")
Back to Journals » Research and Reports in Urology » Volume 10
Unclassified renal cell carcinoma: diagnostic difficulties and treatment modalities
Authors Sirohi D , Smith SC , Agarwal N , Maughan BL
Received 20 June 2018
Accepted for publication 15 October 2018
Published 15 November 2018 Volume 2018:10 Pages 205—217
DOI https://doi.org/10.2147/RRU.S154932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jan Colli
Deepika Sirohi,1 Steven C Smith,2,3 Neeraj Agarwal,4 Benjamin L Maughan4
1Department of Pathology, University of Utah and ARUP Laboratories, Salt Lake City, UT, USA; 2Department of Pathology, Virginia Commonwealth University, School of Medicine, Richmond, VA, USA; 3Department of Urology, Virginia Commonwealth University, School of Medicine, Richmond, VA, USA; 4Division of Medical Oncology, Huntsman Cancer Institute, University of Utah, Salt Lake City, UT, USA
Abstract: Over the past few decades, the classification system of renal cell carcinoma (RCC) variants has witnessed tremendous and ongoing refinement driven by genomic profiling and morphological correlation that have provided valuable insights into tumor biology and characterization of this heterogeneous subset of tumors. The importance of accurate classification cannot be understated given the downstream impact on treatment decisions, risk stratification, and need for genetic testing. While the morphologic heterogeneity across these tumors is increasingly being recognized, all non-clear-cell RCCs are commonly categorized under one therapeutic category with management strategies that largely derive from clear-cell RCCs. As research in metastatic RCC progresses, there is a growing focus on rare subtypes and unclassified tumors, which is rapidly changing the treatment paradigm for non-clear-cell RCC. This review focuses on the histomorphologic diagnostic challenges of unclassified RCCs discussing the utility of contemporary diagnostic tools. It further discusses the current state of knowledge and guidelines for management of this class of tumors.
Keywords: pathology, classification, morphology, management
Introduction
Among the earliest classifications of renal cell carcinoma (RCC), the Mainz Classification was based on morphologic similarities of the tumor to the normal renal cellular compartments.1 Better understanding of the genetics of RCCs prompted the Heidelberg2 and Rochester Classifications3 that laid the modern foundation that evolved through the WHO 2004,4 International Society of Urological Pathology (ISUP) Vancouver 2012,5 and finally the contemporary WHO 2016 classification.6 While the number of recognized subtypes of renal tumors has increased threefold in the process, a subset of tumors that remain to be fully characterized by morphology, immunohistochemistry, and molecular studies were retained as emerging entities. While these classification systems have provided a more robust classification of adult renal epithelial tumors, there remains a class of tumors that defy morphologic and immunohistochemical classification into the recognized more common types of RCC and are grouped into a separate category of unclassified RCC. While frustrating from a diagnostic and management standpoint, retaining this diagnostic category provides an opportunity for future studies and further characterization.
Unclassified RCCs comprise a significant proportion of adult renal epithelial tumors, accounting for 2%–6%7,8 of RCCs. We note that these estimates of relative prevalence come from several referral centers with a population enriched for aggressive, high-grade tumors. However, in recent years, recognizing the diagnostic challenges surrounding RCCs with oncocytic/eosinophilic morphology, an increasing number of cases in this morphologic spectrum are being included in the unclassified category.9
Definition
Unclassified RCC, as defined by WHO 2016 Classification of the kidney,6 is a diagnostic category used for renal tumors that do not fit into any of the well-recognized subtypes and includes admixed patterns of more than one recognized subtype; unclassified oncocytic neoplasms or tumors with pure sarcomatoid histology. Besides these, other morphologic patterns that may be encountered in such lesions include mucin production, mixtures of epithelial and stromal elements and unrecognizable cell types. With increasing recognition of morphologic overlap between clear cell, papillary, and oncocytic subtypes, the spectrum of morphologic patterns across unclassified RCCs has also expanded to be increasingly inclusive of both low- and high-grade histologic tumors.10 As such, unclassified RCC remains a diagnosis of exclusion, entailing meticulous gross evaluation and sampling, careful search for foci of recognizable histologic subtypes, and judicious incorporation of contemporary diagnostic tools in the workup to complement the morphologic assessment.
Clinical significance
While unclassified RCCs with low-grade histology have an indolent behavior, cases with high-grade histologic features comprise a large proportion of this category and are associated with a worse prognosis as compared to conventional clear-cell RCCs (ccRCC). Established histomorphologic features that are used for prognostic stratification for conventional subtypes of RCCs such as ISUP/Fuhrman nuclear grade, sarcomatoid change, coagulative necrosis, vascular invasion, tumor size, and TNM stage have been shown to correlate well with disease-specific survival for this subset of tumors, with tumor size and recurrence being independent predictors of disease-free and cancer-specific survival, respectively. In the current era, similar results are seen across partial nephrectomies wherein histologic subtypes impact recurrence-free and overall survival (OS), with unclassified RCCs demonstrating the lowest OS for all stages.7
Approach to diagnosis
Unclassified RCCs are a histologically heterogeneous category of tumors, many of which are high-grade and high-stage tumors with poor outcomes. However, this tumor also includes low-grade tumors,8 for which the differential diagnosis differs. The diagnostic approach to these tumors is often aided by recognizing the underlying morphologic pattern across the tumor, assessment of the predominant cell type, a search for a minor component showing the pattern of a conventional diagnostic category, and judicious use of ancillary diagnostic assays. While clear cell, oncocytic/eosinophilic, and mucinous are the main cell types that may be seen in these tumors, papillary, sarcomatoid, nested, solid, tubular, and tubulopapillary architectures are also seen.10 These cell types and patterns can be used to guide further workup of these tumors as discussed subsequently. Recognizing that patterns may sometimes overlap across these categories in tumors, to organize this discussion, we will entertain tumors with a 1) clear-cell morphology and/or papillary features, 2) eosinophilic/oncocytic neoplasms, 3) tumors with high-grade distal nephron adenocarcinoma morphology, and 4) sarcomatoid tumors. Characteristic morphologic features and useful diagnostic tests for RCC subtypes are listed in Table 1.
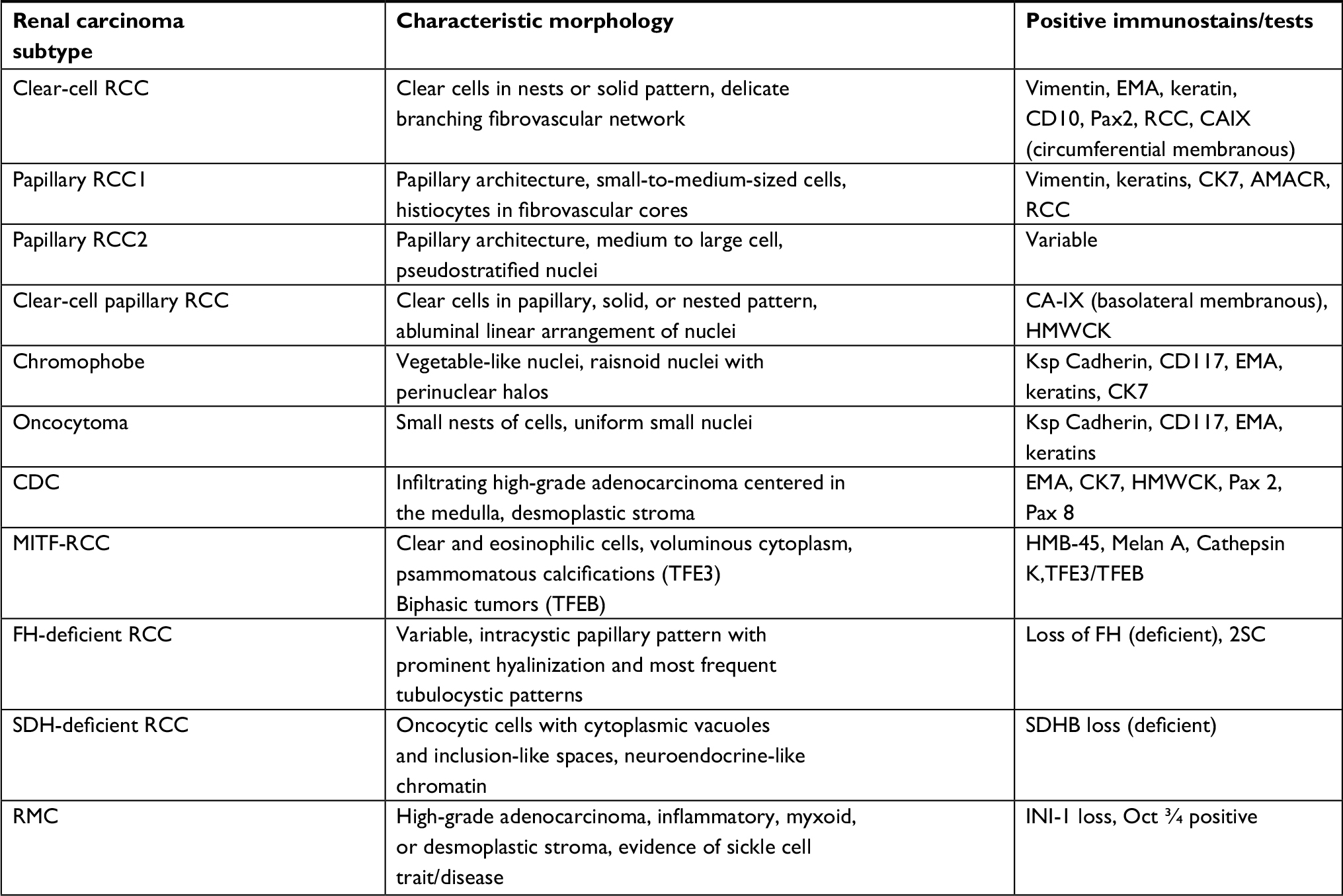 | Table 1 Characteristic morphology and diagnostically useful immunostains for renal cell carcinomas Abbreviations: AMACR, alpha-methylacyl-CoA racemase; CAIX, carbonic anhydrase IX; CDC, collecting duct carcinoma; CK, cytokeratin; EMA, epithelial membrane antigen; FH, fumarate hydratase; HMWCK, high molecular weight cytokeratin; MiTF-RCC, microphthalmia transcription factor family translocation RCC; RCC, renal cell carcinoma; RMC, renal medullary carcinoma; SDH, succinate dehydrogenase. |
Clear-cell morphology and/or papillary features
ccRCC is the most frequent adult renal epithelial tumor.8 From a diagnostic and therapeutic point, its recognition is important, because of inherently aggressive biology with predilection to metastasize to unusual sites years after primary treatment. Most salient currently is the fact that for the purpose of therapeutic management, recent US National Comprehensive Cancer Network (NCCN) guidelines divide renal carcinomas into “clear-cell” and “non-clear-cell” groups.11 Therefore, differentiating ccRCCs from other RCCs is increasingly critical.
