")
Back to Journals » OncoTargets and Therapy » Volume 12
Ubiquitin-specific protease 4 promotes glioblastoma multiforme via activating ERK pathway
Authors Zhou Y, Liang P, Ji W, Yu Z, Chen H, Jiang L
Received 7 June 2018
Accepted for publication 16 December 2018
Published 5 March 2019 Volume 2019:12 Pages 1825—1839
DOI https://doi.org/10.2147/OTT.S176582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
Yudong Zhou,1 Ping Liang,1 Wenyuan Ji,1 Zengpeng Yu,1 Hui Chen,1 Li Jiang1–4
1Department of Neurosurgery, Children’s Hospital of Chongqing Medical University, Chongqing 400014, People’s Republic of China; 2Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing 400014, People’s Republic of China; 3China International Science and Technology Cooperation Base of Child Development and Critical Disorders, Chongqing 400014, People’s Republic of China; 4Chongqing Key Laboratory of Translational Medical Research in Cognitive Development and Learning and Memory Disorders, Chongqing 400014, People’s Republic of China
Background: Glioblastoma multiforme (GBM) is one of the most common brain tumors in adults. Current treatments cannot increase survival to a large extent, as the glioblastoma development mechanisms remain unknown. It has been well documented that ubiquitination contributes to tumor initiation and/or progression in many kinds of cancer. Ubiquitin-specific protease 4 (USP4), a member of deubiquitinating enzymes (DUBs) family, can remove ubiquitin residues and play a role in cancer development.
Methods: In the current study, lentiviruses were used to manipulate the expression of USP4. Real-time PCR and Western blot were used to measure the expression level of USP4. Then, CCK-8 and annexin-V staining were used to detect cell proliferation and cell apoptosis, respectively.
Results: First, we found that USP4 was highly upregulated in GBM tissues in comparison with that in normal tissues and high level of USP4 correlated with poor prognosis. Moreover, knockdown of USP4 could significantly inhibit cell proliferation and increase cell apoptosis in U87 and T98G cells. Cells with stable USP4 reduction exhibited slower tumor growth rate and smaller tumor size than the control group cells in a xenograft mouse model. Inhibition of USP4 downregulated the expression of PCNA, Bcl-2 and p-ERK1/2, but upregulated the expression of Bax both in vitro and in vivo. Inversely, USP4 overexpression could attenuate the effects contributed by ERK inhibitor. TGF-βR inhibition reduced level of TGF-βR1, p-smad2 and p-ERK1/2 which can partially be rescued by USP4 overexpression.
Conclusion: USP4, as a potential novel oncogene, promotes GBM by activation of ERK pathway through regulating TGF-β.
Keywords: USP4, GBM, ERK, ubiquitin
Introduction
Glioblastoma multiforme (GBM) is the most frequent type of incurable brain tumor in adults.1 Each year, around 13,000 cases are diagnosed in the USA.2 Currently, the clinical treatment for GBM includes maximal safe surgical resection, chemotherapy and radiotherapy, or a combination of these three approaches. Despite these treatments, the overall survival of patients with GBM remains poor: 14 months is the average survival time after initial diagnosis.3 To improve the overall survival rate, new biomarkers and genetic alterations need to be identified and may enhance earlier diagnosis and treatment.
Biomarkers and cancer-related genes are considered to play a key role in biological processes, including cell growth, apoptosis and adhesion. TRIM59 has been described as a new oncogene in GBM4 and PAX6 as a suppressor of tumor progression in GBM.5 However, the protein itself and post-translational modifications, such as ubiquitination, usually have a crucial role the biological functions of these genes. Therefore, it is significant to identify the regulation mechanisms of protein post-translational modifications, which may offer a novel approach for GBM treatment.
Among protein post-translational modifications, ubiquitination is very important and regulates various kinds of cellular biological functions, such as cell proliferation, inflammatory response, apoptosis and DNA-damage repair.6–8 Ubiquitin, which is a 76-amino acid protein, completes its functions through covalent attachment to a target protein, and then regulating the stability, localization, or activity of the target protein.9 Ubiquitination processes can be reversed by deubiquitination, which is regulated by deubiquitinating enzymes (DUBs). DUBs are a member of the protease superfamily, which are divided into five classes, including serine, aspartic, metallo, threonine and cysteine proteases. Most DUBs are cysteine proteases. DUBs can be further divided into five DUB subclasses, and ubiquitin-specific proteases (USPs) represent the largest one.10 Recent studies implicated that USPs played an important role in regulating several cancer-related pathways.11
Ubiquitin-specific protease 4 (USP4), a member of the USP family, is involved in diverse human cancers. On the one hand, USP4 operates as a tumor suppressor in breast cancer by inhibiting cell growth,12 and promotes cell apoptosis by deubiquitinating RIP1 in head and neck squamous cell carcinoma.13 On the other hand, USP4 promotes colorectal cancer cell growth by deubiquitinating PRL-3.14 USP4 works as a potential oncogene in colorectal cancers by regulating deubiquitination and stabilization of β-catenin.15 USP4 suppresses p53 and NF-κB activation by stabilizing HDAC2.16 Recently, USP4 is found to be a novel regulator of DNA repair and deubiquitylation of USP4 promotes homologous recombination.17 However, the role in GBM remains unclear and further studies are needed.
In the current study, we tried to elucidate the role of USP4 in GBM and found higher expression levels of USP4 in GBM tissues than in normal tissues. Knockdown of USP4 could inhibit cell proliferation and promote cell apoptosis via ERK signal pathway in vitro and in vivo. Moreover, in response to ERK inhibitor treatment, USP4 significantly reduced cell apoptosis in vitro. Furthermore, USP4 elevated p-ERK through regulating TGF-β. Taken together, these data suggest that USP4 may act as a novel oncogene in GBM and may be a potential therapeutic marker for GBM.
Materials and methods
Clinical tissues
Forty GBM and 20 normal clinical tissues were obtained from Children’s Hospital of Chongqing Medical University. The average age of the 40 patients from whom GBM tissues were taken was 55±4.3 years; 18 of them were male and 22 of them were female; 22 cases were stage IV, 12 cases were stage III and six cases were stage II. Written informed consents were collected from all patients. The study was approved by the Ethics Committee of Children’s Hospital of Chongqing Medical University (Chongqing, People’s Republic of China) and was conducted in compliance with the Declaration of Helsinki.
Cell lines
All the human GBM cell lines U87, U373, T98G, SHG44 and U251 were obtained from Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China) and cultured in high glucose DMEM medium supplemented with 10% fetal bovine serum (FBS, Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA), 100 U/mL penicillin and 100 μg/mL streptomycin. All cells were cultivated at 37°C in 5% CO2 in an incubator.
