")
Back to Journals » OncoTargets and Therapy » Volume 12
Ubiquitin-specific peptidase 28 enhances STAT3 signaling and promotes cell growth in non-small-cell lung cancer
Authors Li P, Huang Z, Wang J, Chen W, Huang J
Received 16 November 2018
Accepted for publication 23 January 2019
Published 26 February 2019 Volume 2019:12 Pages 1603—1611
DOI https://doi.org/10.2147/OTT.S194917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Takuya Aoki
Pengling Li,1,2 Ziming Huang,3 Jipeng Wang,2 Wei Chen,2 Jianan Huang1
1Department of Respiratory Medicine, The First Affiliated Hospital of Soochow University, Suzhou 215006, Jiangsu, China; 2Department of Respiratory Medicine, The Affiliated Huai’an No 1. People’s Hospital, Nanjing Medical University, Huai’an 223300, Jiangsu, China; 3Department of Emergency Surgery, The Affiliated Huai’an No 1. People’s Hospital, Nanjing Medical University, Huai’an 223300, Jiangsu, China
Background and objectives: Ubiquitin-specific peptidase 28 (USP28) has been reported to play significant roles in several tumors, but its roles in non-small-cell lung cancer (NSCLC) is still unknown. In this study, we aimed to investigate the biological function and molecular mechanisms of USP28 in NSCLC.
Materials and methods: Immunoblotting analysis was used to detect relative proteins’ expression. Luciferase assay was performed to explore the activation of signal transducer and activator of transcription 3 (STAT3). Immunoprecipitation was performed to assess whether USP28 interacted with STAT3 or deubiquitinated STAT3. Quantitative real-time PCR was performed to evaluate the relative mRNA levels of STAT3 and USP28. Cycloheximide chase assay was carried out to examine whether USP28 affected the half-life of STAT3 protein. Cell Counting Kit-8 assay and xenograft model were used to assess whether USP28 regulated NSCLC cell growth.
Results: In this study, the deubiquitinating enzyme USP28 was found to mediate STAT3 signaling in NSCLC cells. USP28 interacted with STAT3, and increased the stability of STAT3 by inducing its deubiquitination. Further studies showed that USP28 was upregulated in both the primary tissues and cell lines of NSCLC. The Kaplan–Meier plotter also indicated that USP28 predicted a poor prognosis of NSCLC patients. Moreover, knockdown of USP28 inhibited cell growth of NSCLC cells in vitro and delayed NSCLC tumor growth in vivo.
Conclusion: These results demonstrated that USP28 was functional in NSCLC cells, and promoted NSCLC cell growth by inducing STAT3 signaling. This suggests that USP28 could be a novel target for NSCLC therapy.
Keywords: deubiquitinating enzyme, USP28, non-small-cell lung cancer, STAT3, deubiquitination
Introduction
Deubiquitinating enzymes (DUBs) are a large group of proteases, which can reverse the action of protein ubiquitination by cleaving the peptide or isopeptide bond between ubiquitin and its substrate proteins.1 There are five subfamilies of DUBs, including the cysteine proteases comprise ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases, ovarian tumour proteases, Machado-Joseph domain proteases, and the Jab1/Mov34/Mpr1 Pad1 N-terminal+ (MPN+) (JAMM) domain proteases.2 Ubiquitin-specific peptidase 28 (USP28) belongs to the largest USP DUB family, which was initially identified through homology search for USP25.3 Like USP25, USP28 contains the ubiquitin-associated domain and ubiquitin-interacting motifs in the N-terminal region.4
Recent studies showed that USP28 was involved in cancer-related pathways, and regulated physiological homeostasis of ubiquitination process, DNA-damage response, and cell cycle during genotoxic stress, which suggested that USP28 could be a promising target for cancer therapy.5 USP28 required for Myc function was screened.6 USP28 bound to Myc through an interaction with Fbw7α, and catalyzed the deubiquitination of Myc, thereby promoting its stabilization and contributing to tumor cell growth in colon and breast cancers.6,7 USP28 can also bind to and deubiquitinate some proteins involved in DNA-damage pathways. USP28 was reported to be required to stabilize Chk2 and 53BP1 in response to DNA damage.8 Intriguingly, 53BP1 and USP28 mediated p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis.9