A clear-cell pattern (Figure 1A–H), while most frequently seen in conventional ccRCCs, may also be seen in other tumor types such as clear-cell papillary RCC (pRCC), microphthalmia transcription factor family translocation RCC (MiTF-RCC), chromophobe RCC, tuberous sclerosis (TSC)-associated RCC, urothelial carcinomas (UCs) of the upper urothelial tract, and metastatic tumors, and as nonspecific clearing in other tumors. Perivasular epitheloid cell tumors (PEComas), though not epithelial neoplasms, frequently show a clear-cell change and are in the differential. The morphological features of these are discussed in subsequent paragraphs.
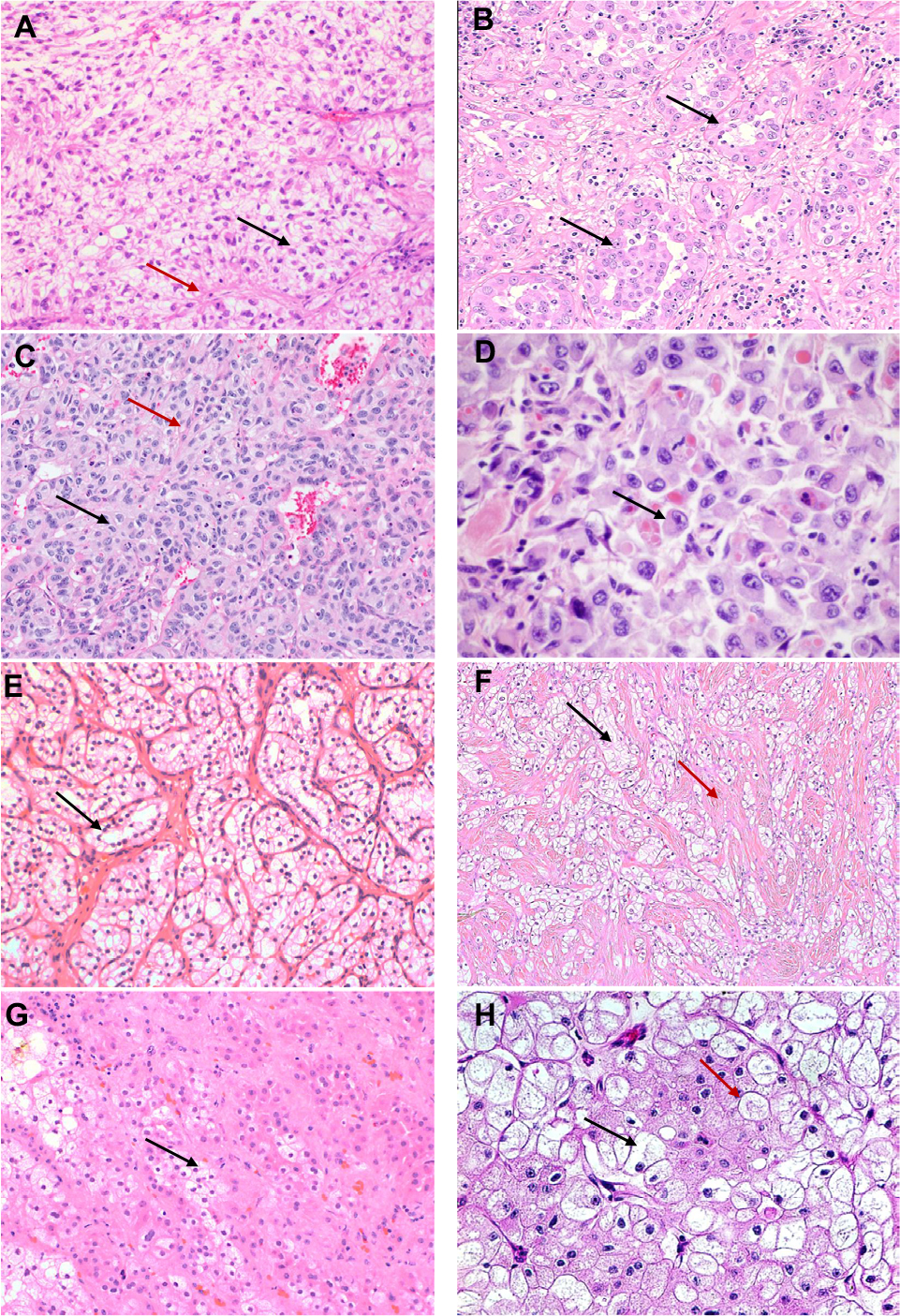 | Figure 1 (A–H) Morphological spectrum of RCC with clear-cell change. (A) Conventional clear-cell RCC, WHO/ISUP nuclear grade 2, with characteristic clear tumor cells (black arrow) in nests and sheets separated by a fine fibrovascular network (red arrow). (B) The clear-cell RCC demonstrated a glandular morphology (black arrow) with eosinophilic cytoplasm. Characteristic morphology of clear-cell RCC was seen in other areas. (C) Clear-cell RCC with cells with eosinophilic cytoplasm arranged in nests (black arrow). The fine fibrovascular network is retained (red arrow). (D) Clear-cell RCC with rhabdoid features. The cells have eosinophilic cytoplasm with eccentric nuclei (black arrow). (E) Characteristic pattern of clear-cell papillary RCC is seen in this case with cells with optically clear cytoplasm arranged in small nests. The nuclei are low grade and linearly arranged apically along the luminal side (black arrow). (F) In this case of RCC with leiomyomatous stroma neoplastic clear cells (black arrow) are seen in a dense fibromuscular stroma (red arrow). (G) Clear cells with vacuolated foamy cytoplasm (black arrow) are often also seen in MITF-associated RCCs, where they are often seen alongside cells with eosinophilic cytoplasm. 1 hour: Clear cells (black arrow) may be a prominent component of Chromophobe RCCs. Typical cellular and nuclear features of prominent cell borders (red arrow) and perinuclear halos and wrinkled nuclear membranes are helpful in making the diagnosis. Abbreviations: ISUP, International Society of Urological Pathology; MiTF, microphthalmia transcription factor family translocation; RCC, renal cell carcinoma. |
ccRCC
ccRCCs demonstrate a diverse morphological spectrum (Figure 1A–D), though the classic and distinctive morphology of ccRCCs is best seen in low-grade (ISUP) tumors. Tumor cells with optically clear cytoplasm are arranged in nests, or solid sheets surrounded by a delicate branching fibrovascular network. The vascular pattern is very characteristic of ccRCCs and is preserved in many high-grade tumors and even metastatic lesions. In fact, recognition of this vascular pattern is often an important clue in establishing a diagnosis of ccRCC in the absence of other diagnostic features. These features may be lost in high-grade tumors which can exhibit a spectrum of changes overlapping with other subtypes including granular/eosinophilic cytoplasm, papillary/pseudopapillary architecture, and sarcomatoid or rhabdoid features. It may be retained in cases where many of the tumor cells have undergone necrosis or otherwise involuted, leaving a vascular pseudohemangiomatous pattern.12 Diagnosis in challenging cases can be aided by immunoreactivity for broad-spectrum keratins, vimentin, CD10, and diffuse circumferential expression of CA-IX; while being negative for CD117 and Ksp Cadherin.13 Circumferential membranous CA-IX staining may be retained, albeit focally even in high-grade and sarcomatoid tumors and lends support to the diagnosis.