Bioinformatics analysis
USP4 gene expression data in GBM patients were obtained from The Cancer Genome Atlas website (TCGA). Survival analysis was performed using a standard Kaplan–Meier curve. A median USP4 expression was used as cutoff.
Lentivirus infection and inhibitor treatment
The knockdown and overexpression lentiviruses of human USP4 were synthesized by Shanghai Genechem Co., Ltd (Shanghai, China). Four knockdown lentiviruses were synthesized to determine the knockdown efficiency and two of them were chosen to study the knockdown effect. The sequences used are as below:
shUSP4-1: 5′-GCGTGGAATAAACTACTAA-3′;
shUSP4-2: 5′-CCTTTCTTCTAGATGGATT-3′;
shUSP4-3: 5′-GGTGGTCGCAGATGTGTAT-3′;
shUSP4-4: 5′-GCTGAACATGTCCGAGTTT-3′.
Cells were seeded into 6-well plates and cultured overnight. Appropriate lentivirus was added into the well respectively, according to lentivirus colony forming unit (CFU). Some of the cells were used to measure infection efficiency by real-time PCR and Western blot, and the remaining cells were cultured further. For generation stable cell line U87, 0.75 mg/mL G418 (Sigma-Aldrich Co., St Louis, MO, USA) was added to culture medium after 48 hours transduction of USP4 knockdown lentivirus. The culture medium was replaced every 2 days. After 2 weeks the medium was replaced with regular culture medium.
For USP4 overexpression experiments, U251 cells were cultured with 1 μM U0126 (Selleck, Houston, TX, USA), a common inhibitor of ERK1/2, after 48 hours transduction of USP4 overexpression lentivirus.
RNA extraction and real-time PCR
Total RNA of tissues and GBM cell lines were extracted using Trizol reagent kit (Invitrogen, Thermo Fisher Scientific). cDNA synthesis kit (Promega Corporation, Fitchburg, WI, USA) was used to reverse transcribe total RNA. SYBR Green qPCR Mixes (Thermo Fisher Scientific) was used to perform real-time PCR according to the user’s manual. This reaction was completed on ABI 7300 system (Applied Biosystems, Thermo Fisher Scientific). 2−ΔΔct method was used to calculate the expression of target genes and GAPDH was used as endogenous controls. Real-time PCR primers for target genes were as below:
USP4 Primer F 5′-ACCGAGGCGTGGAATAAAC-3′
Primer R 5′-TGGCAACTCAGCACATTGG-3′
PCNA Primer F 5′-GCCTGACAAATGCTTGCTGAC-3′
Primer R 5′-TTGAGTGCCTCCAACACCTTC-3′
Bcl-2 Primer F 5′-GCAGTGTGGTCTCCGAATGTC-3′
Primer R 5′-CATTGCCTCTCCTCACGTTCC-3′
Bax Primer F 5′-CTGAGCGAGTGTCTCAAG-3′
Primer R 5′-CAGCCCATGATGGTTCTG-3′
GAPDH Primer F 5′-AATCCCATCACCATCTTC-3′
Primer R 5′-AGGCTGTTGTCATACTTC-3′
Western blot
Protein of tissues and GBM cell lines was extracted using RIPA lysis buffer (Solarbio, Beijing, People’s Republic of China) containing protease inhibitors. Protein concentration was measured using bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Protein samples were separated by SDS-PAGE using 10% polyacrylamide gels and then transferred onto a nitrocellulose membrane (EMD Millipore, Billerica, MA, USA). The membranes were blocked in TBST buffer containing 5% BSA and incubated sequentially with primary antibody at 4°C overnight. After three times washing in TBST buffer for 15 minutes each time, the membranes were incubated with horseradish peroxidase (HRP)-linked secondary antibody for 1 hour at room temperature. Then the blots were washed twice with TBST and were developed by enhanced chemiluminescence (ECL) kit (EMD Millipore). The bands were quantified by ImageJ and normalized to GAPDH. The primary antibodies were as follows:
JNK1/2 CST 1:800 dilution; p-JNK1/2 CST 1:800 dilution; USP4 Abcam 1:1,000 dilution; PCNA Abcam 1:1,000 dilution; Bcl2 Abcam 1:1,000 dilution; Bax Abcam 1:1,000 dilution; P21 Abcam 1:1,000 dilution; P27 Abcam 1:1,000 dilution; ERK1/2 CST 1:1,000 dilution; p-ERK1/2 CST 1:800 dilution; p38 CST 1:1,000 dilution; p-p38 CST 1:800 dilution; GAPDH Abcam 1:2,000 dilution.
Cell proliferation and apoptosis
Cell Counting Kit-8 (CCK-8, Beyotime, Shanghai, China) and annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) were used to measure cell proliferation and apoptosis, respectively. For cell proliferation assay, treated cells were cultured for each time point and then CCK-8 was added into each well and incubated for 1 hour. The results were analyzed according to 450 nm absorbance on a microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA). For cell apoptosis assay, treated cells were stained with annexin-V and/or PI for 5 minutes in the dark according to the instruction and measured by flow cytometry (BD Biosciences).
In vivo tumor growth
Four- to six-week-old male BALB/c nude mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. Mice were fed with free access to food and water and were maintained under a 12-hour light/dark cycle at 22–24°C and 40%–60% humidity. The mice were randomly divided into two groups, each group with six mice. For in vivo tumor growth assay, treated or control cells were injected subcutaneously into the groin of mice at the density of 2×106 cells/mL. Tumor size including length and width were measured every 3 days after injection. Tumor volume was calculated as length × (width2/2).
The experimental procedures for xenografted nude mouse model and care were approved by the Committee on Ethical Use of Animals of Chongqing Medical University (Chongqing, People’s Republic of China).
Histologic examination and TUNEL assay
After 48 hours fixing in a 10% formalin solution, tumor tissues of mice were used for histologic examination. Sections (5 mm; Leica RM2125, Leica Microsystems, Wetzlar, Germany) were stained with hematoxylin and eosin (H&E) according to standard methods. Then light microscopy (Olympus Corporation, Tokyo, Japan) was used to collect the images at ×200 magnification.
For apoptosis detection, sections from the tumor tissues were stained with the reaction buffer applied from the TUNEL Kit (Hoffman-La Roche Ltd., Basel, Switzerland) following the manual instructions. Percentages of apoptotic cells were evaluated in five randomly selected fields.
Statistical analysis
All values are mean ± SD. Data sets in groups were analyzed using one-way ANOVA. Student’s t-test was used for two groups. Experiments were performed in triplicate. P<0.05 was considered as statistically significant.
