On the other hand, growing evidence exists that uncontrolled activation of BCR-ABL is directly linked to the genetic instability observed in disease progression, initiated by reactive oxygen species (ROS) and oxidative DNA damage. BCR-ABL kinase activity seems to promote various DNA repair mechanisms but with inefficient, unfaithful DNA repair in return, facilitating the accumulation of additional cytogenetic abnormalities that cause irreversible changes in the phenotype of CML-CP cells toward that in BP and selection of BP clones.24–27 Multiple mechanisms, such as oncogene activation, loss of tumor suppressor genes, blockade of cell differentiation, malfunction of apoptosis, telomere theory, and abnormal epigenetic modification, may be involved in the disease progression of CML (Table 1, Figure 1).7,19
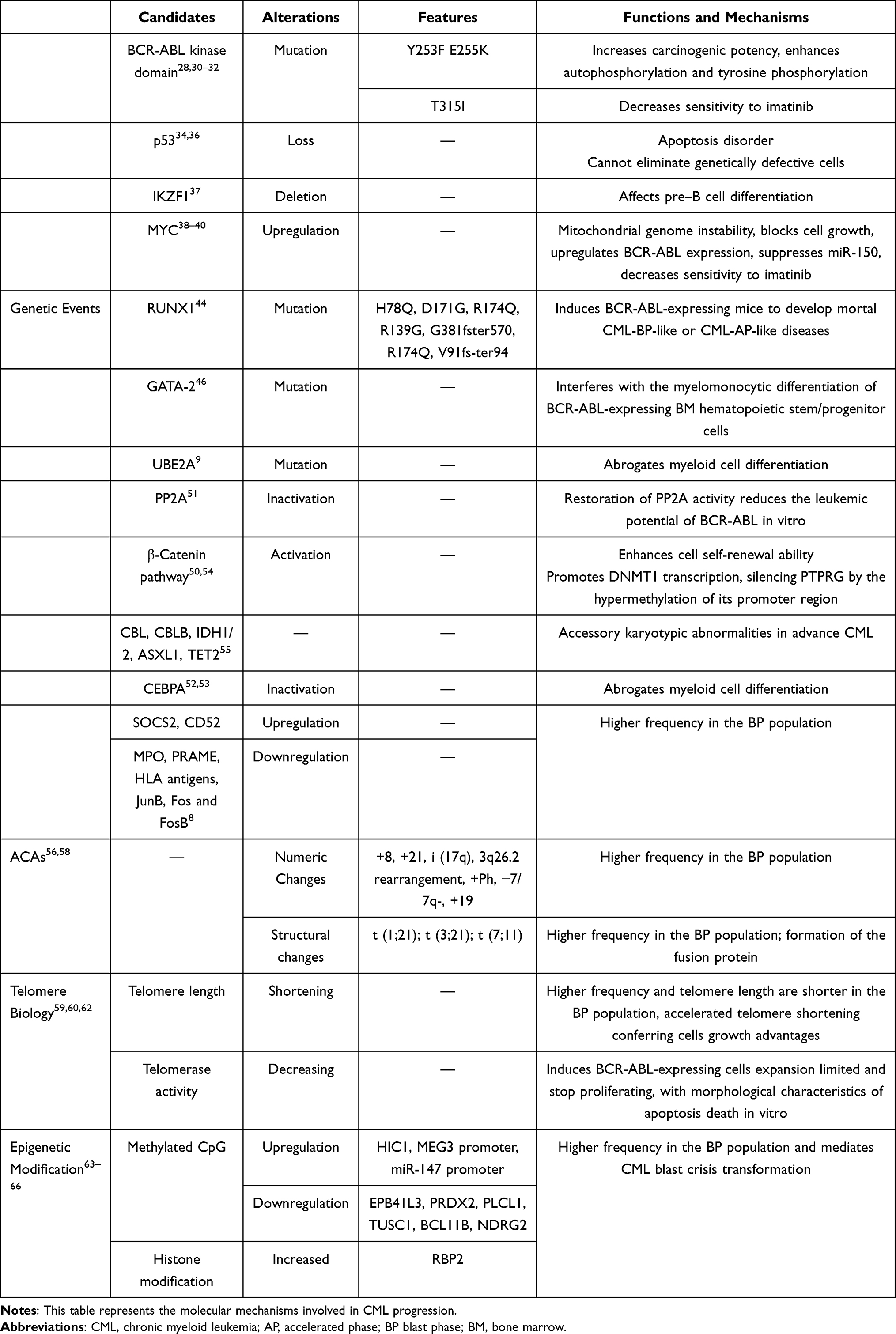 |
Table 1 Genetic, Chromosomal, Telomere Biology and Epigenetic Modification Changes Associated with BP Transformation |
Genetic Events
Fortunately, with the advent of microarray gene expression profiling and next-generation sequencing (NGS) technologies, a realistic opportunity exists to identify suitable biomarkers that alter gene expression and profile disease progression gene candidates.
Mutations in the BCR-ABL kinase domain (KD) are the most frequent events that are closely associated with the reactivation of clonal BCR-ABL leukemia cells28 and are reported to account for roughly 70%-80% in BP patients.29 Notably, low platelet counts (estimated odds ratio [OR], 19.7), clonal cytogenetic evolution (estimated OR, 14.163), and prior exposure to 6-thioguanine (estimated OR, 43.3) were associated with KD mutation detection but not the imatinib treatment response, event-free survival (EFS) or OS.26 However, a study on CML by the GIMEMA Working Party identified the presence of BCR-ABL KD mutations associated with a particularly poor prognosis in terms of time to progression (p=0.0002) and survival (p=0.001). This finding is similar to that of Shah et al. Additionally, the Working Party concluded that a specific subset of mutations that fall within the ATP binding loop (P-loop) of ABL confers a worse outcome, such as a shorter time to progression (p=0.032) and shorter survival (p=0.045). It has not yet been intensively investigated why the P-loop mutation seems to be associated with this aggressive manifestation; however, a previous study reported that P-loop mutations such as Y253F and E255K represent pronounced increasing carcinogenicity and transformation potency. Griswold et al suspected that diversity in intrinsic kinase activity might partially explain why differences in the transformation potential exist.28,30 Additionally, compared with wild-type BCR-ABL, when the mutation is located in E255K, along with the hydrogen bond location with imatinib (T315I), the abilities for the autophosphorylation and tyrosine phosphorylation of many important cellular proteins are enhanced, including STAT5.28,31 Mutations in the abovementioned sites have also significantly decreased sensitivity to imatinib at both the biochemical and cellular levels.32 There are many other mutations in BCR-ABL KD, such as V299L, F359V/I/C, and F317L,6,23 which have not been enumerated, and we must further explore their relationship with CML blast crisis. Additionally, Etienne et al showed that the disease phase at the time of mutation testing affects prognosis, regardless of whether a mutation is present. Thus, early mutation analysis together with accessibility to 2nd- and 3rd-generation TKIs are recommended because in preclinical studies, the worst outcome associated with BCR-ABL kinase domain mutations was reversed.33
In addition to BCR-ABL kinase domain mutations, other genetic defects are also associated with the progression of chronic myeloid leukemia, including MYC, RUNX1, IKZF1, PP2A, PTEN, p53, and GATA-2 genes, resulting in various outcomes.