Differential diagnosis of low-grade ccRCCs includes two entities associated with excellent prognosis: multilocular cystic renal neoplasm of low malignant potential and clear-cell pRCC.6,14 The former is distinguished from a predominantly cystic ccRCC based on the absence of expansile nests of tumor cells on evaluation of entire lesional tissue.
Clear-cell pRCC
Clear-cell pRCCs share overlapping morphology with clear cell and pRCCs. As the name suggests, it is composed of clear cells arranged in a papillary configuration, although other patterns such as cystic, solid, acinar, and tubular patterns may be seen (Figure 1E). The diagnostic hallmark of this lesion is the linear arrangement of low-grade nuclei, displaced apically away from the basement membrane, although this feature may be subtle. Cytologic atypia, necrosis, foamy macrophages, and aggressive features such as vascular invasion are characteristically absent. A diffuse cuplike or basolateral membranous staining pattern of CA-IX and diffuse cytokeratin (CK) 7 and high molecular weight cytokeratin (HMWCK) positivity with negative racemase are supportive of the diagnosis.13 Recent studies have also described potential utility of Cyclin D115 and GATA 316 as markers for this RCC subtype.
Clear-cell pRCC shares overlapping morphologic and immunohistochemical features with TCEB1-mutated RCC,17 RCC with leiomyomatous stroma, and renal angiomyoadenomatous tumor (RAT). The latter two are characterized by proliferation of clear cells with varied architectural pattern in a dense fibromuscular stroma or angioleiomyoma-like stroma (Figure 1F). Although there is extensive morphologic overlap among these entities, the underlying genetic alterations are different, with tumors with RAT morphology sharing a frequent mutation in TCEB1 gene. With the current understanding of these tumors, it is uncertain at present if these are distinct entities and are considered to be tumors along a spectrum.18
Renal carcinomas with papillary architecture
While a papillary architecture is, on its face, quite suggestive of conventional pRCC, it is not unique to them and may be seen in other subtypes such as MiTF-RCC, collecting duct carcinoma (CDC), fumarate hydratase (FH)-deficient RCCs such as that arise in hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC-RCC) and unclassified RCC. Focal papillary and pseudopapillary patterns may also be seen in high-grade ccRCCs. Conventional pRCCs are well circumscribed and encapsulated tumors that are classified into so-called type 1 and type 2 patterns that have been shown to be distinct molecular entities. While gains of chromosomes 7 and 17 as well as MET alterations are seen in type 1 tumors, type 2 is increasingly recognized to represent a very distinct (from conventional pRCC) but quite heterogeneous group containing at least three distinct molecular clusters. These include tumors with molecular alterations involving the NRF2-ARE pathway, chromatin-modifying genes, TFE3 fusions, CDKN2A silencing, and CpG island methylator phenotype.19
Types 1 and 2 pRCC
Type 1 tumors, increasingly recognized to represent the typical and conventional archetype of pRCC, characteristically show a papillary pattern with delicate fibrovascular cores, lined by small-to-medium-sized cuboidal cells with irregular nuclear membranes and variably prominent nucleoli (Figure 2A). Fused papillae may impart a solid appearance, and oncocytic cells may be seen occasionally. Histiocytes and macrophages are frequently seen within the fibrovasular cores. In contrast, type 2 pRCCs are a morphologically and molecularly heterogeneous category as elucidated by the TCGA sequencing datasets. As such, type 2 pRCC remains a diagnosis of exclusion. In prototypical cases, the papillary structures are lined by medium-to-large cells with abundant eosinophilic cytoplasm that may demonstrate clearing, irregular pseudostratified nuclei, and prominent nucleoli (Figure 2B, C). pRCCs are positive for racemase, generally including both type 1 and type 2, with CK7 positive in lower grade type 1 tumors. Variable positivity for CD10 is noted, while pRCCs are negative for CA-IX.
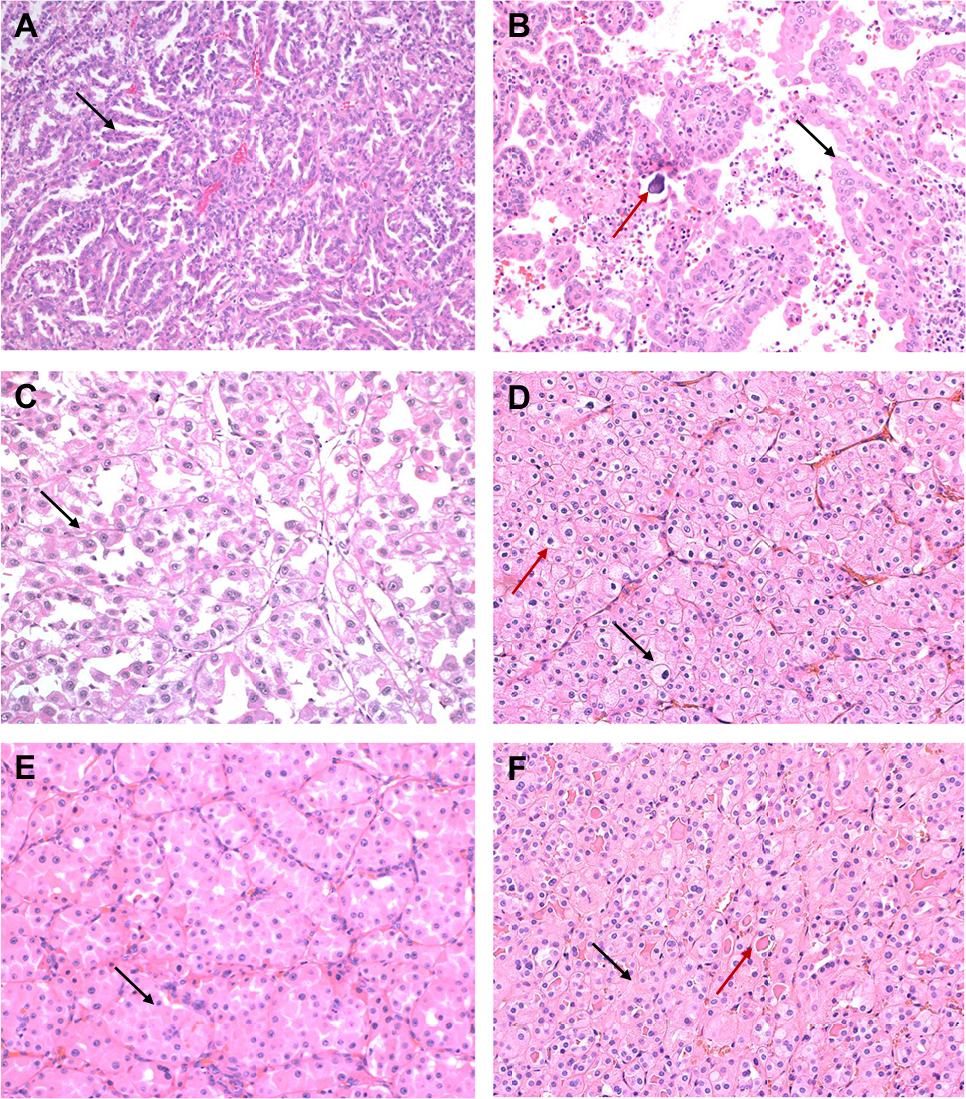 | Figure 2 (A–C) Papillary RCCs. (A) Type 1 papillary RCC with papillae (black arrow) lined by small-to-medium-sized cells. (B) In this papillary type 2 pattern of RCC, cells have an oncocytic appearance (black arrow). Psammoma bodies (red arrow) are seen. (C) Type 2 papillary pattern seen in an MITF-RCC. A papillary architecture with cells having abundant eosinophilic cytoplasm (black arrow) is identified. (D–F) RCCs with oncocytic cytoplasm. (D) Chromophobe RCC with eosinophilic cytoplasm. Distinct cellular borders (black arrow) and wrinkled nuclei with perinuclear halos (red arrow) are seen. (E) Oncocytoma with small nests of cells (black arrow) with abundant eosinophilic cytoplasm and uniform round nuclei. (F) A succinate dehydrogenase-deficient RCC is also composed of oncocytic cells with eosinophilic cytoplasm (black arrow). Cytoplasmic vacuoles and eosinophilic globules (red arrow) are seen. Abbreviations: MiTF, microphthalmia transcription factor family translocation; RCC, renal cell carcinoma. |
MiTF-family translocation RCC (MiTF-RCC)
MiTF-RCCs are a group of tumors characterized by recurrent rearrangements of TFE3 (at the Xp11 locus) or TFEB (at the 6p21 locus) genes, both of which are members of the MiT family of transcription factors. They share variable morphologic features that overlap considerably with other subtypes, including both clear cell and pRCCs. Hence, they are frequently considered in the differential diagnosis of renal tumors with clear cell and/or papillary features. Xp11.2 RCCs were originally described in children but are increasingly diagnosed in adults, a setting in which some series have observed more aggressive clinical behavior.20 These tumors demonstrate a varied morphologic spectrum, which seems somewhat correlated with which fusion gene partner is involved in the rearrangement. ASPL-TFE3 translocation RCC have papillary/pseudopapillary, nested or alveolar appearance with high-grade nuclei, voluminous clear to granular cytoplasm, and psammomatous calcifications (Figure 2B, C). On the other hand, tumors with PRCC-TFE3 rearrangement tend to have more of a nested, papillary, or compact architecture, with less abundant cytoplasm and lower grade nuclei.20,21 TFEB or t(6;11) translocation RCCs are more indolent tumors characterized by TFEB gene fusion with Alpha gene. They are usually biphasic tumors composed of large epithelioid cells with clear to eosinophilic cells and a “second population” of smaller cells with eosinophilic cytoplasm and hyperchromatic nuclei forming rosette-like structures within basement membrane-like material.21
The immunohistochemical profile of this family of tumors includes negative/patchy staining with broad-spectrum epithelial markers (pancytokeratin AE1/3, epithelial membrane antigen), negative/focal CA-IX, and positive staining with melanocytic markers HMB45, Melan-A, MiTF, and less frequently S-100. This overall pattern is seen irrespective of whether carcinomas show TFE3 or TFEB rearrangements, though often TFEB-rearranged carcinomas show quite strong expression of melanocytic markers. Cathepsin K, an osteoclast-associated protein that is also expressed with melanocytic differentiation, is an emerging marker that among renal epithelial neoplasms is quite specific for translocation RCCs, with the caveat that it is also positive in PEComas. Although immunostains for TFE3 and TFEB are available, they lack sensitivity and are technically difficult to optimize. Dual-color break-apart fluorescence in situ hybridization assays for TFE3 and TFEB rearrangements are very sensitive and specific for confirming the diagnosis.22,23
To summarize, a judicious morphological and/or immunohistochemical approach incorporating broad-spectrum cytokeratins and CAIX should guide the evaluation of tumors with clear cell and/or papillary features before designating a renal neoplasm as unclassified. While working up these tumors, it is important to be cognizant of the fact that while ccRCCs and MiTF-RCC are at the aggressive end of the clinical spectrum, clear-cell pRCCs and multilocular cystic renal neoplasm of low malignant potential behave more like benign neoplasms and are considered as neoplasms of uncertain malignant potential. Moreover, as discussed subsequently, ccRCCs stand apart from the non-clear cell carcinomas from a treatment standpoint.