Results
USP4 was upregulated and correlated with poor prognosis in GBM
To understand the role of USP4 in GBM, USP4 expression level was analyzed in 40 GBM tissues and 20 normal tissues with real-time PCR. As shown in Figure 1A, the mRNA level of USP4 was much higher in GBM tissues than in normal tissues, which was consistent with the results from the TCGA database (Figure 1B). To validate our finding, Western blot was performed to detect protein level of UPS4 in the same specimens as well. As shown in Figure 1C, higher level of UPS4 was observed in GBM than in normal tissues. To illustrate the relationship between USP4 and GBM prognosis, we then analyzed survival probability associated with USP4 level in TCGA database. As shown in Figure 1D, log-rank test showed that patients with higher level of USP4 exhibited shorter survival time than those with lower level of USP4.
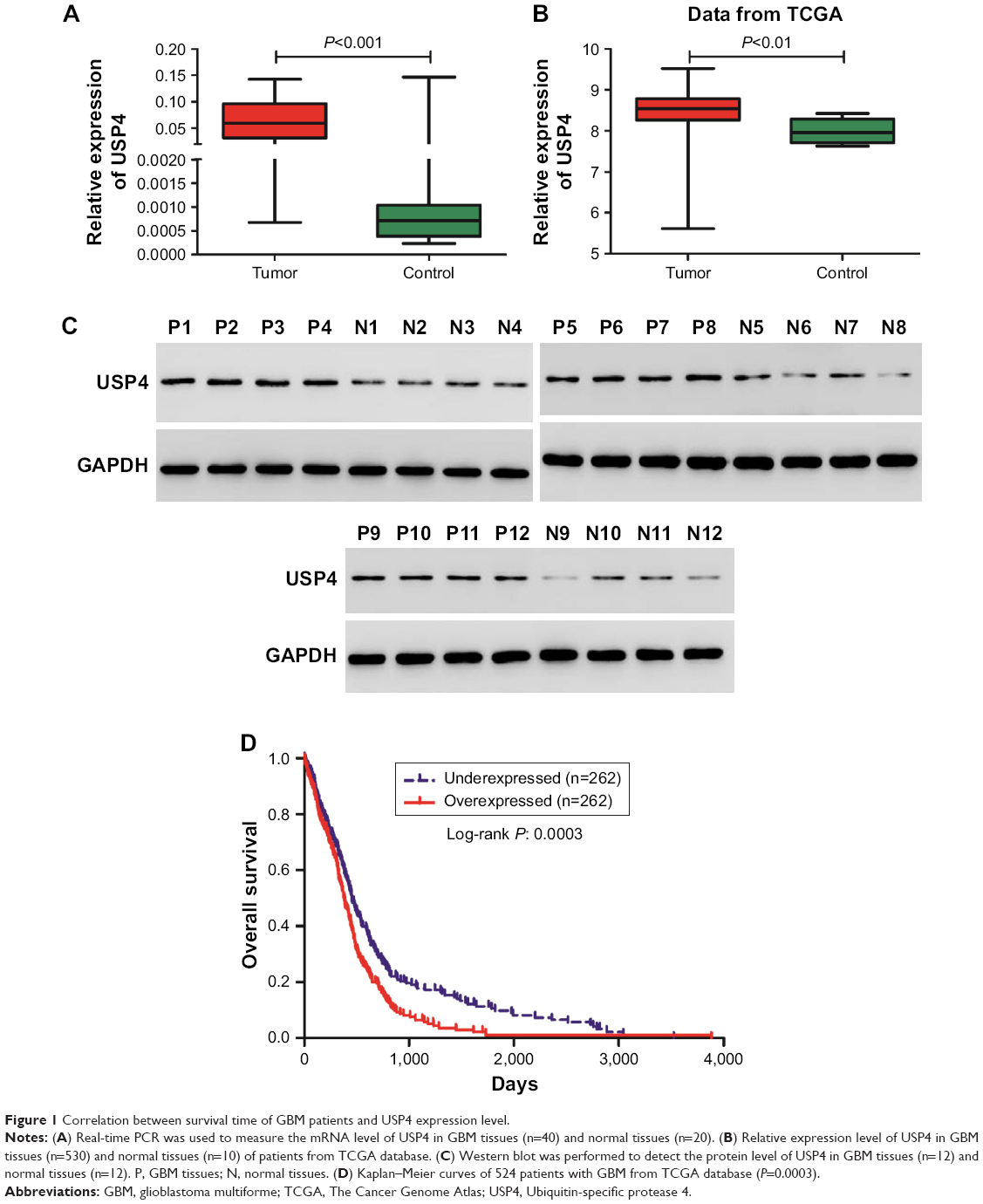 | Figure 1 Correlation between survival time of GBM patients and USP4 expression level. |
USP4 knockdown inhibited GBM cell proliferation
To study function of USP4 in GBM cells, first we detected transcription and protein level of USP4 in five GBM cell lines, including U87, T98G, U373, SHG44 and U251. Our results showed that U87 and T89G expressed higher expression level of USP4 than others, whereas U251 had the lowest USP4 level (Figure 2A and B).