The p53 gene, which maps to chromosome 17, is one of the most frequent tumor suppressor genes associated with human cancers. Accumulated evidence has shown that changes in the p53 gene are usually associated with blast crisis in CML, where they manifest as rearrangements, deletions, and point mutations.34 p53 gene deletion is uncommon in CP-CML, whereas it can be detected in 25%–30% of the BP-CML population.35 In 1996, Stuppia et al reported that the loss of the p53 gene is correlated with a reduction in the apoptosis process and blast transformation of CML.36 When BCR/ABL-expressing mice were injected with p53-deficient bone marrow cells, in contrast with the wild-type p53 counterpart, a fatal acute leukemia-like disease developed. The cells were homogeneously morphologically and immunophenotypically undifferentiated, had stronger resistance to apoptosis induced by growth factor deprivation, and maintained high clonal potential in the growth factor deprivation culture environment.20,35
Indeed, wild-type p53 works as a “molecular policeman” in the normal cell cycle. When DNA is injured by either irradiation or reagents, p53 mediates cell cycle arrest and the DNA damage response by increasing self-overexpression or inducing the expression of the cyclin-dependent kinase (CDK) inhibitors p21 WAF1/CIP1 and p16 to stop the transition of G1 to S phase simultaneously; thus, the cells can repair the damage or undergo apoptosis to eliminate cells with irreparable genetic defects.34 In the absence of a functional p53 gene, genomic instability occurs, and aberrant cells develop reproductive advantages.
The IKZF1 (Ikaros) gene is another tumor suppressor gene in human hematopoietic cells that was originally identified in genetic target studies of mice. The Ikaros protein, encoded by IKZF1, belongs to the zinc finger DNA-binding protein family and is an essential transcription factor for the development of the blood lymphatic system.23 Deletions of the IKZF1 gene were noted in 50%–55% of patients with lymphoid BP.7 A convincing study concluded that Ikaros activity could be affected by the overexpression of dominant-negative Ik-6, a transcription factor gene and non-DNA-binding splice isoform of Ikaros, involved in pre-B cell differentiation, through interacting with larger Ikaros isoforms, such as Ik-1, Ik-2, and Ik-3. Additionally, when this aberrant Ik-6 is silenced in Ph-positive pre-B cells using siRNA or reduced expression by imatinib treatment, differentiation along the pre-B cell lineages that have ceased before experimental management is partially restored,16,37 suggesting that genetic lesions resulting in the loss of Ikaros function are an important event in the development of BP.
The MYC proto-oncogene is upregulated in blast crises of CML patients.21 Albajar et al showed that MYC overexpression is a key event in mitochondrial genome instability, such as unscheduled DNA replication and aberrant DNA synthesis in the presence of imatinib.38 By recognizing the SKP2 subunit of the SCF-SKP2 complex, MYC reduces p27KIP1 expression to block cell growth.39,40 Lower levels of miR-150 were identified in BP-CML; in fact, the MYC oncogene suppresses miR-150, and this process is enhanced by BCR-ABL. More importantly, MYC upregulates BCR-ABL expression by activating the partner MYB gene and then combines with the BCR promoter.40,41 This hypothesis is confirmed by inhibiting MYC with antisense oligonucleotides or dominant-negative constructs to inhibit BCR-ABL transformation or leukemia in vitro.27 These findings, along with the established role of MYC in myeloid differentiation,42 imply that dysregulation of the MYC gene is a powerful promoter of BC transformation in CML.
In addition to MYC, mutations of RUNX1 (also known as AML1) are aggressive drivers of hematologic malignancies,23,43 followed by a poor prognosis. A high frequency of RUNX1 mutations of 12.9% to 33.3% is found in the advanced-phase population.44 RUNX1 mutants such as H78Q and V91fs-ter94 (located in the runt homology domain selected) can abrogate granulocytic differentiation in vitro. When BCR-ABL-expressing mice were injected with H78Q or V91fs-ter94 BM cells, interestingly, 80% of H78Q- and 33.3% of V91fs-ter94-harboring mice developed mortal CML-BP-like or CML-AP-like diseases and were characterized by many amplification myeloid mature granulocytes. Other RUNX1 mutations, such as D171G, R174Q, R139G, G381fster570, and R174Q, also result in abnormal function.45 Recalling earlier studies on GATA-2, similar experiments found that GATA-2 mutants are mostly recognized in advanced-phase CML but are not detected among CP patients. GATA-2 mutants interfered with the myelomonocytic differentiation of BCR-ABL-expressing BM hematopoietic stem/progenitor cells by inhibiting the main hematopoiesis regulator of PU.1.46 Awad et al revealed characteristic transcriptional programming in RUNX1mut cases, including B-lymphoid markers such as CD19 and CD7. In a newly published study, CD19 CAR T cells revealed potent activity against RUNX1mut BP-CML patient cells in vivo. Additionally, CD19-CAR T cells showed enhanced killing of RUNX1mut BP-CML blasts when combined with TKI. Consequently, CAR-T cell immunotherapy in BP-CML individuals, particularly those who harbor CD19-positive RUNX1 mutations, represents a potentially promising therapeutic option.44,47
To shed light on the transcriptional signatures of chronic myeloid leukemia, a study that analyzed gene expression profiles in CD34+ BCR-ABL-positive BP patients has yielded some new findings. Suppressor of cytokine signaling 2 (SOCS2) was the most obviously upregulated in BP-CML, followed by CAMPATH-1 antigen (CD52), while MPO declined the most; others, such as HLA antigens, PRAME, JunB, Fos, and FosB, showed varying degrees of dysregulation.8,48 Recently, a newly defined ubiquitin-conjugating enzyme E2A gene (UBE2A) mutation was recurrently obtained when evolving to fatal advanced-stage CML. Decreased UBE2A activity abrogates myeloid cell differentiation.9 The tumor suppressor PP2A is a major cellular serine-threonine phosphatase that provides negative feedback to signal triggers and is sustained by protein kinases. Inactivation of PP2A is mainly induced by BCR-ABL, increasing the expression of the phosphoprotein SET, which is a critical element of disease progression. CCAAT/enhancer binding protein (CEBPA), a major regulator of granulocytic differentiation, is also suppressed by elevated expression of BCR-ABL.23,27,49–53 β-Catenin, a pivotal regulator of leukemic stem cell (LSC) maintenance and CML proliferation, is overexpressed in primary BP-CML cell samples, correlating with enhanced self-renewal of CML cells. Additionally, activation of β-catenin promotes DNMT1 transcription, silencing protein tyrosine receptor type γ (PTPRG, a crucial tumor suppressor gene) through hypermethylation of its promoter region.50,54 Mutations such as CBL, CBLB, TET2, ASXL1, and IDH1/2 have been extensively reviewed elsewhere and have only been detected in accelerated and blast phase CML, but they may play an accessory role in the pathogenesis of evolution into the aggressive phase of CML.55
Chromosomal Abnormalities