Oncocytic/eosinophilic tumors
Tumors with oncocytic/eosinophilic cells (Figure 2D–F) span a range of conventional and recently described entities with low- and high-grade morphologies that include oncocytoma, eosinophilic variant of chromophobe RCC, ccRCC with eosinophilic/granular cells, high-grade renal epithelial neoplasms with rhabdoid differentiation, succinate dehydrogenase (SDH)-deficient RCCs, TSC-associated RCCs, eosinophilic, solid, and cystic (ESC)-RCCs, tubulocystic RCCs, and epithelioid angiomyolipomas (PEComas). Besides these, some low-grade oncocytic neoplasms remain unclassified and differ from oncocytoma in having a denser nested or solid architecture without the characteristic myxoid/hyalinized background. In recent studies and in our experience, renal neoplasms with oncocytic/eosinophilic cytoplasm are among the most challenging, diagnostically.10
The prototypic oncocytic renal tumor, oncocytoma, requires a stringent set of morphologic criteria, of small nests of cells in a myxoid or hyalinized stroma, with uniform round nuclei that may have prominent nucleoli, while lacking significant areas of clear cells, papillary formation, or necrosis24 (Figure 2E). Although degenerative-type atypia and areas of increased cellularity and proliferation may be seen, mitoses are usually absent or rare. Benign renal tubules may be entrapped at the periphery.
Oncocytomas show significant morphologic overlap with eosinophilic variant of chromophobe RCC and tumors with hybrid chromophobe and oncocytic morphology that augment the diagnostic challenges in borderline tumors and small biopsies. Differences in diagnostic criteria and reporting practices for these tumors are also seen among experts.25 While understanding the limitations around their diagnosis, the differentiation between oncocytoma from chromophobe carcinomas largely relies on cytologic features. The cells of chromophobe tumors have abundant cytoplasm with prominent cell borders, imparting a plant cell wall-like appearance. Also, very characteristics are perinuclear halos and wrinkled nuclear membranes with “raisinoid” or pseudokoilocytic nuclei (Figures 1H and 2D). Both oncocytoma and chromophobe RCCs are positive for CD117. CK7 is typically diffusely positive in chromophobe, while negative or focal in scattered cells in oncocytoma. Hybrid tumors with overlapping features of renal oncocytoma and eosinophilic chromophobe RCCs, though recognized to be a part of the spectrum of Birt–Hogg–Dubé syndrome and renal oncocytosis, are not a distinct category in the WHO 2016 classification.
Rhabdoid differentiation, which is a high-grade (ISUP grade 4) pattern that may be demonstrated by many different types of RCC, consists of cells, usually enlarged with eccentric, high-grade nuclei with prominent nucleoli displaced by dense cytoplasmic eosinophilic inclusions (Figure 1D) and may mimic oncocytic change. The result is a cytologic pattern that resembles cells (substantially enlarged) as of a rhabdoid tumor of the kidney or even rhabdomyoblasts. Finding even focal areas showing characteristic morphology can establish the RCC type of origin, and additional sampling may be useful in arriving at a classification.
More recently described entities in this differential include SDH-deficient RCCs, such as that arise in the hereditary paraganglioma/pheochromocytoma syndromes (PGL1-4) or Carney Stratakis syndrome (with GISTs), in patients harboring germline mutation of SDH subunit genes. These are generally eosinophilic tumors, solid, nested, or tubular, with prominent cytoplasmic vacuoles, some of which show almost inclusion-like spaces containing eosinophilic fluid or flocculent material (26) (Figure 2F). They have round nuclei with neuroendocrine-like chromatin, intratumoral mast cells, and entrapped benign renal tubules.26,27 Although microcysts may be seen, macrocysts such as those seen in ESC-RCCs and tubuolocystic RCCs are unusual. Biphasic morphology overlapping with t(6;11) RCC9 may be seen. In clinical practice, screening for SDH deficiency is done by SDHB immunohistochemical stain which also identifies deficiencies in other subunits of the enzyme (SDHA, SDHC, SDHD, SDHAF2) that result in destabilization of the SDH complex. A specific antibody against SDHA is also available to detect deficiency of this enzyme.28
TSC complex (TSC) is a multiorgan neoplastic predisposition syndrome with numerous manifestations that include, in the kidney, angiomyolipomas (AML) and RCCs. The RCCs arising in Tuberous Sclerosis can have three distinct patterns: RAT-like, chromophobe-like, and a granular eosinophilic type.29 RAT-like and granular eosinophilic type may be considered in the differential of translocation RCCs.
Based on experience with the granular eosinophilic tumors of TSC, an indolent tumor morphologically similar to TSC-associated RCC has been recently described in female patients with no clinical features/associations of TSC. Named after the morphologic spectrum seen in this entity, ESC-RCCs30 are characterized by solid and cystic architecture with cysts lined by cells with eosinophilic/granular cytoplasm with hobnailing protrusion into lumina. An acinar or nested growth pattern and microcysts may be seen. Voluminous eosinophilic cytoplasm, coarse cytoplasmic precipitates or stippling, and admixed histiocytes, lymphocytes, and multinucleate giant cells are other features of these tumors. Clear-cell features with voluminous cytoplasm, and papillary and nested architecture that are seen in MiTF-RCC are typically not seen in ESC-RCC. These tumors are at least focally positive for CK20, with a characteristic CK20-positive/CK7-negative immunophenotype.
Although not an epithelial neoplasm, epithelioid AML (PEComa) can have clear and eosinophilic cells arranged in nests, alveolar-like patterns, or solid sheets, and may demonstrate atypia and mitoses. Significant amounts of adipose, smooth muscle, and vascular differentiation are lacking, essentially by definition, as authors have generally used a high threshold (>80%) in terms of predominance of epithelioid pattern for their diagnosis. Similar to conventional AMLs, they express HMB-45, Melan-A, and Cathepsin K, and therefore can have significant morphologic and immunophenotypic overlap with MiTF-RCC. However, epithelioid AMLs are negative for keratins and PAX8 and express smooth muscle actin.
Thus, in the end, oncocytic renal neoplasms remain a significant area of challenge between new entities and unclassified oncocytic tumors. The overall approach should focus on documentation that the tumor represents bona fide RCC using markers such as PAX8 and keratin, excluding PEComa and other tumors, and then on establishing a tumor in the oncocytic class, especially with markers such as CD117 or KSP-cadherin. Especially in young patients, pathologists should consider recently described entities, consider any evidence of a syndromal context, and have a low threshold for performing FH, SDHB, and CK20 IHC, with consideration of FH- and SDH-deficient RCCs, or ESC-RCCs, respectively.9
High-grade distal nephron adenocarcinoma morphology
A group of high-grade renal cell tumors, centered in the renal medulla with overlapping adenocarcinomatous morphology, includes CDC and renal medullary carcinoma (RMC). Even more challenging, these tumors share striking morphologic overlap with recently characterized FH-deficient RCCs. The latter tumors generally arise in the syndrome of HLRCC, with cutaneous and uterine leiomyomatosis and germline mutations in the FH gene. The high-grade kidney tumors in this syndrome show an infiltrative adenocarcinomatous pattern overlapping with CDC and RMC, though their relation to the collecting system of the distal nephron, per se, remains unclear.