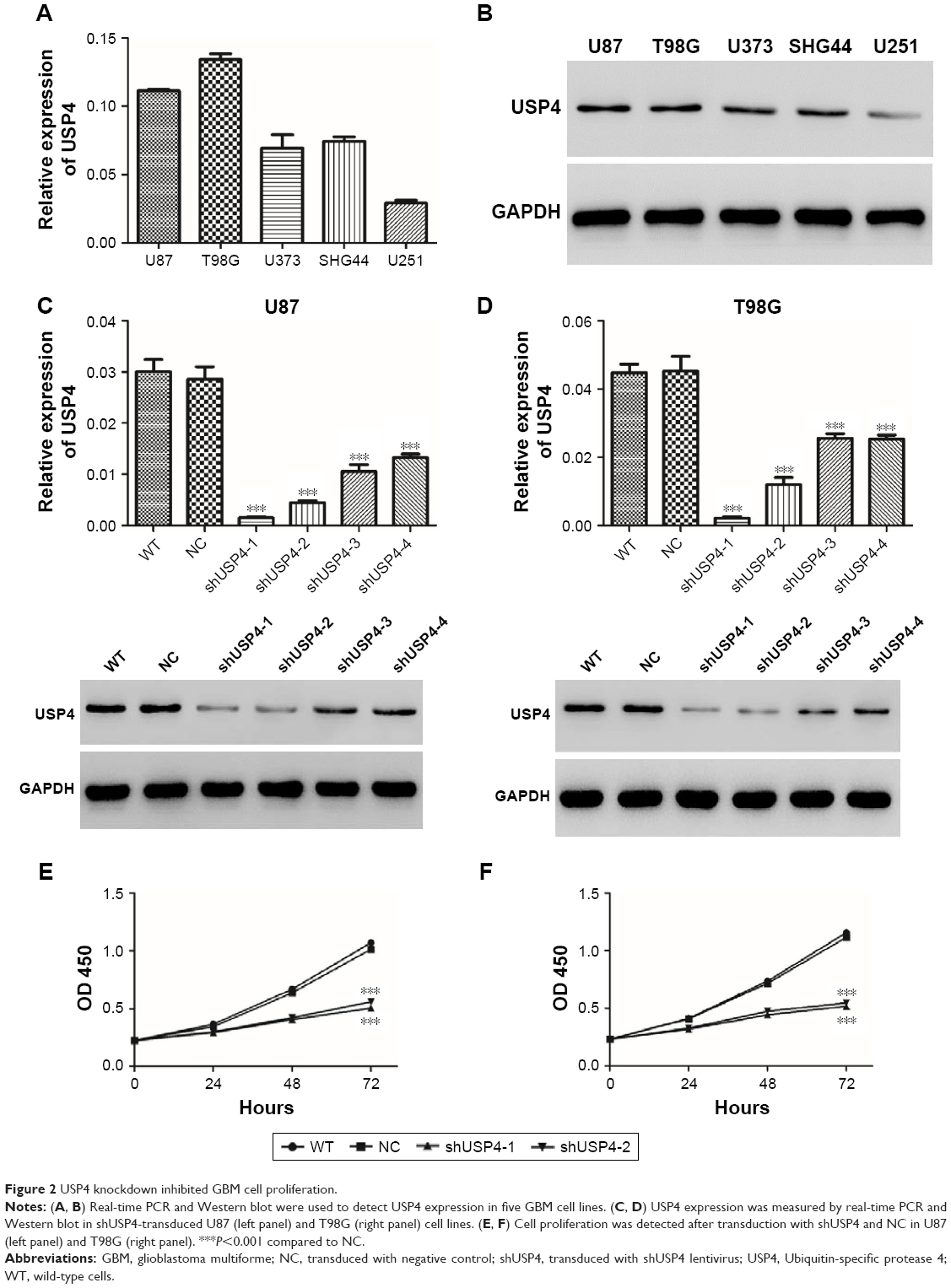 | Figure 2 USP4 knockdown inhibited GBM cell proliferation. |
To further investigate the potential role of USP4, four shRNAs targeting USP4 were used to silence USP4 in U87 and T98G cells. As shown in Figure 2C and D, all the shRNA inordinately suppressed the expression level of USP4 in U87 and T98G cells. shUSP4-1 and shUSP4-2 were chosen for further assay as they showed better knockdown efficiency. After that, CCK-8 assay was performed to measure proliferation of cells. Silencing of USP4 significantly inhibited the proliferation of U87 and T98G cells (Figure 2E and F, P<0.001). These results suggested that USP4 knockdown inhibited GBM cell proliferation.
Silencing of USP4 promoted cell apoptosis in GBM cell lines
To further assess the function of USP4 on GBM cells, we then performed annexin-V/PI staining assay to explore cell apoptosis. The results showed that USP4 knockdown increased cell apoptosis in U87 and T98G cell lines (Figure 3A). For further studying apoptotic effect of USP4 silence, real-time PCR and Western blot were used to analyze the expression level of related targets including PCNA, Bcl-2, Bax and cleaved caspase3. As shown in Figure 3B and C, silencing of USP4 downregulated the expression of PCNA and Bcl-2, but upregulated the expression of Bax and cleaved caspase3 in both U87 and T98G cell lines. The collective data suggest that USP4 knockdown inhibits cell proliferation by promoting cell apoptosis in GBM cell lines.
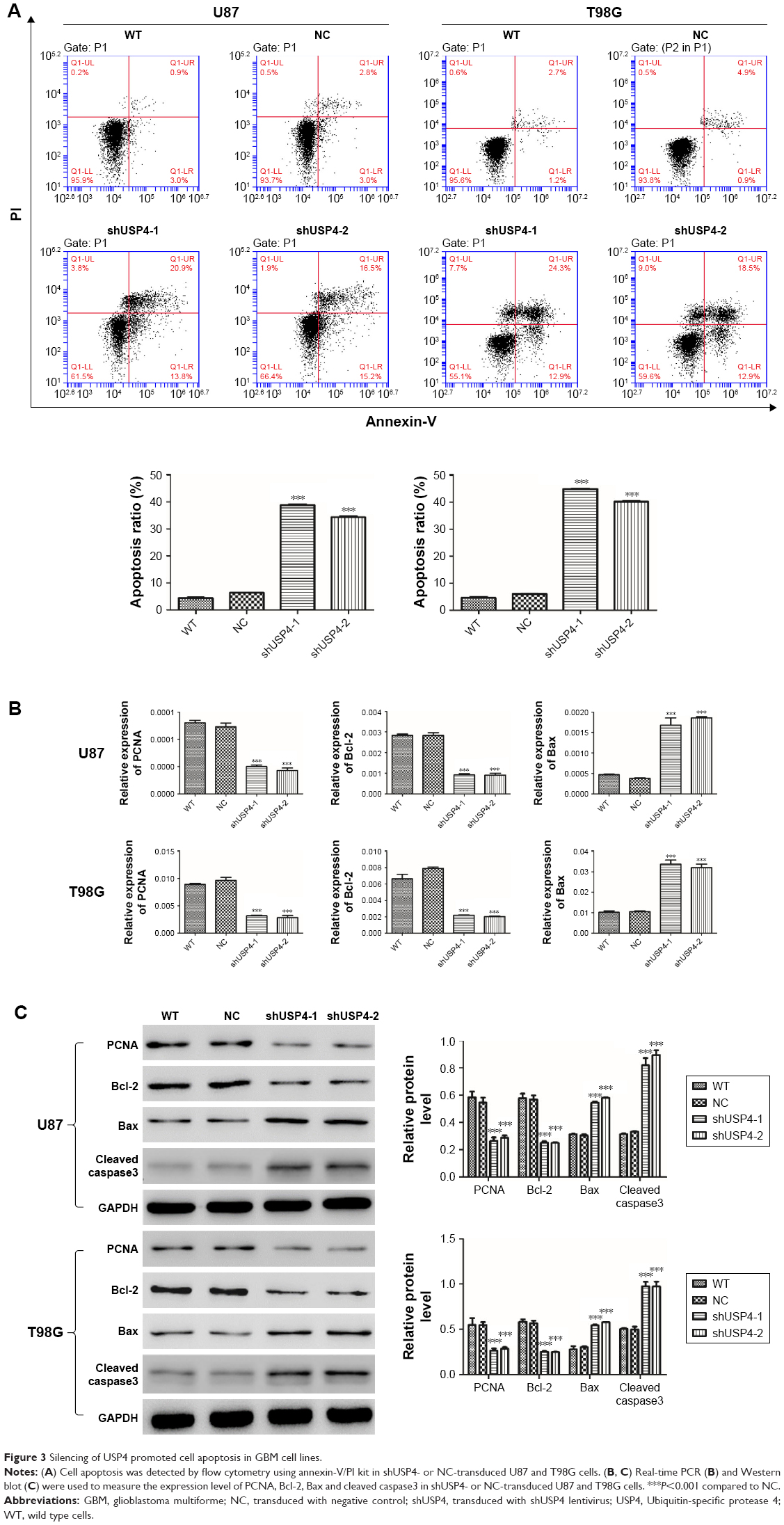 | Figure 3 Silencing of USP4 promoted cell apoptosis in GBM cell lines. |
Knockdown of USP4 exerted complex roles in MAPK pathway
As reported, MAPK signaling pathway is involved in cell proliferation and cell apoptosis in many kinds of tumors, including GBM.18–20 DUBs are supposed to be a novel anticancer therapeutic strategy via MAPK signaling pathway.21 MAPK has four main components, extracellular-signal regulated kinase (ERK1/2), Jun-N-terminal kinase (JNK1/2/3), p38, and ERK-5.22 We hypothesized that USP4 performs its role in GBM through regulating MAPK pathway. To better understand the mechanisms through which USP4 was involved in GBM cell proliferation and apoptosis, we chose ERK, JNK and P38 to study. As shown in Figure 4A and B, silencing of USP4 downregulated the expression of p-ERK1/2 and p-JNK, but upregulated the expression of p-P38 in both U87 and T98G cell lines. Consistently, p-c-fos was found to be inhibited by USP4 silence in both U87 and T98G cell lines (Figure 4C and D). Collectively, USP4 knockdown activates ERK and JNK but inactivates P38.