Compared with CML-CP, numeric chromosomal changes are observed at a 50-fold higher frequency, and structural changes are observed at a 12-fold higher frequency in CML-BP. More sensitive comparative genomic hybridization (CGH) and single-nucleotide polymorphism (SNP) analyses have detected multiple genetic aberrations in CP, but the karyotype of BP patients is much more complex.7,16 Existing evidence suggests that BCR-ABL fusion is the root of acquired genetic instability and leads to the continued acquisition of additional chromosomal aberrations (ACAs) and mutations.56 The incidence of ACAs among CP-CML patients is 5%, while 60%-80% ACAs are detected in BP-CML.2,57,58 Previously, the European Leukemia Network (ELN) recommended that ACAs present at the time of a new diagnosis be defined as a warning signal and that newly emerging ACAs for treatment be defined as a failure. However, it was later found that the ACA subtype could be used to predict the patient outcome. A randomized German CML Study IV showed a significantly negative prognostic impact of major-route ACAs at diagnosis on the time to complete cytogenetic response (CCyR) and MMR (major molecular response) and progression-free survival (PFS) and OS.56 For example, 3q26.2 rearrangement, a newly defined major-route ACA with a high frequency of ABL1 mutations, indicates a poor prognosis. Other major-route changes, such as +8, +19, +21, and +Ph alone, are less powerful predictors, but a strong possibility of promoting the progression to BP exists when other cytogenetic alterations coexist. However, 3q26.2 rearrangements, −7/7q- and i(17q), particularly 3q26.2 rearrangements and −7/7q-, are more potent. The coincidence of +8 and other ACAs, despite the nature of other concurrent ACAs, suggests a poor treatment response and poor survival during TKI therapy, whereas +8 alone is associated with an optimal response to TKI therapy and good survival.23,57 This finding is consistent with a retrospective study of 2,326 CML patients. However, it also revealed that all ACAs have a risk of evolving to myeloid BP (MyBP), whereas only −7/7q- had a higher incidence with progression to lymphoid BP (LyBP); and the underlying reason for the difference between blast transformation and phenotype is not well understood. Importantly, the main determinant of prognosis is the time from ACA emergence to the onset of BP, rather than from the initial diagnosis to ACA development or after the onset of BP to ACA occurrence, which largely depends on the subtype of ACA.58
In addition to t(1;21), which involves RUNX1 expression, structural chromosomal changes known to occur in the blast phase are relatively infrequent (<5%). The most notable recurrent translocation is the acquisition of t(3;21) and t(7;11), resulting in the formation of dominant-negative transcription factors, such as the AML-1/EVI-1 and NUP98-HOXA9 fusion proteins, respectively. Coexpression of BCR-ABL1 and both transcription factors can cause CML-BP–like disease in mouse models by intervening with the signal required for the correct activation of self-renewal and differentiation programs.16,19,27
Telomere Biology
Telomere biology is closely related to the disease evolution of human cancers and plays a pivotal role in the progression of chronic myelogenous leukemia.59 Furthermore, by establishing a CML-like cell culture of telomerase knockout (mTR-/-) BMCs in a mouse model, experts have shown that accelerated telomere shortening, rather than critically shortened telomeres, is frequently found in the advanced phase. Intriguingly, murine CML-like bone marrow cells with critically short telomeres (CML-iG4) underwent senescence and growth limitation, whereas preshortened (generation G2) CML-like BMCs expanded rapidly and expressed a significantly upregulated telomere-associated secretory phenotype (TASP), comprising complex chemokines, cytokines, and other growth factors, thereby conferring selective growth advantages on cells driven by the BCR-ABL oncogene.19,60 Consistent with an earlier retrospective study in 2004, the telomere length in BP cells was significantly shortened (P<0.0001) compared with that in early CP. Additionally, in de novo CP CML patients, telomere shortening proceeds rapidly and may be considered a precursor to early disease evolution because of telomere dysfunction and serious genetic instability.61
However, telomerase maintains the normal structure of chromosome ends in eukaryotic cells, but telomerase activity is upregulated in many human tumors, including late-phase CML. To investigate the impact of telomerase inhibition by dominant-negative human telomerase reverse transcriptase (DN-hTERT) in BCR-ABL-positive BMCs, Tauchi et al introduced DN-hTERT into BCR-ABL-transformed BMCs compared with their control counterparts injected with wild-type (WT)-hTERT and expressed only a puromycin resistance marker. The experimental cells showed a significant decline in telomerase activity and a shortened telomere length. Cell expansion was limited, and the cells finally stopped proliferating, with morphological characteristics of apoptosis death. More importantly, they also showed enhanced sensitivity to imatinib, suggesting that imatinib combined with telomerase inhibition may be effective in treating BCR-ABL-positive leukemia,62 even in the advanced phase of CML.
Epigenetic Modification
Previous studies have indicated that epigenetic anomalies, including DNA methylation and histone modification, play an important role in the progression of CML from CP to BP.24 Heller et al identified 10-fold more differentially methylated CpG sites in BP patients than in CP patients, and methylation is increased in most of the affected CpG sites. Methylated genes that are tumor suppressor genes (EPB41L3 and PRDX2), putative tumor suppressor genes (PLCL1 and TUSC1), regulators of cell proliferation (BCL11B, NDRG2, and PID1), or regulators of drug metabolism (CYP1B1) are downregulated.63 In a larger methylation group study, the number of methylated CpG sites increased 100-fold.64 HIC1 hypermethylation was detected in 100% of blast crisis CML.65 Long noncoding RNAs MEG3 and miR-147 showed lower expression levels, MEG3 and miR-147 promoter methylation was detected, and the expression levels of DNMT1, DNMT3B, MBD2, MECP2, and HDAC1 were higher. Low MEG3 expression levels result in hyperphosphorylation of the JAK2 and STAT pathways, which promote proliferation, inhibit apoptosis, and affect genetically related genes. Treatment with chidamide increased MEG3 and miR-147 expression levels while decreasing the expression levels of DNMT1, DNMT3B, MBD2, MECP2, and HDAC1.66
Next, we concentrated on histone modification. Retinoblastoma-binding protein 2 (RBP2) is a member of the KDM5 family. Underexpressed RBP2 in CML-BP mediates CML blast crisis transition in a BCR-ABL-independent pathway and BCR-ABL-dependent pathway at the transcriptional level. The former pathway downregulates miR-21 expression, while the latter activates an RBP2/PTEN/BCR-ABL cascade. RBP2 reduces the expression of PTEN by binding to its promoter directly. At the protein level, PTEN targets the protein phosphatase activity of BCR-ABL to mediate the dephosphorylation of BCR-ABL, which finally regulates the downstream signaling pathways of BCR-ABL such as the p-STAT5 and p-ERK signaling pathways.24
What is a Better Method of Management?