In FH-deficient RCCs, enzymatic FH deficiency results in intracellular accumulation of aberrantly succinated proteins, driving pseudohypoxic signaling and aberrant chromatin and DNA methylation.31 Most cases with FH mutation demonstrate loss of expression of FH itself by immunohistochemistry, as well as aberrant expression of 2 succinyl-cysteine (2SC) on cytoplasmic and nuclear proteins, useful in confirming the diagnosis. FH-deficient RCCs have a wide spectrum of morphologic patterns32 that overlap with CDC and RMC, with intracystic papillary pattern with prominent stromal hyalinization (Figure 3A) and tubulocystic patterns (Figure 3B) being the most specific to this tumor type. Prominent viral inclusion-like nucleoli with perinuclear halos have been described as a very sensitive but not specific feature characteristic of the FH-deficient RCCs of HLRCC, and may also be focally seen in RMC and CDC.
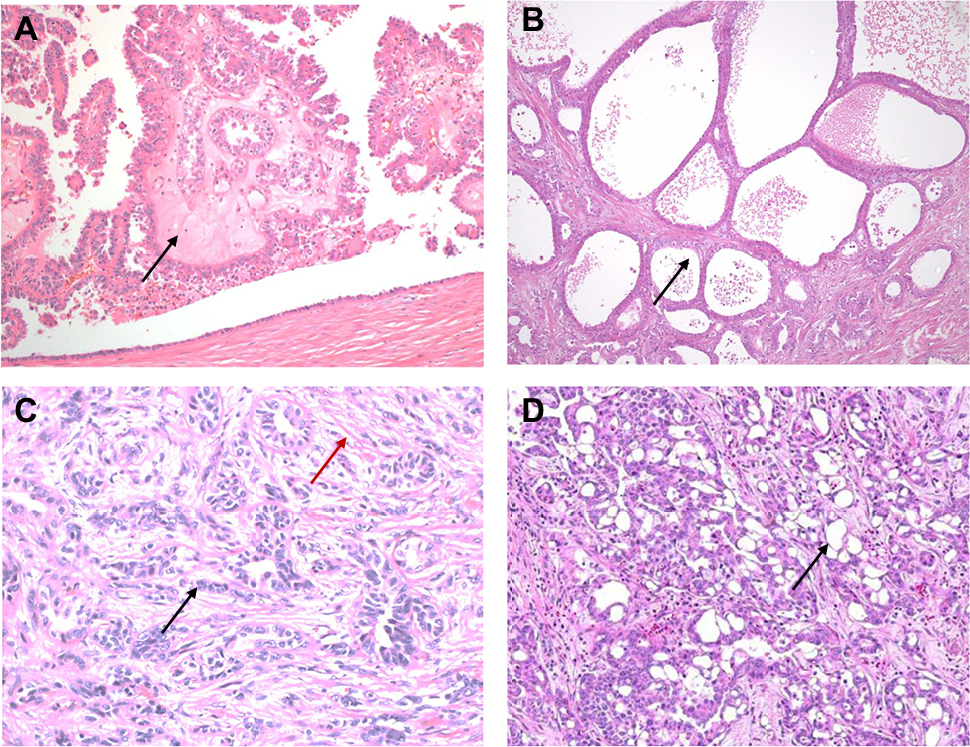 | Figure 3 (A–D) RCCs with high-grade distal nephron adenocarcinoma morphology. (A) An intracystic papillary pattern with hyalinized cores (black arrow) is seen in this case of fumarate hydratase-deficient RCC. (B) Tubulocystic RCC with infiltrating tubules and cystic spaces (black arrow) lined by atypical cells. (C) A collecting duct carcinoma with infiltrating angulated glands (black arrow) in a desmoplastic stroma (red arrow). (D) Renal medullary carcinoma with a reticular yolk sac-like pattern (black arrow). Abbreviation: RCC, renal cell carcinoma. |
CDC is an aggressive, infiltrative, and high-grade neoplasm arising in the medulla. A diagnosis of CDC requires a tubular, solid, or papillary adenocarcinoma, with high-grade cytology, infiltrative growth pattern, and a desmoplastic stromal reaction (Figure 3C), and at least focally involving the renal medulla in the absence of any other recognizable RCC subtype.6 Renal intratubular dysplasia involving adjacent tubules may be identified and seems to be a relatively specific feature. A diagnosis of CDC requires careful exclusion of RMC, FH-deficient RCC, UCs of upper tract, and metastatic carcinomas. Specifically, exclusion of UC of the upper urothelial tract is important because of treatment implications necessitating a nephroureterectomy with bladder cuff for UCs vs just a nephrectomy alone for primary RCC.
RMC are high-grade tumors with poorly differentiated glands infiltrating an inflammatory, myxoid, or desmoplastic stroma in individuals with sickle cell trait/disease. Other patterns that may be seen include reticular yolk sac and sieve-like/cribriform patterns (Figure 3D). Evidence of sickle cell disease or trait either as sickled erythrocytes (drepanocytes) in histologic sections or clinical or laboratory evidence of a hemoglobinopathy is supportive of the diagnosis. Further confirmation of the diagnosis may be supported by biallelic inactivation of SMARCB1 (INI-1) through deletion and/or genomic rearrangements, with overexpression of POU5F1 (Oct3/4) with widely available immunohistochemical assays. Although INI-1 loss is characteristically seen in RMC, it has been reported to occur in up to 15% of CDC, though the degree of exclusion of hemoglobinopathy in such cases is unclear.33 As such, when considering a diagnosis of CDC in this setting, the possibility of a clinically occult hemoglobinopathy should be raised with the clinical team to ensure further evaluation.34
Differentiating a primary renal carcinoma from a metastatic or UC of the upper urothelial tract requires careful clinicopathologic correlation incorporating a good history and judicious use of contemporary immunostains. While PAX8 positivity favors a renal primary, especially if gynecologic and thyroidal tumors are excluded, it may also be seen in up to 10%–20% of upper tract UCs, although it is usually weak and patchy.35 p63, GATA3, and uroplakin 2 positivity, on the other hand, favors a primary UC.36 Overall, it is our experience that correlation with clinical findings, any endoscopic examinations, and cytology samples, taken with extensive sampling of the pelvicalyceal mucosa, can resolve nearly all cases providing diagnostic challenge between RCC and UC.36
Thus, in consideration of diagnosis of an unclassified RCC with poorly differentiated adenocarcinomatous morphology vs CDC and RMC, we recommend rigorous exclusion of UC and metastasis and clinical correlation with any hemoglobinopathy. Use of SMARCB1 immunohistochemistry can be quite helpful, but we have noted that bona fide unclassified RCCs with medullary phenotype occur.37 To guide resource-conscious workup, we have recently studied the morphologic overlap of eight patterns across CDC, RMC, and FH-deficient RCC, highlighting the importance of recognition of a sieve-like/cribriform and reticular/yolk sac tumor-like pattern favoring a diagnosis of RMC, while intracystic papillary and tubulocystic patterns favored FH-deficient RCCs, and multinodular infiltrating pattern with dysplastic intratubular in situ changes favoring CDC.32 After exclusion of these recognized types, and unclassified carcinoma, remains the favored diagnosis.
Sarcomatoid RCCs
Although not a distinct diagnostic entity, sarcomatoid morphology can be seen across different subtypes of RCCs, being present in about 26% of patients with T4 disease.38 The differential diagnosis for predominantly spindled renal carcinomas includes sarcomatoid carcinomas arising in a background of one of the known subtypes, angiomyolipoma and retroperitoneal sarcomas, secondarily involving the kidney. These tumors require adequate sampling for identification of an underlying epithelial component and use of immunohistochemical stains to support a renal epithelial origin while excluding the possibility of primary retroperitoneal sarcomas that may extend to involve the kidney. Angiomyolipoma may have a predominantly spindled morphology and is previously discussed.
In conclusion, a diagnosis of unclassified RCC requires a comprehensive and systematic workup to exclude established diagnostic categories. With increasing availability of next-generation sequencing assays, a number of aggressive, high-grade, and unclassified renal tumors are being submitted to identify therapeutic targets and/or for diagnostic purpose. While clarifying the underlying alterations across different subtypes, in some studies, these have also proved to be efficacious in identifying molecular alterations across 76%39–41 of previously unclassified renal carcinomas.
Treatment modalities
Molecular characterization of unclassified RCCs demonstrates many shared underlying genomic alterations compared with ccRCCs such as TP53 alterations, MTOR mutations, and NF2 mutations, while there are distinct differences such as lack of VHL alterations with unclassified RCC compared to ccRCC.39–41 These molecular differences support the variation in prognosis between ccRCC and non-clear-cell RCC in clinical practice. Despite this difference, NCCN guidelines generally suggest similar treatment recommendations between these two categories.