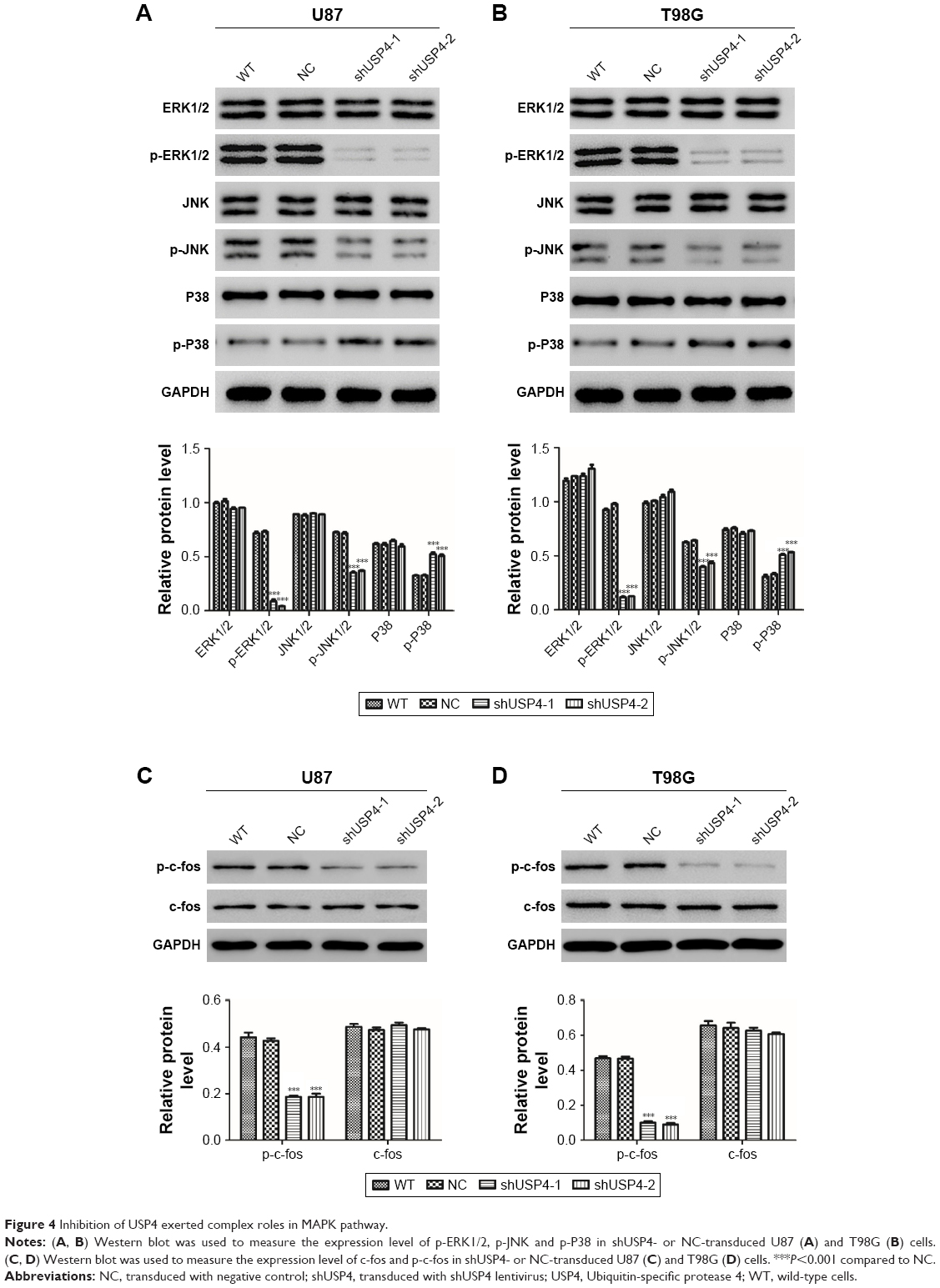 | Figure 4 Inhibition of USP4 exerted complex roles in MAPK pathway. |
Knockdown of USP4 inhibited GBM cell tumorigenesis in vivo
To validate effects exerted in GBM cell lines, we then switched to U87-derived xenograft tumor model. A stable genetic approach (USP4 shRNA) was used. Equal number of U87-shUSP4 cells and -NC cells were injected into flank of nude mice subcutaneously and the diameters of the tumors were measured every 3 days. As shown in Figure 5A, USP4 depletion significantly inhibited tumor growth (P<0.001). Consistently, USP4 depletion showed smaller tumor than in NC cells at the end point of experiment. Subsequently, tumors were sliced and stained with H&E. As shown in Figure 5B, tumors from NC group showed typical characteristics of tumor cells, such as big and deeply stained nuclei, higher nuclear/cytoplasm ratio and densely arranged cells, whereas tumors from USP4 depletion group shown partial necrosis, apoptosis, cell shrinkage and vacuole (Figure 5B). Furthermore, TUNEL assay was used to detect the late apoptosis of tumor xenografts. We found there were more apoptosis cells in shUSP4 group than in NC group (Figure 5C). In addition, we found that shUSP4 tumor xenografts showed lower level of USP4, PCNA and Bcl-2, but higher level of Bax and cleaved caspase3, which means inhibition of tumor proliferation and promotion of tumor apoptosis (Figure 5D). Of note, p-ERK1/2 was decreased in shUSP4 tumor xenografts compared with NC groups (Figure 5D). These results demonstrated that knockdown of USP4 could suppress GBM growth and tumorigenesis in vivo.