BCR-ABL Monitoring to Prevent the Onset of CML Blast Crisis Transformation
Patients with CML-CP have a comparable prognosis to the normal population in the TKI era, although some patients who respond initially later become resistant, most of whom may at some stage acquire additional genetic and/or epigenetic changes that cause leukemia transformation.67 Notably, progression in patients who progressed mainly occurred during the first 3 years of treatment. A more rapid decline in the leukemic clone burden is linked to an optimized outcome as well as reduced rates of transformation.68,69 Hence, even if a good curative response is achieved, monitoring must be performed frequently and regularly to prevent blast crises, particularly in the first 3 years.
Detection of BCR-ABL transcripts in patients with CML by quantitative reverse transcription-polymerase chain reaction (QR-PCR) is clinically important for determining the treatment response and selecting the optimal treatment strategy (as well as for providing prognostic information).4,70 For example, leukemia cell lines expressing high levels of BCR-ABL are less sensitive to imatinib; when a response occurs, it is rarely sustained, and the development of refractoriness is often rapid because it yields mutant subclone resistance in chronic myeloid leukemia.3
Furthermore, several “molecular response milestones” at specific time points might help predict the outcome and select treatment drugs. The first crucial molecular milestone is an early molecular response (EMR), defined as the achievement of BCR-ABL IS (international scale) transcript levels <10% (1-log reduction) at 3 months and 6 months after the onset of TKI therapy. This approach has been standard of care because it is highly predictive of long-term outcomes, including OS, PFS, and event-free survival (EFS). Approximately 70% of patients face a 5-year OS of 95% after achieving EMR within 3 months of imatinib treatment. In 2014, Branford et al showed that, although patients did not achieve EMR, those with BCR-ABL that declined from their baseline to a halving time of no more than 76 days had significantly superior outcomes (OS: 95% vs 58%, P=0002; PFS: 92% vs 63%, P=0.008; MMR: 54% vs 5%, P=0.008). Last year, in 2020, a similar outcome was shown in the halving time in the ≤40 days group. More importantly, the German C. M. L. Study Group demonstrated precisely that a 3-month BCR-ABL transcript elimination rate of 0.46 log (approximately equivalent to a half log) from baseline accurately identified patients at risk for disease progression (high risk: 48/301 (16%) patients; 5-year OS: 83% vs 98%; hazard ratio (HR): 6.3; P=0.001). The second crucial time point is after 6 months of therapy, when the BCR-ABL IS transcript levels are expected to be <1% (2-log reduction). This time point helps identify a higher-risk subgroup of patients who failed to achieve EMR at 3 months after imatinib treatment. For example, group 2, who achieved BCR-ABL IS transcripts <1% IS at 6 months after failing to achieve an EMR at 3 months, did well in the absence of a significant difference in the short- or long-term outcomes compared with group 1, who achieved an EMR at 3 months. The higher-risk patients in group 4, who failed to attain levels <10% IS at 3 and 6 months, showed the worst short- and long-term outcomes, supporting the panel of the new ELN recommendations that a single failure to achieve the BCR/ABL transcript level <10% at 3 months is insufficient to define “failure”. Therefore, it is recommended to recheck molecular response more frequently in case of early failure, even and preferably before 6 months.
MMR is the third time point, defined as a reduction in the BCR-ABL IS transcript levels by at least three logs after 12 months of imatinib treatment. More recently, the more effective second-generation TKI allows for a deep molecular response (DMR), defined as a transcription level of BCR-ABL ≤0.01%. BCR-ABL monitoring sorted CML patients into “optimal” for continuously using the same TKI, “warning” to be considered for a possible TKI alteration, and “failed patients” who do not reach EMR, MMR, or DMR, and an immediate switch of treatment is required. When evaluating patients who are eligible for TKI discontinuation, more stringent TFR criteria should include typical BCR-ABL transcripts (b2a2 or b3a2), a minimal duration of TKI treatment of 4–5 years, and a duration of DMR (MR4 or better) of more than 2 years. BCR-ABL monitoring should be performed monthly for the first 6 to 12 months during TKI discontinuation and may be tapered off after that to avoid recurrence.4,6,69,71–77
Finally, in the posttransplant population, persistently positive BCR-ABL levels after 6–12 months can be regarded as a precursor for disease relapse. The BCR-ABL transcripts also differed significantly between molecular and cytogenetic recurrence (0.004% VS 0.4%; P< 0.001), and the clinical significance is that unequivocal therapeutic decisions are allowed, such as the withdrawal of immunosuppressive agents to prevent and treat graft-versus-host disease (GVHD), application of donor leukocyte infusion, or therapy with interferon (IFN)-a. Notably, patients with very low or undetectable BCR-ABL transcripts (BCR-ABL <0.1%) after 10 years of transplantation are least likely to experience relapse.78,79
Prospects of Immunologic Surveillance in the Warning of CML Blast Crisis Transformation
Dysregulation of the immunological response turns out to be critical pathogenesis of CP-CML. Additionally, approximately half of the patients who have achieved an optimal therapy response can discontinue their treatment without relapse, partly because of the immunological changes observed during TKI treatment.80 The immune system, notably natural killer (NK) cell-mediated immune surveillance, is now the most consistently identified predictor of TFR success following TKI cessation. This is due to the decreased NK cell cloning frequency in CP at diagnosis and worsening as the disease progresses to AP and BC, whereas successful TFR patients are recorded to have elevated NK cells at TKI discontinuation.77,81,82 Conversely, immune suppressor cells such as regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSCs) expand during disease progression or relapse and are reduced following TKI therapy.77,80 Other immunological factors, such as dendritic cells (DCs),83 plasmacytoid dendritic cells (pDCs),84 CD8+ cytotoxic T-cells (CTLs),85 leukemia-associated antigens (LAAs),86 and B-cells,87 also play a pivotal role in contributing to CML; however, further investigation should be launched to explore their impact on therapy responses. Additionally, studies have also indicated that imatinib and dasatinib treatment generates a more active immune system, and changes were not observed during bosutinib treatment.82,86,88,89
Thus, immunologic surveillance, similar to BCR-ABL monitoring, is a promising method to prevent CML progression. Additionally, imatinib and dasatinib have potent immunomodulatory effects in CML.
Selecting Optimal Endpoints During Treatment
Patients in the chronic phase who achieve CCyR have a life expectancy close to that of those achieving MMR or the general population.6,90 In contrast, the 5-year survival rate of CML-BP patients with CCyR (12.2%) is similar to that of patients with HR (11.0%) and significantly lower than that of patients with MMR. CML-BP patients who achieve MMR have a poor 5-year survival rate of only 34.4%. Therefore, achieving CCyR and MMR is not the optimal endpoint for optimal survival. By contrast, the survival advantage of molecularly undetectable leukemia (MUL) is easily appreciated, with a 5-year survival rate of 71.8%.90 Consequently, MUL should be the ultimate goal for the best long-term outcome in CML-BP patients.