Treatment strategies for unclassified RCC are determined by tumor stage, amenability to resection, and comorbidities, similar to ccRCC. For localized disease, defined as stages I–III, surgical resection remains the preferred treatment irrespective of the histologic type, including unclassified RCC. For localized disease, partial nephrectomy (PN) has been shown to have comparable outcomes to radical nephrectomy (RN). As such, a PN is recommended for small, localized tumors amenable to this approach. PN offers the advantage of preserving renal function and decreased overall mortality42 regardless of tumor histology and reduced frequency of chronic kidney disease. RN is the treatment of choice for locally advanced tumors that are not surgically amenable to PN and tumors extending into inferior vena cava. Alternate options for localized disease include active surveillance or nephron-sparing approaches such as cryoablation for patient unfit for surgery. Cryoablation has a relatively low rate of complications even in patients unfit for surgery. It does have a higher rate of local treatment failure compared with nephrectomy, so it is not recommended for patients who are surgical candidates.42–44
There is no defined role for adjuvant therapy in patients with localized non-clear-cell RCC as there have not been any studies conducted in this setting. Active surveillance has not been specifically studied in the context of non-clear-cell RCC. However, Lane et al45 evaluated the OS of 537 patients with localized RCC who were ≥75 years old with tumors ≤7 cm and were treated with nephrectomy vs any nephron-sparing approach vs active surveillance. In their study, the 5-year OS was 72% (95% CI, 63%–80%) with nephrectomy, 76% (95% CI, 69%–81%) with a nephron-sparing intervention, and 58% (95% CI, 46%–69%) with active surveillance for T1a or T1b tumors. There was no association identified between treatment type and OS in all patients, regardless of tumor stage (P=0.3) in the multivariate model after accounting for age and comorbidities. In their study, only 2.3% of patients underwent a biopsy so many of these patients presumably had non-clear-cell RCC. This suggests that active surveillance may be an appropriate treatment strategy for non-clear-cell RCC in selected patients.
Neoadjuvant therapy with vascular endothelial growth factor receptor-tyrosine kinase inhibition (VEGFR-TKI) therapy has been tested in phase II clinical trials for localized ccRCC as a method of facilitating partial or complete nephrectomies in patients who otherwise are ineligible for these surgeries. Patients consistently have experienced relatively small reductions in tumor volume,46–48 but this may be effective in facilitating these surgeries in some patients who otherwise would not be eligible. However, this strategy has not been validated in randomized phase III clinical trials, nor has it been conducted in the setting of non-clear-cell RCC. Additionally, this strategy has not been demonstrated to increase the metastasis-free survival or OS. Therefore, neoadjuvant therapy with VEGFR-TKI therapy is not a recommended treatment strategy at this time. No studies have been completed in the neoadjuvant setting using immunotherapy approaches to date, so this is also not a recommended treatment strategy at this time.
The role for cytoreductive nephrectomy in the setting of metastatic disease is controversial. There are no randomized phase III studies on non-clear-cell RCC evaluating this issue. Recently, the CARMENA study was reported, addressing this question in ccRCC. Patients treated with nephrectomy in combination with sunitinib has inferior OS (13.9 months) compared to patients treated with sunitinib alone (18.4 months), though the two arms were found to be non-inferior (stratified hazard ratio for death, 0.89; 95% CI, 0.71–1.10; upper boundary of the 95% CI for noninferiority, ≤1.20). The role for cytoreductive nephrectomy in patients with non-clear-cell RCC is unclear and should be approached with caution. It should only be considered for highly selected patients,49 especially since many of these patients (for example, those with sarcomatoid or unclassified RCC) have a relatively poor prognosis compared to patients with ccRCC.
Systemic therapy is the cornerstone for surgically unresectable or advanced disease and for metastatic disease. The selection of systemic therapy is largely determined by the tumor histology categorized as ccRCC or non-clear-cell RCC (including unclassified RCC).11 Clear cell is the predominant histologic type, so most clinical trials thus so far have included only patients with clear cell histology. Recently, a few clinical trials have been conducted in patients exclusively with non-clear-cell RCC, which provide valuable guidance into the optimal treatment for these patients. Selected ongoing prospective clinical trials in non-clear cell renal carcinoma are listed in Table 2.
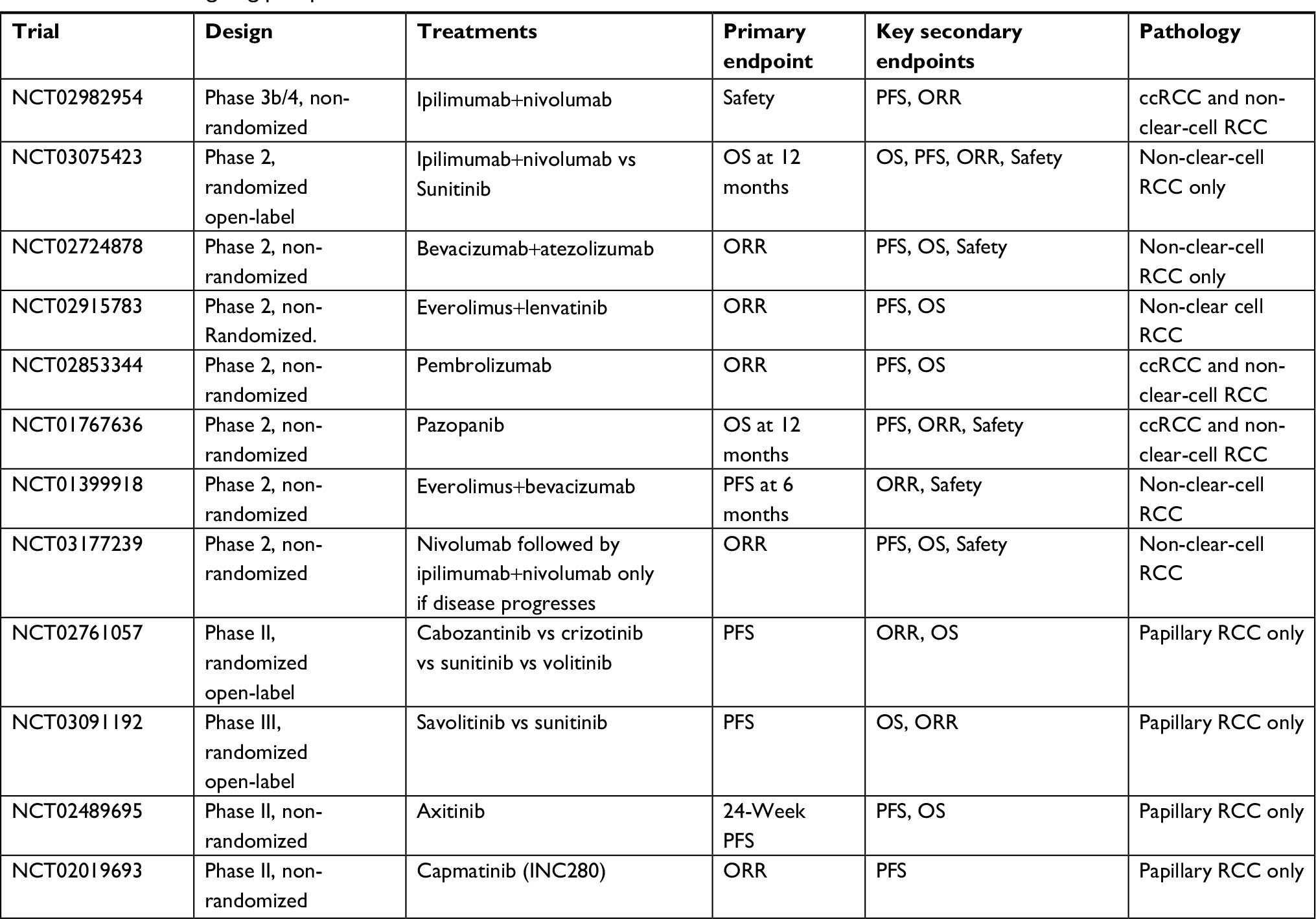 | Table 2 Selected ongoing prospective clinical trials in non-clear-cell renal carcinoma Abbreviations: ORR, overall response rate; OS, overall survival; PFS, progression-free survival; RCC, renal cell carcinoma. |
Sunitinib is a VEGFR-TKI inhibitor targeting platelet-derived growth factor receptor, VEGF receptor, stem cell factor receptor (c-KIT), FMS-like tyrosine kinase (FLT-3), colony-stimulating factor (CSF-1R), and neurotrophic factor receptor (RET). It has been compared with everolimus (an mTOR antagonist) in two randomized studies, the ASPEN50 and ESPN51 trials. Both demonstrated improved progression-free survival (PFS) and OS for sunitinib compared with everolimus and both studies included a significant proportion of patients with unclassified RCC (17.9% and 14.7%, respectively). The RECORD-3 study52 also confirmed these findings. It is a cross-over study of 471 patients treated with sunitinib followed by everolimus compared with the reverse sequence. The everolimus–sunitinib arm was found to be inferior to the sunitinib–everolimus sequence regarding combined PFS with a median of 21.1 months vs 25.8 months, respectively (HR 1.3; 95% CI, 0.9–1.7) and OS with a median of 22.4 months and 32.0 months (HR, 1.2; 95% CI, 0.9–1.6). This study included ccRCC and non-clear-cell patients (only 14%). Sunitinib is the preferred first-line option for advanced non-clear-cell RCC according to the NCCN recommendations.11,53 Other VEGFR-TKI therapies are also recommended for use in these patients and are supported by less robust data. For instance, pazopanib has also demonstrated activity in the first-line setting for non-clear-cell RCC in an Italian retrospective54 study of 37 patients, 8 of which had unclassified RCC (22%). Overall, 81% of patients had disease control defined as achieving a partial response or stable disease. The median PFS and OS were 15.9 months and 17.3 months, respectively. Cabozantinib, axitinib, bevacizumab, erlotinib, lenvatinib+everolimus, and sorafenib are also listed as approved VEGF-directed therapies for non-clear-cell RCC.