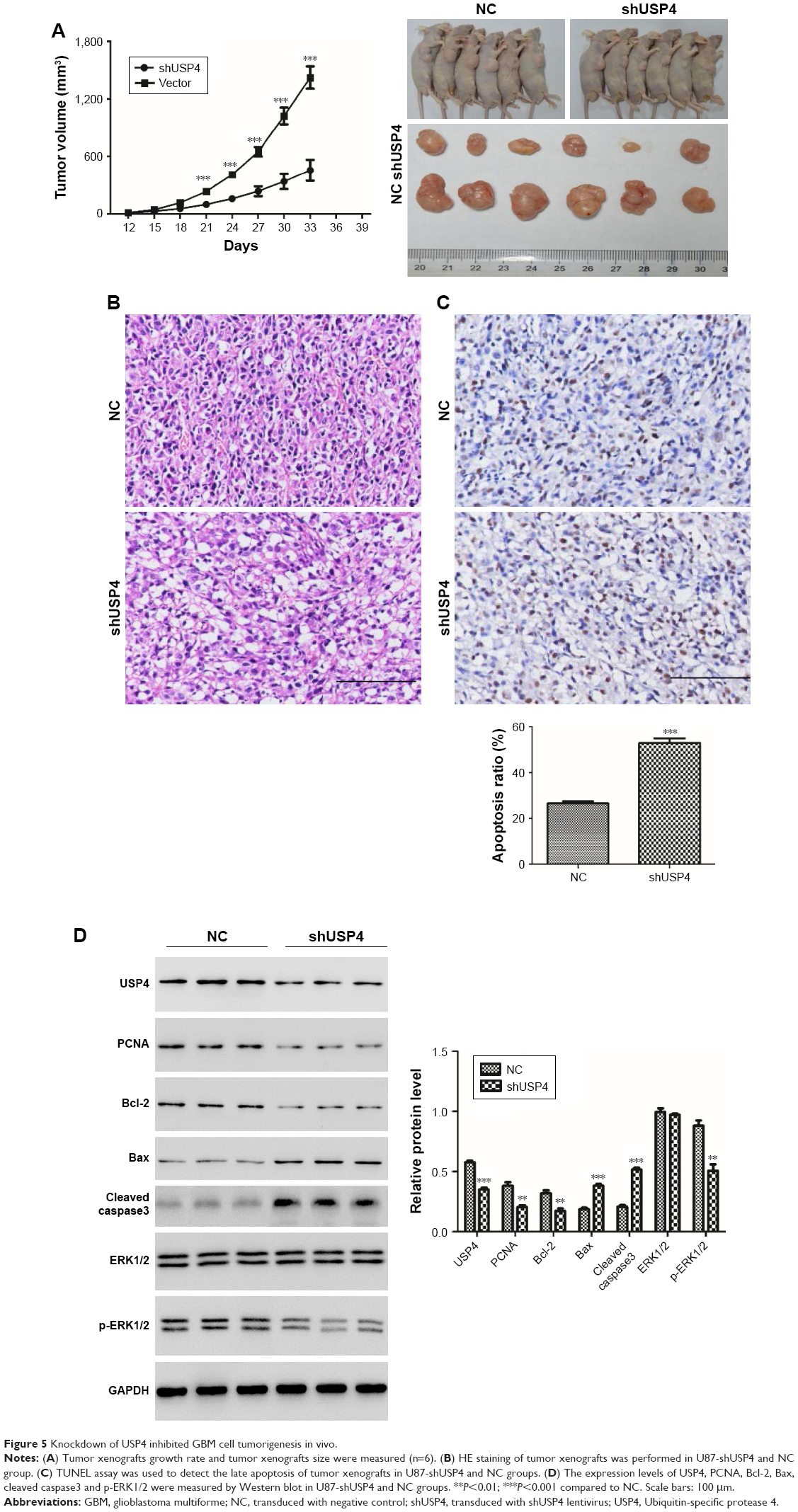 | Figure 5 Knockdown of USP4 inhibited GBM cell tumorigenesis in vivo. |
USP4 overexpression attenuated apoptosis induced by ERK inhibitor
Among MAPK pathways, the MAPK/ERK pathway is the most extensively studied and perhaps the most relevant to cancer pathogenesis.23–25 To further test the mechanisms involved, USP4-overexpressing lentivirus and ERK inhibitor were used. To further validate our findings, U251, with lower USP4 endogenous level, were transduced with USP4 overexpression or vector lentivirus (Figure 6A). After that, cells were treated with U0126, an inhibitor of ERK1/2, followed with cell proliferation assay. As shown in Figure 6B, U0126 treatment inhibited cell proliferation which could be attenuated by USP4 overexpression. Flow cytometry was then used to detect apoptosis. As observed in Figure 6C, U0126 treatment increased apoptosis proportion which can be rescued by overexpression of USP4. Of note, USP4 overexpression can only overcome partial apoptosis induced by U0126. Consistent with this finding, U0126 treatment decreased the expression level of PCNA, Bcl-2 and p-ERK1/2, but increased the expression level of Bax and cleaved caspase3 (Figure 6D). To further explore alterations of PCNA, Bcl2 and Bax contributed by USP4 or p-ERK1/2, we performed time-course experiment with/without U0126 treatment. As expected, we observed a decrease of phosphor ERK1/2 for 30 and 60 minutes after treatment with p-ERK1/2 inhibitor. Intriguingly, significant upregulation of p-ERK1/2 can be found upon USP4 overexpression. Consistently, U0126 dramatically reduced phosphor ERK1/2 which is still higher that than in the vector group (Figure 6E). However, we observed no alteration of PCNA, Bcl2 and Bax with ERK1/2 activation alone or plus USP4 ectopic expression (Figure 6E). In summary, apoptosis induced by inhibition of ERK1/2 can be impaired by USP4.