Treatment-free remission is emerging as an important motivation in patients with CML to reduce the long-term complications of TKIs, such as relieving the financial burden and having fewer late adverse effects, particularly in younger patients. DMR is the most relevant clinical endpoint for CP-CML patients and is currently required to consider a test of TFR. More importantly, 40%-60% of patients who attained deep and durable molecular responses successfully maintained remission after TKI discontinuation.12,22,23,68 Numerous studies have identified correlations between TFR and the cellular immune system, including a higher number of NK cells and a lower Treg proportion, as previously mentioned, compared with patients who show disease relapse or transformation, suggesting that TFR is based on immune regulation and should be considered to identify promising immunotherapeutic targets, allowing more patients to achieve successful TKI cessation.81,82,91,92 However, to further evaluate the impact of the immune system on TFR, additional immunological parameters must be studied, preferably quantified, to determine the optimal therapeutic endpoint for successful TFR.
Identifying Prognostic Factors
BP-CML is treatment refractory and has a poor prognosis. Although there is an initial response in most patients, CCyR is relatively rare, and less than 30% of patients may achieve CCyR after extensive chemotherapy; however, it is generally short-lived, with a median duration of response of approximately 6 months.93 Allogeneic hematopoietic stem-cell transplantation (HSCT) is the sole curative approach and is recommended for patients who have a complete hematologic response (CHR). However, with transient responses less than 10%,94 more than 50% eventually undergo leukemic relapse,78 only 5%–10% of patients show long-term leukemia-free survival,16 and the 2-year and 5-year OS rates are only 18% and 6%, respectively.13,49,95 Risk factors must be identified and risk-adapted therapeutic options must be developed, such as upfront HSCT once a second chronic phase is achieved. In a single-institution survey, BP-CML patients who had undergone lymphocyte transformation and were treated with tyrosine kinase inhibitors were two independent positive prognostic factors for CHR, and age >60 years, a hemoglobin level <100 g/L, and a complex chromosomal karyotype were associated with shorter survival.96 However, no favorable evidence was found on the relationship between combined chemotherapy with TKIs and OS. A hemoglobin level <100 g/L, no second CP after TKI therapy and TKI therapy alone rather than combination therapy are independent adverse predictors of OS and EFS.97 Another larger-scale study in 477 BP-CML patients suggested that lymphoid immunophenotype, no previous TKI treatment, age less than 58 years, LDH <1227 IU/L, platelet count >102 K/μL, stem cell transplantation (SCT) recipients, and de novo BP predicted a superior prognosis but that chromosome 15 aberrations (a type of minor route ACA), hemoglobin <130 g/dL, and 5% blast cells in BM often indicate a worse clinical outcome.17 BP patients with lymphoid phenotype have a superior prognosis likely because they respond effectively to vincristine and prednisone. They have certain unique clinical, hematological, and other laboratory features, including a higher proportion who progress to BP directly without the intermediate AP (P=0.002), limited enlargement of organs such as the liver (P=0.02) and spleen (P=0.03), a higher percentage of bone marrow blasts (P <0.0001), fewer circulating basophils (P <0.0001), and a higher level of serum albumin (P=0.001).98 Another study also reported other features, such as a significantly younger patient population, less anemia, lower white blood cell and peripheral blood protocell counts, and lower serum lactate dehydrogenase levels.99 Guidelines call for TKI therapy based on the mutation profile for blast crisis patients combined with chemotherapy or TKI alone, with the goal of achieving a second chronic phase and proceeding to allogeneic stem cell transplantation as quickly as possible.48
Clinically, flow cytometry is rarely employed to diagnose CP-CML because of differentiated leukemia cells. However, a cohort study indicated that flow cytometry can be applied to predict early blast lineage transformation in CP-CML by detecting the surface markers of leukemia cells. Among 33 CP patients, 2 expressed lymphoid markers and 31 expressed myeloid markers; both lymphoid patients rapidly progressed to lymphoid blast crisis after 6 and 11 months, and three of the 31 myeloid patients progressed to lymphoid BP at 4, 20, and 72 months after therapy.100 Furthermore, flow cytometry was performed to identify that spleen tyrosine kinase (Syk) was significantly overexpressed, constitutive phosphorylation of Syk Y348 only appeared at the time of disease progression, and further clinical studies strongly indicated that SYK activation is a relevant biomarker of CML progression.101 Flow cytometry can be applied as a valuable tool to predict patients who may progress to blast crisis early.
Studies on CML found that the expression of PD-1 in CD8+ T cells in CHR is lower than that in newly diagnosed, AP and BP patients. PD-1 is positively correlated with the BCR-ABL gene; furthermore, PD-1 expression in CD4+ T cells in BP is higher than that in patients who achieve CHR, emphasizing the role of detecting PD-1 expression in CD8+ T cells in predicting disease recurrence or progression.77,102 Thus, PD-LI inhibitors may be considered.
In conclusion, the mechanism underlying chronic myeloid leukemia blast crisis is complicated, resulting from synthetic anomalies at different levels. Recognizing these underlying mechanisms contributes to the identification of promising target medicines for precise treatment. By discerning high-risk prognostic factors, monitoring effectively for posttreatment patients, alerting for minor discrepancies and adopting timely countermeasures, we believe that disease relapse or transformation can be prevented.
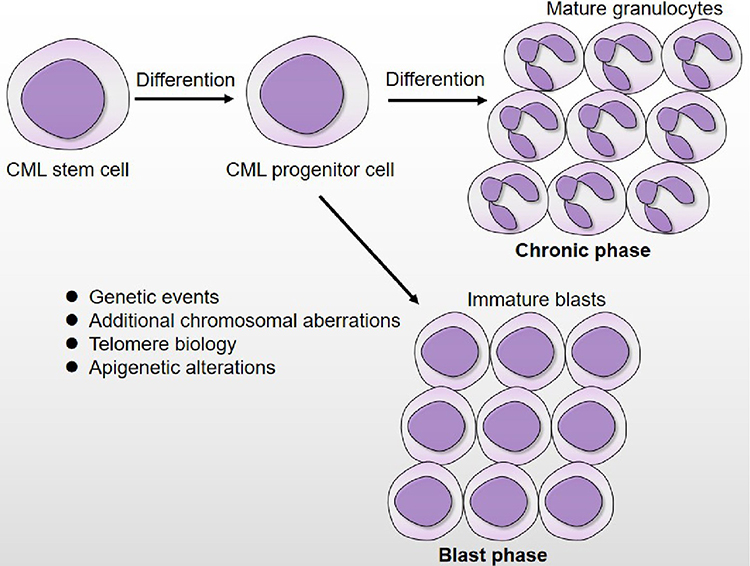 |
Figure 1 Disease progression of CML. During chronic phase, the CML stem/progenitor cells remain capable of differentiation and result in over production of mature granulocytes. In blast phase, differentiation of CML stem/progenitor has become arrested, leading to excessive accumulation of immature blasts that spill into the circulation. The biological mechanism that are responsible for CML blast transformation involves in genetic events, additional chromosomal aberrations, telomere biology and epigenetic alterations. |
Acknowledgments
This study was supported by the Natural Science Foundation of China (NO. 81900152), the Natural Science Foundation of Zhejiang Province (NO. LQ19H080005 and NO. LY19H080005), and the Health Department of Zhejiang Province (NO. 2020KY113).