Temsirolimus is the only therapy listed in the NCCN guidelines which has been tested in a randomized phase 3 clinical trial for patients with non-clear-cell RCC.55 Temsirolimus was compared with interferon-α in the ARCC trial. In a subgroup analysis evaluating 73 patients with non-clear-cell histology (18/73 with unclassified histology, 25%), treatment with temsirolimus compared with interferon-α demonstrated improvement in tumor reduction of any amount (68% vs 14%; no difference in objective response rate, however, at 12% vs 12%), improved PFS (7.0 months vs 1.8 months; HR 0.38, 95% CI 0.23–0.62) and improved OS (11.6 months vs 4.3 months; HR 0.49, 95% CI 0.29–0.85). NCCN guidelines thus list temsirolimus as a category 1 recommendation for patients with non-clear-cell carcinoma with poor prognosis features (according to Memorial Sloan Kettering Cancer Center risk criteria), while it remains a category 2A recommendation for patients belonging to other prognostic non-clear-cell risk groups. As discussed previously in the ASPEN50 and ESPN51 studies, everolimus does have some activity in this disease and is also listed as an approved therapy for this disease.
Like many other tumors, immunotherapy has shown efficacy in renal tumors and is approved for treatment of metastatic ccRCC.56 Nivolumab, a monoclonal antibody directed against programmed death 1 receptor (PD-1), has demonstrated improved OS in previously treated patients with advanced RCC compared with everolimus.57,58 The role for immunotherapy in non-clear-cell carcinoma is less well defined. Some data suggest that immunotherapy may be effective for unclassified as well as other non-clear-cell RCC histologies. For instance, the ARCC study55 demonstrated some activity for interferon-α in this disease, though with modest benefits only. High-dose interleukin-2 has also demonstrated modest activity in patients with sarcomatoid RCC, with an overall response rate of 10% and a complete response rate of 5%.59 Case reports60–62 also support the use of nivolumab in non-clear RCC patients. Recently, a more comprehensive retrospective analysis was reported from six centers in the United States, evaluating 41 patients treated with nivolumab of which 14 had unclassified RCC. A total of 35 patients were evaluable for response, 7 (20%) had a partial response, and 10 (29%) had stable disease. No patients experienced a complete response. These data support the NCCN guidelines which include nivolumab therapy for non-clear RCC.
Chemotherapy plays a role in the management of a subset of aggressive non-clear-cell RCCs including sarcomatoid RCCs, CDCs, and RMCs. The potential role for chemotherapy in unclassified RCC is untested and at this time is not recommended outside of a clinical trial.
Increasingly the non-clear-cell RCCs are being further subdivided into separate, pathologically distinct groups regarding treatment recommendations. The pathogenic drivers appear to be distinctly different, leading to differences in optimal treatment strategies. For instance, MET alterations drive pRCC63 and MET-targeted treatments have demonstrated significant clinical activity in pRCC.64 This led to the development of SWOG1500 which is designed to identify the optimal first-line MET-targeted therapy in pRCC.
Conclusion
While the unclassified RCCs present a formidable diagnostic approach, the genomic data available from The Cancer Genome Atlas and other studies provide insights into the molecular characteristics of this disease and potentially actionable targets. The molecular analysis of 62 unclassified renal cancers by Chen et al41 found recurrent alterations in many key and potentially targetable pathways in unclassified RCCs. These include CHECK2 alterations or PBRM1 alterations (potentially respond to checkpoint inhibitors), ALK translocations (ALK-targeted therapies), and ATM/BRCA2 alterations (PARP inhibitors), as examples. For unclassified RCCs, underlying molecular alterations may guide treatment decisions, expanding the scope of precision medicine for unclassified RCCs in the future, though these targets need to be tested in clinical trials.
Disclosure
Dr Neeraj Agarwal reports consultancy to Pfizer, Novartis, Merck, Genentech, Eisai, Exelixis, Clovis, EMD Serono, BMS, Astra Zeneca, Astellas, Eli Lilly and Armo Biosciences. Dr Benjamin Maughan reports consultancy for Janssen Oncology on an advisory board. The authors report no other conflicts of interest in this work.
References
Thoenes W, Störkel S, Rumpelt HJ. Histopathology and classification of renal cell tumors (adenomas, oncocytomas and carcinomas).The basic cytological and histopathological elements and their use for diagnostics. Pathol Res Pract. 1986;181(2):125–143. | ||
Kovacs G, Akhtar M, Beckwith BJ, et al. The Heidelberg classification of renal cell tumours. J Pathol. 1997;183(2):131–133. | ||
Störkel S, Eble JN, Adlakha K, et al. Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 1997;80(5):987–989. | ||
Eble JNSG, Epstein JI, Sesterhenn IA. Pathology and Genetics of Tumors of the Urinary System and Male Genital Organs. Lyon: IARC Press; 2004. | ||
Srigley JR, Delahunt B, Eble JN, et al; ISUP Renal Tumor Panel. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol. 2013;37(10):1469–1489. | ||
Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-part a: renal, penile, and testicular tumours. Eur Urol. 2016;70(1):93–105. | ||
Nguyen DP, Vertosick EA, Corradi RB, et al. Histological subtype of renal cell carcinoma significantly affects survival in the era of partial nephrectomy. Urol Oncol. 2016;34(6):259.e1–e8. | ||
Amin MB, Amin MB, Tamboli P, et al. Prognostic impact of histologic subtyping of adult renal epithelial neoplasms: an experience of 405 cases. Am J Surg Pathol. 2002;26(3):281–291. | ||
Li Y, Reuter VE, Matoso A, Netto GJ, Epstein JI, Argani P. Re-evaluation of 33 “unclassified”eosinophilic renal cell carcinomas in young patients. Histopathology. 2018;72(4):588–600. | ||
Perrino CM, Grignon DJ, Williamson SR, Idrees MT, Eble JN, Cheng L. Morphological spectrum of renal cell carcinoma, unclassified: an analysis of 136 cases. Histopathology. 2018;72(2):305–319. | ||
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines)-Kidney Cancer; 2018. Available from: https://www.nccn.org/. Accessed November 13, 2018. | ||
Kryvenko ON, Roquero L, Gupta NS, Lee MW, Epstein JI. Low-grade clear cell renal cell carcinoma mimicking hemangioma of the kidney: a series of 4 cases. Arch Pathol Lab Med. 2013;137(2):251–254. | ||
Reuter VE, Argani P, Zhou M, Delahunt B; Members of the ISUP Immunohistochemistry in Diagnostic Urologic Pathology Group. Best practices recommendations in the application of immunohistochemistry in the kidney tumors: report from the International Society of Urologic Pathology consensus conference. Am J Surg Pathol. 2014;38(8):e35–e49. | ||
Rohan SM, Xiao Y, Liang Y, et al. Clear-cell papillary renal cell carcinoma: molecular and immunohistochemical analysis with emphasis on the von Hippel-Lindau gene and hypoxia-inducible factor pathway-related proteins. Mod Pathol. 2011;24(9):1207–1220. | ||
Leroy X, Camparo P, Gnemmi V, et al. Clear cell papillary renal cell carcinoma is an indolent and low-grade neoplasm with overexpression of cyclin-D1. Histopathology. 2014;64(7):1032–1036. | ||
Mantilla JG, Antic T, Tretiakova M. GATA3 as a valuable marker to distinguish clear cell papillary renal cell carcinomas from morphologic mimics. Hum Pathol. 2017;66:152–158. | ||
Hakimi AA, Tickoo SK, Jacobsen A, et al. TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Mod Pathol. 2015;28(6):845–853. | ||
Deml KF, Schildhaus HU, Compérat E, et al. Clear cell papillary renal cell carcinoma and renal angiomyoadenomatous tumor: two variants of a morphologic, immunohistochemical, and genetic distinct entity of renal cell carcinoma. Am J Surg Pathol. 2015;39(7):889–901. | ||
Cancer Genome Atlas Research Network; Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med. 2016;374(2):135–145. | ||
Argani P, Olgac S, Tickoo SK, et al. Xp11 translocation renal cell carcinoma in adults: expanded clinical, pathologic, and genetic spectrum. Am J Surg Pathol. 2007;31(8):1149–1160. | ||
Magers MJ, Udager AM, Mehra R. MiT family translocation-associated renal cell carcinoma: a contemporary update with emphasis on morphologic, immunophenotypic, and molecular mimics. Arch Pathol Lab Med. 2015;139(10):1224–1233. | ||