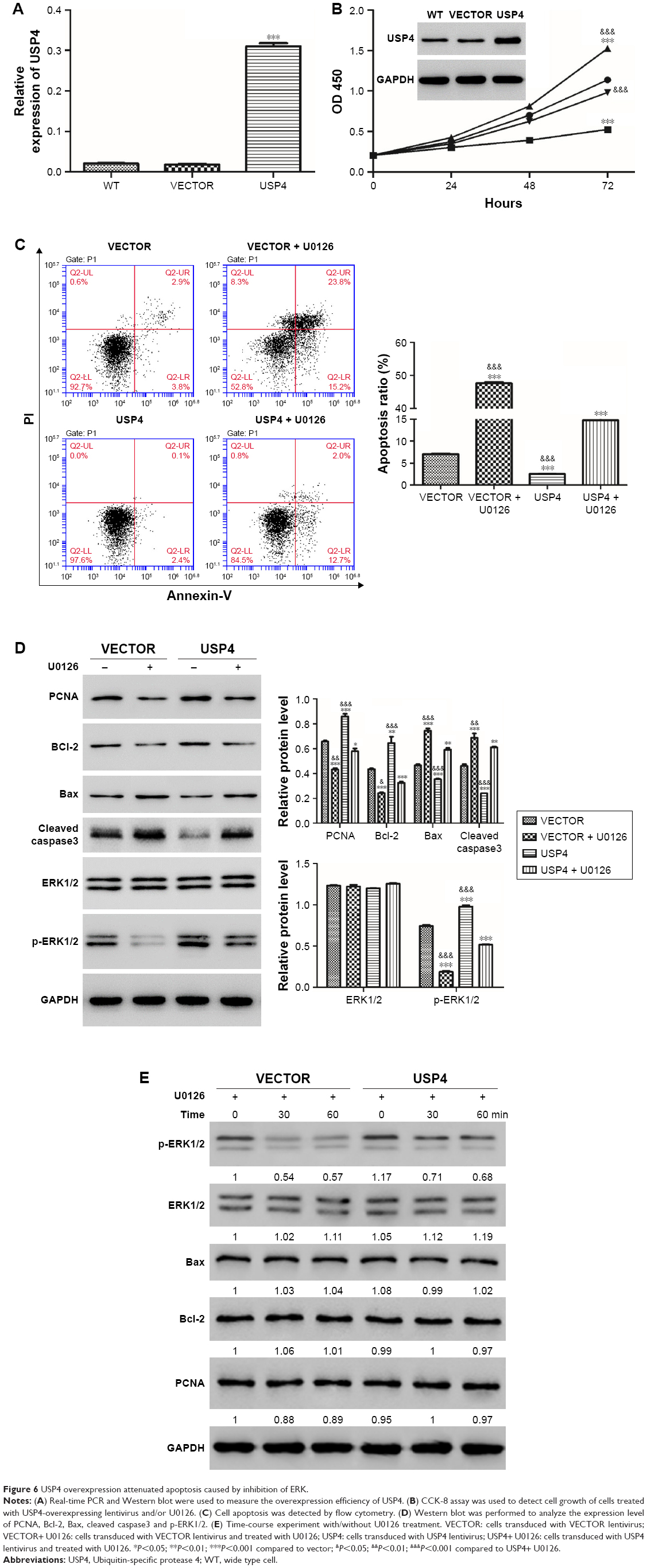 | Figure 6 USP4 overexpression attenuated apoptosis caused by inhibition of ERK. |
USP4 elevated p-ERK through regulating TGF-β
According to a previous study, USP4 is found to directly deubiquitylate TGF-β type I receptor.26 TGF-β receptor (TGF-βR) has been shown to activate the MAPK pathway.27 We assumed that TGF-βR may be the substrate of USP4 and the bridge of USP4 and MAPK pathway. To verify our hypothesis, USP4 overexpressed lentivirus and/or LY2157299, an inhibitor of TGF-βR, were used to treat U251 cells. As shown in Figure 7A, cells treated with LY2157299 showed lower levels of TGF-βR1, p-smad2 and p-ERK1/2. However, cells with USP4 overexpression showed higher level of TGF-βR1, p-smad2 and p-ERK1/2 than that in VECTOR group. Of note, cells treated with both LY2157299 and USP4 overexpression partially overcame lower level of TGF-βR1, p-smad2 and p-ERK1/2 contributed by LY2157299 treatment. These results indicated that USP4 elevated p-ERK through regulating TGF-β.
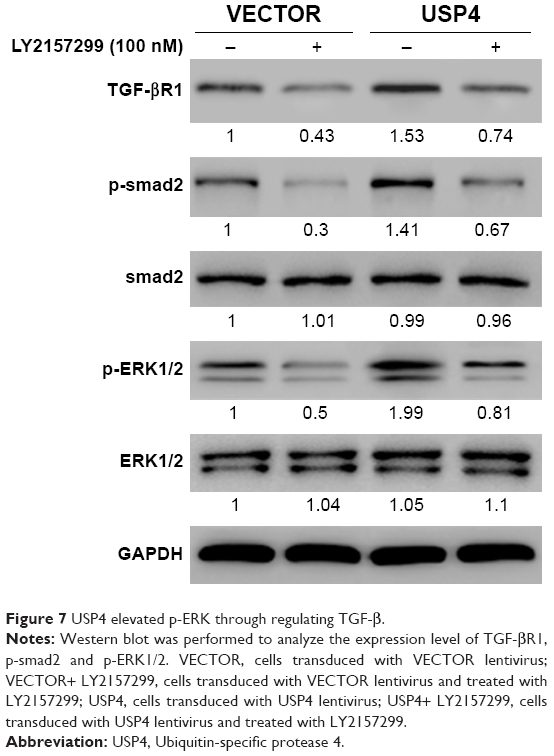 | Figure 7 USP4 elevated p-ERK through regulating TGF-β. |
Discussion
Some studies demonstrated that USP4 has an important effect in tumor progression, but the function of USP4 in tumors is still controversial. USP4 is found to act as tumor suppressor in breast cancer and increase cell apoptosis in head and neck squamous cell carcinoma.12,13 Otherwise, USP4 has been recognized as an oncogene by other studies.14,28,29 However, the functions of USP4 in GBM remain unclear.
In the current study, we found USP4 was upregulated in GBM tissues compared with that in normal tissues (Figure 1A), which means that USP4 may play an important role in GBM. High expression level of USP2 was also found in U87, T98G, U373 and SHG44 GBM cell lines. In support, clinical data from TCGA database indicated overexpression of USP4 was correlated with poor prognosis (Figure 1C). Thus, overexpression of USP4 correlated with initiation and/or progression of GBM and poor prognosis indicates that targeting USP4 may be a possible approach to treat this disease. However, to this end, we need to understand the underlying mechanisms contributing to GBM. We revealed that knockdown of USP4 could significantly inhibit cell proliferation and increase cell apoptosis via MAPK/ERK pathway both in vitro (Figures 2–4) and in vivo (Figure 5). The mitochondrial pathway is an intrinsic model of apoptosis, including the MAPKs (JNK, ERK, and p38).30 The MAPK signaling pathway, especially the MAPK/ERK pathway, plays a key role in tumor progression including cell proliferation, cell apoptosis, and cell survival.31,32 Here, we found the level of ERK phosphorylation was decreased with USP4 knockdown. Inversely, USP4 overexpression could attenuate the effects induced by ERK inhibitor, U0126 (Figure 6), which also supports our finding that USP4 may act as a novel oncogene in GBM. What needs to be noted is that even though we found manipulation of USP4 could induce activation of the ERK pathway, we still did not know the exact mechanism by which USP4 induced these alterations. As reported, USP4 is a deubiquitinase for TGF-β type I receptor,26 which has been shown to activate the MAPK pathway.27 TGF-β plays an essential role in embryogenesis and tissue homeostasis.33 Perturbations in TGF-β signal pathway have been found in many kinds of human diseases, including cancer.34 Ubiquitin modification of TGF-β signaling components is emerging as a key mechanism of TGF-β pathway control. As reported, USP4 can directly deubiquitylate TGF-β R1 to enhance TGF-β signaling.26 In our study, we found USP4 elevated p-ERK through regulating TGF-β and finally activated the MAPK pathway (Figure 7).