Author Contributions
XJY and XBH conceived the study, LL and RRC collected and selected the references and revised the manuscript, and LLW wrote the paper. All the authors have read and approved the manuscript. All the authors have contributed to conceiving, drafting or revising the manuscript, have agreed on the journal to which the article will be submitted, have provided final approval of the version to be published, and have agreed to be accountable for all aspects of the work. Lulu Wang and Li Li are co-first authors.
Disclosure
The authors declare that they have no competing financial interests.
References
1. Lee S-T, Ji Y, Kim H-J, et al. Sequential array comparative genomic hybridization analysis identifies copy number changes during blastic transformation of chronic myeloid leukemia. Leuk Res. 2012;36(4):418–421. doi:10.1016/j.leukres.2011.12.021
2. Popp HD, Kohl V, Naumann N, et al. DNA damage and DNA damage response in chronic myeloid leukemia. Int J Mol Sci. 2020;21:4.
3. Barnes DJ, Palaiologou D, Panousopoulou E, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65(19):8912–8919.
4. Cumbo C, Anelli L, Specchia G, Albano F. Monitoring of Minimal Residual Disease (MRD) in chronic myeloid leukemia: recent advances. Cancer Manag Res. 2020;12:3175–3189.
5. Gao S, Hu J, Li Y. Targeting of the Alox12-12-HETE in blast crisis chronic myeloid leukemia inhibits leukemia stem/progenitor cell function. Cancer Manag Res. 2020;12:12509–12517.
6. Morita K, Sasaki K. Current status and novel strategy of CML. Int J Hematol. 2021;113(5):624–631.
7. Skorski T. Genetic mechanisms of chronic myeloid leukemia blastic transformation. Curr Hematol Malig Rep. 2012;7(2):87–93.
8. Zheng C, Li L, Haak M, et al. Gene expression profiling of CD34+ cells identifies a molecular signature of chronic myeloid leukemia blast crisis. Leukemia. 2006;20(6):1028–1034.
9. Magistroni V, Mauri M, D’Aliberti D, et al. De novo UBE2A mutations are recurrently acquired during chronic myeloid leukemia progression and interfere with myeloid differentiation pathways. Haematologica. 2019;104(9):1789–1797.
10. Yamaguchi H, Takezako N, Ohashi K, et al. Treatment-free remission after first-line dasatinib treatment in patients with chronic myeloid leukemia in the chronic phase: the D-NewS Study of the Kanto CML Study Group. Int J Hematol. 2020;111(3):401–408. doi:10.1007/s12185-019-02801-z
11. Russo D, Garcia-Gutierrez JV, Soverini S, Baccarani M. Chronic myeloid leukemia prognosis and therapy: criticisms and perspectives. J Clin Med. 2020;9:6.
12. Cortes J, Lang F. Third-line therapy for chronic myeloid leukemia: current status and future directions. J Hematol Oncol. 2021;14(1):44.
13. Karbasian Esfahani MM, Evelyn L, Dutcher JP, Wiernik PH. Blastic phase of chronic myelogenous leukemia. Curr Treat Options Oncol. 2006;7(3):189–199.
14. Zhou L, Shi H, Shi W, et al. Durable molecular remission in a lymphoid BP-CML patient Harboring T315I mutation treated with Anti-CD19 CAR-T therapy. Onco Targets Ther. 2019;12:10989–10995.
15. Grossmann V, Kohlmann A, Zenger M, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25(3):557–560.
16. Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–2264.
17. Jain P, Kantarjian HM, Ghorab A, et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: cohort study of 477 patients. Cancer. 2017;123(22):4391–4402.
18. Gaiger A, Henn T, Hörth E, et al. Increase of bcr-abl chimeric mRNA expression in tumor cells of patients with chronic myeloid leukemia precedes disease progression. Blood. 1995;86(6):2371–2378.
19. Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7(6):441–453.
20. Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103(11):4010–4022.
21. Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol. 2015;94(Suppl 2):S107–121.
22. Lee H, Basso IN, Kim DDH. Target spectrum of the BCR-ABL tyrosine kinase inhibitors in chronic myeloid leukemia. Int J Hematol. 2021;113(5):632–641.
23. Osman AEG, Deininger MW. Chronic myeloid leukemia: modern therapies, current challenges and future directions. Blood Rev. 2021;100825.
24. Yin X, Zhou M, Fu Y, et al. Histone demethylase RBP2 mediates the blast crisis of chronic myeloid leukemia through an RBP2/PTEN/BCR-ABL cascade. Cell Signal. 2019;63:109360.
25. Skorski T. Chronic myeloid leukemia cells refractory/resistant to tyrosine kinase inhibitors are genetically unstable and may cause relapse and malignant progression to the terminal disease state. Leuk Lymphoma. 2011;52(Suppl 1):23–29.
26. Willis SG, Lange T, Demehri S, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106(6):2128–2137.
27. Radich JP. The biology of chronic myelogenous leukemia progression: who, what, where, and why? Hematol Oncol Clin North Am. 2011;25(5):967–980, v.
28. Soverini S, Martinelli G, Rosti G, et al. ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol. 2005;23(18):4100–4109.
29. Bavaro L, Martelli M, Cavo M, Soverini S. Mechanisms of disease progression and resistance to tyrosine kinase inhibitor therapy in chronic myeloid leukemia: an update. Int J Mol Sci. 2019;20:24.
30. Griswold IJ, MacPartlin M, Bumm T, et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol. 2006;26(16):6082–6093.
31. Yamamoto M, Kurosu T, Kakihana K, Mizuchi D, Miura O. The two major imatinib resistance mutations E255K and T315I enhance the activity of BCR/ABL fusion kinase. Biochem Biophys Res Commun. 2004;319(4):1272–1275.
32. Corbin AS, La Rosée P, Stoffregen EP, Druker BJ, Deininger MW. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood. 2003;101(11):4611–4614.
33. Etienne G, Dulucq S, Huguet F, et al. Incidence and outcome of BCR-ABL mutated chronic myeloid leukemia patients who failed to tyrosine kinase inhibitors. Cancer Med. 2019;8(11):5173–5182.