Rao Q, Williamson SR, Zhang S, et al. TFE3 break-apart FISH has a higher sensitivity for Xp11.2 translocation-associated renal cell carcinoma compared with TFE3 or cathepsin K immunohistochemical staining alone: expanding the morphologic spectrum. Am J Surg Pathol.2013;37(6):804–815. | ||
Argani P, Yonescu R, Morsberger L, et al. Molecular confirmation of t(6;11)(p21;q12) renal cell carcinoma in archival paraffin-embedded material using a break-apart TFEB FISH assay expands its clinicopathologic spectrum. Am J Surg Pathol. 2012;36(10):1516–1526. | ||
Amin MB, Crotty TB, Tickoo SK, Farrow GM. Renal oncocytoma: a reappraisal of morphologic features with clinicopathologic findings in 80 cases. Am J Surg Pathol. 1997;21(1):1–12. | ||
Williamson SR, Gadde R, Trpkov K, et al. Diagnostic criteria for oncocytic renal neoplasms: a survey of urologic pathologists. Hum Pathol. 2017;63:149–156. | ||
Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol. 2015;28(1):80–94. | ||
Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol. 2014;38(12):1588–1602. | ||
Miettinen M, Sarlomo-Rikala M, McCue P, et al. Mapping of succinate dehydrogenase losses in 2258 epithelial neoplasms. Appl Immunohistochem Mol Morphol. 2014;22(1):31–36. | ||
Guo J, Tretiakova MS, Troxell ML, et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014;38(11):1457–1467. | ||
Trpkov K, Abou-Ouf H, Hes O, et al. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC): Further Morphologic and Molecular Characterization of ESC RCC as a Distinct Entity. Am J Surg Pathol. 2017;41(10):1299–1308. | ||
Bardella C, El-Bahrawy M, Frizzell N, et al. Aberrant succination of proteins in fumaratehydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011;225(1):4–11. | ||
Ohe C, Smith SC, Sirohi D, et al. Reappraisal of morphologic differences between renal medullary carcinoma, collecting duct carcinoma, and fumaratehydratase-deficient renal cell carcinoma. Am J Surg Pathol. 2018;42(3):279–292. | ||
Gupta R, Billis A, Shah RB, et al. Carcinoma of the collecting ducts of Bellini and renal medullary carcinoma: clinicopathologic analysis of 52 cases of rare aggressive subtypes of renal cell carcinoma with a focus on their interrelationship. Am J Surg Pathol. 2012;36(9):1265–1278. | ||
Singh JA, Ohe C, Smith SC. High grade infiltrative adenocarcinomas of renal cell origin: New insights into classification, morphology, and molecular pathogenesis. Pathol Int. 2018;68(5):265–277. | ||
Carvalho JC, Thomas DG, McHugh JB, Shah RB, Kunju LP. p63, CK7, PAX8 and INI-1: an optimal immunohistochemical panel to distinguish poorly differentiated urothelial cell carcinoma from high-grade tumours of the renal collecting system. Histopathology. 2012;60(4):597–608. | ||
Amin MB, Trpkov K, Lopez-Beltran A, Grignon D; Members of the ISUP Immunohistochemistry in Diagnostic Urologic Pathology Group. Best practices recommendations in the application of immunohistochemistry in the bladder lesions: report from the International Society of Urologic Pathology consensus conference. Am J Surg Pathol. 2014;38(8):e20–34. | ||
Sirohi D, Smith SC, Ohe C, et al. Renal cell carcinoma, unclassified with medullary phenotype: poorly differentiated adenocarcinomas overlapping with renal medullary carcinoma. Hum Pathol. 2017;67:134–145. | ||
Shuch B, Said J, La Rochelle JC, et al. Cytoreductive nephrectomy for kidney cancer with sarcomatoid histology--is up-front resection indicated and, if not, is it avoidable? J Urol. 2009;182(5):2164–2171. | ||
Hu ZY, Pang LJ, Qi Y, et al. Unclassified renal cell carcinoma: a clinicopathological, comparative genomic hybridization, and whole-genome exon sequencing study. Int J Clin Exp Pathol. 2014;7(7):3865–3875. | ||
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma.Nature.2013;499(7456):43–49. | ||
Chen YB, Xu J, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131. | ||
Pierorazio PM, Johnson MH, Patel HD. Management of renal masses and localized renal cancer. Rockville (MD): AHRQ Comparative Effectiveness Reviews No. 167; 2016. Available from: https://www.ncbi.nlm.nih.gov/books/NBK350333/. Accessed November 11, 2018. | ||
Georgiades CS, Hong K, Bizzell C, Geschwind JF, Rodriguez R. Safety and efficacy of CT-guided percutaneous cryoablation for renal cell carcinoma. J Vasc Interv Radiol. 2008;19(9):1302–1310. | ||
Kunkle DA, Uzzo RG. Cryoablation or radiofrequency ablation of the small renal mass: a meta-analysis. Cancer. 2008;113(10):2671–2680. | ||
Lane BR, Abouassaly R, Gao T, et al. Active treatment of localized renal tumors may not impact overall survival in patients aged 75 years or older. Cancer. 2010;116(13):3119–3126. | ||
Rini BI, Plimack ER, Takagi T, et al. A Phase II Study of Pazopanib in Patients with Localized Renal Cell Carcinoma to Optimize Preservation of Renal Parenchyma. J Urol. 2015;194(2):297–303. | ||
Karam JA, Devine CE, Urbauer DL, et al. Phase 2 trial of neoadjuvantaxitinib in patients with locally advanced nonmetastatic clear cell renal cell carcinoma. Eur Urol. 2014;66(5):874–880. | ||
Powles T, Sarwar N, Stockdale A, et al. Safety and efficacy of pazopanib therapy prior to planned nephrectomy in metastatic clear cell renal cancer. JAMA Oncol. 2016;2(10):1303–1309. | ||
Motzer RJ, Russo P. Cytoreductive nephrectomy - patient selection is key. N Engl J Med. 2018;379(5):481–482. | ||
Armstrong AJ, Halabi S, Eisen T, et al. Everolimus versus sunitinib for patients with metastatic non-clear cell renal cell carcinoma (ASPEN): a multicentre, open-label, randomised phase 2 trial. Lancet Oncol. 2016;17(3):378–388. | ||
Tannir NM, Jonasch E, Albiges L, et al. Everolimus Versus Sunitinib Prospective Evaluation in Metastatic Non-Clear Cell Renal Cell Carcinoma (ESPN): A Randomized Multicenter Phase 2 Trial. Eur Urol. 2016;69(5):866–874. | ||
Motzer RJ, Barrios CH, Kim TM, et al. Phase II randomized trial comparing sequential first-line everolimus and second-line sunitinib versus first-line sunitinib and second-line everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2014;32(25):2765–2772. | ||
Ciccarese C, Iacovelli R, Brunelli M, et al. Addressing the best treatment for non-clear cell renal cell carcinoma: A meta-analysis of randomised clinical trials comparing VEGFR-TKis versus mTORi-targeted therapies. Eur J Cancer. 2017;83:237–246. | ||
Buti S, Bersanelli M, Maines F, et al. First-Line PAzopanib in NOn-clear-cell RenalcArcinoMA: The Italian Retrospective Multicenter PANORAMA Study. Clin Genitourin Cancer. 2017;15(4):e609–e614. | ||
Dutcher JP, de Souza P, McDermott D, et al. Effect of temsirolimus versus interferon-alpha on outcome of patients with advanced renal cell carcinoma of different tumor histologies. Med Oncol. 2009;26(2):202–209. | ||
Xu JX, Maher VE, Zhang L, et al. FDA Approval summary: nivolumab in advanced renal cell carcinoma after anti-angiogenic therapy and exploratory predictive biomarker analysis. Oncologist. 2017;22(3):311–317. | ||
Motzer RJ, Escudier B, McDermott DF, et al; CheckMate 025 Investigators. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1803–1813. | ||
Escudier B, Sharma P, McDermott DF, et al; CheckMate 025 investigators. CheckMate 025 Randomized Phase 3 Study: outcomes by key baseline factors and prior therapy for nivolumab versus everolimus in advanced renal cell carcinoma. Eur Urol. 2017;72(6):962–971. | ||
Achkar T, Arjunan A, Wang H, et al. High-dose interleukin 2 in patients with metastatic renal cell carcinoma with sarcomatoid features. PLoS One.2017;12(12):e0190084. | ||
Noguchi G, Tsutsumi S, Yasui M, et al. Significant response to nivolumab for metastatic chromophobe renal cell carcinoma with sarcomatoid differentiation: a case report. BMC Urol. 2018;18(1):26. | ||
Rouvinov K, Osyntsov L, Shaco-Levy R, Baram N, Ariad S, Mermershtain W. Rapid response to nivolumab in a patient with sarcomatoid transformation of chromophobe renal cell carcinoma. Clin Genitourin Cancer. 2017;15(6):e1127–e1130. | ||
Mizutani K, Horie K, Nagai S, et al. Response to nivolumab in metastatic collecting duct carcinoma expressing PD-L1: A case report. Mol Clin Oncol. 2017;7(6):988–990. | ||
Shuch B, Hahn AW, Agarwal N. Current Treatment Landscape of Advanced Papillary Renal Cancer. J Clin Oncol. 2017;35(26):2981–2983. | ||
Choueiri TK, Plimack E, Arkenau HT, et al. Biomarker-based Phase II trial of savolitinib in patients with advanced papillary renal cell cancer. J Clin Oncol. 2017;35(26):2993–3001. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.