In summary, our study is the first exploration of USP4 in GBM. We found that inhibition of USP4 suppressed cell proliferation and promoted cell apoptosis by activating the ERK pathway. Intriguingly, effects contributed by the inhibitor of ERK pathway, U0126, give us the possibility to introduce ERK inhibitors into treatment of GBM in future.
Acknowledgments
The authors are grateful for the help of Professor Xuan Zhai. This research was funded by the Key Project of the Chongqing National Science Foundation (cstc2015jcyjBX0144).
Disclosure
The authors report no conflicts of interest in this work.
References
Sathornsumetee S, Rich JN. New treatment strategies for malignant gliomas. Expert Rev Anticancer Ther. 2006;6(7):1087–1104. | ||
Reardon DA, Mitchell DA. The development of dendritic cell vaccine-based immunotherapies for glioblastoma. Semin Immunopathol. 2017;39(2):225–239. | ||
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | ||
Sang Y, Li Y, Song L, et al. TRIM59 promotes gliomagenesis by inhibiting TC45 dephosphorylation of STAT3. Cancer Res. 2018;78(7):1792–1804. | ||
Hegge B, Sjøttem E, Mikkola I. Generation of a PAX6 knockout glioblastoma cell line with changes in cell cycle distribution and sensitivity to oxidative stress. BMC Cancer. 2018;18(1):496. | ||
Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014;74(18):4955–4966. | ||
Liu J, Zhang C, Zhao Y, et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun. 2017;8(1):1823. | ||
Yan FJ, Zhang XJ, Wang WX, et al. The E3 ligase tripartite motif 8 targets TAK1 to promote insulin resistance and steatohepatitis. Hepatology. 2017;65(5):1492–1511. | ||
Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 2004;1695(1–3):55–72. | ||
Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. | ||
Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20(11):1242–1253. | ||
Li Y, Jiang D, Zhang Q, Liu X, Cai Z. Ubiquitin-specific protease 4 inhibits breast cancer cell growth through the upregulation of PDCD4. Int J Mol Med. 2016;38(3):803–811. | ||
Hou X, Wang L, Zhang L, Pan X, Zhao W. Ubiquitin-specific protease 4 promotes TNF-α-induced apoptosis by deubiquitination of RIP1 in head and neck squamous cell carcinoma. FEBS Lett. 2013;587(4):311–316. | ||
Xing C, Lu X-X, Guo P-D, et al. Ubiquitin-specific protease 4-mediated deubiquitination and stabilization of PRL-3 is required for potentiating colorectal oncogenesis. Cancer Res. 2016;76(1):83–95. | ||
Yun SI, Kim HH, Yoon JH, et al. Ubiquitin specific protease 4 positively regulates the Wnt/β-catenin signaling in colorectal cancer. Mol Oncol. 2015;9(9):1834–1851. | ||
Li Z, Hao Q, Luo J, et al. USP4 inhibits p53 and NF-κB through deubiquitinating and stabilizing HDAC2. Oncogene. 2016;35(22):2902–2912. | ||
Wijnhoven P, Konietzny R, Blackford AN, et al. USP4 Auto-Deubiquitylation promotes homologous recombination. Mol Cell. 2015;60(3):362–373. | ||
Zhang Y, Ren XIA, Shi M, et al. Downregulation of STAT3 and activation of MAPK are involved in the induction of apoptosis by HNK in glioblastoma cell line U87. Oncol Rep. 2014;32(5):2038–2046. | ||
Che XF, Moriya S, Zheng CL, Abe A, Tomoda A, Miyazawa K. 2-Aminophenoxazine-3-one-induced apoptosis via generation of reactive oxygen species followed by c-Jun N-terminal kinase activation in the human glioblastoma cell line LN229. Int J Oncol. 2013;43(5):1456–1466. | ||
Cha JH, Choi YJ, Cha SH, Choi CH, Cho WH. Allicin inhibits cell growth and induces apoptosis in U87MG human glioblastoma cells through an ERK-dependent pathway. Oncol Rep. 2012;28(1):41–48. | ||
Kumari N, Jaynes PW, Saei A, Iyengar PV, Richard JLC, Eichhorn PJA. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim Biophys Acta Rev Cancer. 2017;1868(2):456–483. | ||
Avruch J. MAP kinase pathways: the first twenty years. Biochim Biophys Acta. 2007;1773(8):1150–1160. | ||
Mccubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–1284. | ||
Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4(12):937–947. | ||
Tortora G, Bianco R, Daniele G, et al. Overcoming resistance to molecularly targeted anticancer therapies: rational drug combinations based on EGFR and MAPK inhibition for solid tumours and haematologic malignancies. Drug Resist Updat. 2007;10(3):81–100. | ||
Zhang L, Zhou F, Drabsch Y, et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat Cell Biol. 2012;14(7):717–726. | ||
Tinoco-Veras CM, Santos A, Stipursky J, et al. Transforming growth factor β1/SMAD signaling pathway activation protects the intestinal epithelium from Clostridium difficile toxin A-induced damage. Infect Immun. 2017;85(10):1–13. | ||
Hwang SJ, Lee HW, Kim HR, et al. Ubiquitin-specific protease 4 controls metastatic potential through β-catenin stabilization in brain metastatic lung adenocarcinoma. Sci Rep. 2016;6:21596. | ||
Heo MJ, Kim YM, Koo JH, et al. microRNA-148a dysregulation discriminates poor prognosis of hepatocellular carcinoma in association with USP4 overexpression. Oncotarget. 2014;5(9):2792–2806. | ||
Eum KH, Lee M. Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem. 2011;348(1–2):61–68. | ||
Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17(1–4):287–296. | ||
Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. | ||
Kang JS, Liu C, Derynck R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 2009;19(8):385–394. | ||
Ikushima H, Miyazono K. Tgfbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415–424. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.