34. Francesco Lanza SB. Role of p53 in leukemogenesis of chronic myeloid leukemia. Stem Cells. 1995;13(4):445–452.
35. Skorski T, Nieborowska-Skorska M, Wlodarski P, et al. Blastic transformation of p53-deficient bone marrow cells by p210bcr/abl tyrosine kinase. Proc Natl Acad Sci U S A. 1996;93(23):13137–13142.
36. Liborio Stuppia GC, Peila R, Guanciali-Franchi P, Morizio E, Spadano A, Palka G. p53 loss and point mutations are associated with suppression of apoptosis and progression of CML into myeloid blastic crisis. Cancer Genetic Cytogenetics. 1997;98(1):28–35.
37. Hiroyuki Nakayama FI, Avitahl N, Sezaki N, et al. Decreases in ikaros activity correlate with blast crisis in patients with chronic myelogenous leukemia. Cancer Res. 1999;59(16):3931–3934.
38. Albajar M, Gomez-Casares MT, Llorca J, et al. MYC in chronic myeloid leukemia: induction of aberrant DNA synthesis and association with poor response to imatinib. Mol Cancer Res. 2011;9(5):564–576.
39. Gomez-Casares MT, Garcia-Alegria E, Lopez-Jorge CE, et al. MYC antagonizes the differentiation induced by imatinib in chronic myeloid leukemia cells through downregulation of p27(KIP1.). Oncogene. 2013;32(17):2239–2246.
40. Pippa R, Odero MD. The role of MYC and PP2A in the initiation and progression of myeloid leukemias. Cells. 2020;9:3.
41. Srutova K, Curik N, Burda P, et al. BCR-ABL1 mediated miR-150 downregulation through MYC contributed to myeloid differentiation block and drug resistance in chronic myeloid leukemia. Haematologica. 2018;103(12):2016–2025.
42. Delgado MD, León J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010;1(6):605–616.
43. Branford S, Wang P, Yeung DT, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132(9):948–961.
44. Adnan Awad S, Dufva O, Ianevski A, et al. RUNX1 mutations in blast-phase chronic myeloid leukemia associate with distinct phenotypes, transcriptional profiles, and drug responses. Leukemia. 2020.
45. Zhao LJ, Wang YY, Li G, et al. Functional features of RUNX1 mutants in acute transformation of chronic myeloid leukemia and their contribution to inducing murine full-blown leukemia. Blood. 2012;119(12):2873–2882.
46. L-ym S-JZ, Huang Q-H, Guo L, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. pnas. 2007;105(6):2076–2081.
47. Adnan Awad S, Dufva O, Ianevski A, et al. RUNX1 mutations in blast-phase chronic myeloid leukemia associate with distinct phenotypes, transcriptional profiles, and drug responses. Leukemia. 2021;35(4):1087–1099.
48. How HR. I treat CML blast crisis. Blood. 2012;120(4):737–747.
49. Chandran RK, Geetha N, Sakthivel KM, et al. Genomic amplification of BCR-ABL1 fusion gene and its impact on the disease progression mechanism in patients with chronic myelogenous leukemia. Gene. 2019;686:85–91.
50. Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–667.
51. Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood. 2011;117(24):6660–6668.
52. Perrotti D, Cesi V, Trotta R, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30(1):48–58.
53. Guerzoni C, Bardini M, Mariani SA, et al. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood. 2006;107(10):4080–4089.
54. Tomasello L, Vezzalini M, Boni C, et al. Regulative loop between β-catenin and protein tyrosine receptor type γ in chronic myeloid leukemia. Int J Mol Sci. 2020;21:7.
55. Makishima H, Jankowska AM, McDevitt MA, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011;117(21):e198–206.
56. Fabarius A, Leitner A, Hochhaus A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760–6768.
57. Chen Z, Shao C, Wang W, et al. Cytogenetic landscape and impact in blast phase of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia. 2017;31(3):585–592.
58. Gong Z, Medeiros LJ, Cortes JE, et al. Cytogenetics-based risk prediction of blastic transformation of chronic myeloid leukemia in the era of TKI therapy. Blood Adv. 2017;1(26):2541–2552.
59. Braig M, Pallmann N, Preukschas M, et al. A ‘telomere-associated secretory phenotype’ cooperates with BCR-ABL to drive malignant proliferation of leukemic cells. Leukemia. 2014;28(10):2028–2039.
60. Jackie Boultwood SH, Fidler C, Kusec R, et al. Telomere length shortening is associated with disease evolution. Am J Hematol. 1999;61(1):5–9.
61. Drummond M, Lennard A, Brummendorf T, Holyoake T. Telomere shortening correlates with prognostic score at diagnosis and proceeds rapidly during progression of chronic myeloid leukemia. Leuk Lymphoma. 2004;45(9):1775–1781.
62. Tetsuzo Tauchi AN, Sashida G, Takashi Shimamoto JH, Ohyashiki KA, Yamamoto K, Ohyashiki K. Inhibition of human telomerase enhances the effect of the tyrosine kinase inhibitor, imatinib, in BCR-ABL-positive leukemia cells. Clin Cancer Res. 2002;8(11):3341–3347.
63. Heller G, Topakian T, Altenberger C, et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia. 2016;30(9):1861–1868.
64. Ruggiu M, Oberkampf F, Ghez D, et al. Azacitidine in combination with tyrosine kinase inhibitors induced durable responses in patients with advanced phase chronic myelogenous leukemia. Leuk Lymphoma. 2018;59(7):1659–1665.
65. Issa JP, Zehnbauer BA, Kaufmann SH, Biel MA, Baylin SB. HIC1 hypermethylation is a late event in hematopoietic neoplasms. Cancer Res. 1997;57(9):1678–1681.
66. Li ZY, Yang L, Liu XJ, Wang XZ, Pan YX, Luo JM. The long noncoding RNA MEG3 and its Target miR-147 Regulate JAK/STAT pathway in advanced chronic myeloid leukemia. EBioMedicine. 2018;34:61–75.
67. Bonifacio M, Stagno F, Scaffidi L, Krampera M, Di Raimondo F. Management of chronic myeloid leukemia in advanced phase. Front Oncol. 2019;9:1132.
68. Shah NP, García-Gutiérrez V, Jiménez-Velasco A, et al. Dasatinib discontinuation in patients with chronic-phase chronic myeloid leukemia and stable deep molecular response: the DASFREE study. Leuk Lymphoma. 2019;61(3):650–659.
69. Hanfstein B, Shlyakhto V, Lauseker M, et al. Velocity of early BCR-ABL transcript elimination as an optimized predictor of outcome in chronic myeloid leukemia (CML) patients in chronic phase on treatment with imatinib. Leukemia. 2014;28(10):1988–1992.
70. Zhang Z, Chen Z, Jiang M, et al. Heterogeneous BCR-ABL1 signal patterns identified by fluorescence in situ hybridization are associated with leukemic clonal evolution and poorer prognosis in BCR-ABL1 positive leukemia. BMC Cancer. 2019;19(1):935.
71. Hughes NSTP. Molecular monitoring in CML: how deep? How often? How should it influence therapy? Hematol Am Soc Hematol Educ Prog. 2018;168–176.
72. Kim DD, Hamad N, Lee HG, Kamel-Reid S, Lipton JH. BCR/ABL level at 6 months identifies good risk CML subgroup after failing early molecular response at 3 months following imatinib therapy for CML in chronic phase. Am J Hematol. 2014;89(6):626–632.
73. Branford S, Yeung DT, Parker WT, et al. Prognosis for patients with CML and >10% BCR-ABL1 after 3 months of imatinib depends on the rate of BCR-ABL1 decline. Blood. 2014;124(4):511–518.
74. Cai Z, Jia X, Zi J, et al. BCR-ABL1 transcript decline ratio combined BCR-ABL1IS as a precise predictor for imatinib response and outcome in the patients with chronic myeloid leukemia. J Cancer. 2020;11(8):2234–2240.
75. Branford S. Molecular monitoring in chronic myeloid leukemia-how low can you go? Hematol Am Soc Hematol Educ Prog. 2016;2016(1):156–163.
76. Cortes JE, Talpaz M, Giles F, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. 2003;101(10):3794–3800.
77. Hsieh YC, Kirschner K, Copland M. Improving outcomes in chronic myeloid leukemia through harnessing the immunological landscape. Leukemia. 2021;35(5):1229–1242.
78. Elmaagacli AH, Beelen DW, Opalka B, Seeber S, Schaefer UW. The amount of BCR-ABL fusion transcripts detected by the real-time quantitative polymerase chain reaction method in patients with Philadelphia chromosome positive chronic myeloid leukemia correlates with the disease stage. Ann Hematol. 2000;79(8):424–431.
79. Barrett AJ, Ito S. The role of stem cell transplantation for chronic myelogenous leukemia in the 21st century. Blood. 2015;125(21):3230–3235.
80. Irani YD, Hughes A, Clarson J, et al. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. Br J Haematol. 2020;191(3):433–441.
81. Nakajima H, Zhao R, Lund TC, et al. The BCR/ABL transgene causes abnormal NK cell differentiation and can be found in circulating NK cells of advanced phase chronic myelogenous leukemia patients. J Immunol. 2002;168(2):643–650.
82. Rea D, Henry G, Khaznadar Z, et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. 2017;102(8):1368–1377.
83. Dong R, Cwynarski K, Entwistle A, et al. Dendritic cells from CML patients have altered actin organization, reduced antigen processing, and impaired migration. Blood. 2003;101(9):3560–3567.
84. Inselmann S, Wang Y, Saussele S, et al. Development, function, and clinical significance of plasmacytoid dendritic cells in chronic myeloid leukemia. Cancer Res. 2018;78(21):6223–6234.
85. Mumprecht S, Claus C, Schürch C, Pavelic V, Matter MS, Ochsenbein AF. Defective homing and impaired induction of cytotoxic T cells by BCR/ABL-expressing dendritic cells. Blood. 2009;113(19):4681–4689.
86. Kreutzman A, Yadav B, Brummendorf TH, et al. Immunological monitoring of newly diagnosed CML patients treated with bosutinib or imatinib first-line. Oncoimmunology. 2019;8(9):e1638210.
87. de Lavallade H, Khoder A, Hart M, et al. Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood. 2013;122(2):227–238.
88. Najima Y, Yoshida C, Iriyama N, et al. Regulatory T cell inhibition by dasatinib is associated with natural killer cell differentiation and a favorable molecular response-The final results of the D-first study. Leuk Res. 2018;66:66–72.
89. Imagawa J, Tanaka H, Okada M, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre Phase 2 trial. Lancet Haematol. 2015;2(12):e528–535.
90. Chen Z, Medeiros LJ, Kantajian HM, et al. Differential depth of treatment response required for optimal outcome in patients with blast phase versus chronic phase of chronic myeloid leukemia. Blood Cancer J. 2017;7(2):e521.
91. Cayssials E, Jacomet F, Piccirilli N, et al. Sustained treatment-free remission in chronic myeloid leukaemia is associated with an increased frequency of innate CD8(+) T-cells. Br J Haematol. 2019;186(1):54–59.
92. Ohyashiki K, Katagiri S, Tauchi T, et al. Increased natural killer cells and decreased CD3(+)CD8(+)CD62L(+) T cells in CML patients who sustained complete molecular remission after discontinuation of imatinib. Br J Haematol. 2012;157(2):254–256.
93. Breccia M, Serrao A, Salaroli A, Loglisci G, Zacheo I, Alimena G. Dasatinib combined with weekly administration of vincristine as effective therapy in sudden or resistant Ph+ lymphoid blast crisis of chronic myeloid leukaemia. Br J Haematol. 2012;159(5):612–613.
94. Cortes J, Rousselot P, Kim DW, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood. 2007;109(8):3207–3213.
95. Druker BRIANJ, Sawyers CHARLESL, Kantarjian HAGOP, et al. Activity of a specific inhibitor of the Bcr-Abl tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engd J Med. 2001;3(345):232.
96. Perez-Jacobo F, Tuna-Aguilar E, Demichelis-Gomez R, et al. prognostic factors, response to treatment, and survival in patients with chronic myeloid leukemia in blast phase: a single-institution survey. Clin Lymphoma Myeloma Leuk. 2015;15(12):778–784.
97. Jiang H, Xu LP, Liu DH, et al. Allogeneic hematopoietic SCT in combination with tyrosine kinase inhibitor treatment compared with TKI treatment alone in CML blast crisis. Bone Marrow Transplant. 2014;49(9):1146–1154.
98. Francisco Cervantes N, Esteve J, Montoto S, Rives S, Rozman C, Montserrat E. ‘Lymphoid’ blast crisis of chronic myeloid leukaemia is associated with distinct clinicohaematological features. Br J Haematol. 1998;100(1):123–128.
99. Derderian PM, Kantarjian HM, Talpaz M, et al. Chronic myelogenous leukemia in the lymphoid blastic phase: characteristics, treatment response, and prognosis. Am J Med. 1993;94(1):69–74.
100. El Rassi F, Bergsagel JD, Arellano M, et al. Predicting early blast transformation in chronic-phase chronic myeloid leukemia: is immunophenotyping the missing link? Cancer. 2015;121(6):872–875. doi:10.1002/cncr.29142
101. Bourgne C, Janel A, Berger J, et al. Phosphorylation of spleen tyrosine kinase at tyrosine 348 (pSyk348) may be a marker of advanced phase of Chronic Myeloid Leukemia (CML). Leuk Res. 2015;39(3):329–334. doi:10.1016/j.leukres.2014.11.014
102. Lee MY, Park CJ, Cho YU, et al. Differences in PD-1 expression on CD8+ T-cells in chronic myeloid leukemia patients according to disease phase and TKI medication. Cancer Immunol Immunother. 2020;69(11):2223–2232.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.