The signal transducer and activator of transcription 3 (STAT3) is an important signaling mediator for many cytokines and growth factor receptors, which plays significant roles in cell growth, cell survival, cell differentiation, immunity, and inflammatory responses.10 Overexpression or overactivation of STAT3 is required for tumorigenesis, and STAT3 is tightly regulated in mamalian cells.11,12 Recent studies showed that STAT3 could be ubiquitinated for degradation, which indicated that STAT3 protein was regulated by the ubiquitin-proteasome pathway (UPP). A recent study reported that the ubiquitin ligase Fbw7 induced STAT3 ubiquitination for degradation, and that Fbw7 inhibited downstream antiapoptotic targets of STAT3 in diffuse large B-cell lymphoma.13 In glioblastoma stem cell-like cells, Bcl2-interacting cell death suppressor (BIS) depletion increased STAT3 ubiquitination, suggesting that BIS was necessary for STAT3 stabilization.14 Another paper showed that porcine reproductive and respiratory syndrome virus antagonized the STAT3 signaling by accelerating STAT3 degradation via the UPP, which led to perturbation of the host innate and adaptive immune responses.15 However, the ubiquitination mechanism of STAT3 in non-small-cell lung cancer (NSCLC) was still unclear.
In this study, we investigated the function of USP28 in NSCLC. We found that USP28 mediated STAT3 signaling in NSCLC cells. USP28 interacted with STAT3 and decreased the polyubiquitination of STAT3, thereby increasing the stability of STAT3. Moreover, USP28 was highly expressed in NSCLC and predicted a poor prognosis of NSCLC patients. Knockdown of USP28 suppressed the cell growth of NSCLC both in vitro and in vivo. These results indicated that targeting USP28/STAT3 axis could be a potential strategy for NSCLC therapy.
Materials and methods
Cells, culture, and chemicals
NSCLC cell lines (A549, H460, H1299, and H1975), the human bronchial epithelial cell line HBE and HEK293T cell line were purchased from American Type Culture Collection (Manassas, VA, USA). All NSCLC cell lines and human bronchial epithelial cell line were maintained in Roswell Park Memorial Institute1640 medium. HEK293T cells were maintained in DMEM medium. All media were supplemented with 10% FBS, 100 μg/mL of penicillin, and 100 units/mL of streptomycin.
Plasmids construction and gene transfection
The human USP28, USP25, STAT3, and ubiquitin genes were generated by PCR amplification from a cDNA library, and cloned into pcDNA3.1 vector with a Myc, Flag, or influenza hemagglutinin epitope (HA) tag as previously described.11 The site-directed mutant of USP28 (USP28C171A) was generated by using QuickMutation™ Site-Directed Mutagenesis Kit (Beyotime Biotechnology, Nantong, China). A STAT3 luciferase construct (STAT3-Luc) driven by specific STAT3 response elements was purchased from Beyotime Biotechnology.
Plasmids were transiently transfected into HEK293T or H1299 cells by Lipofectamine® 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instruction.
Co-immunoprecipitation (Co-IP) analysis
The whole cell lysates were prepared for Co-IP as described previously.16 In brief, the whole cell lysates were incubated with a specific primary antibody overnight at 4°C, followed by incubating with protein A/G-Sepharose beads (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for 3 hours. Then, the co-precipitated proteins were collected and identified by immunoblotting analysis against specific antibodies.
Immunoblotting
The whole cell lysates were prepared for immunoblotting as described previously.17 The primary antibodies against Flag, Myc, and HA were purchased from Medical & Biological Laboratories (Tokyo, Japan). Anti-p-STAT3, anti-STAT3, anti-immunoglobulin G (IgG), and anti-K48-linkage specific polyubiquitin antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-USP28 primary antibody was purchased from Proteintech Group (Wuhan, China). Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), anti-mouse IgG, and anti-rabbit IgG horseradish peroxidase-conjugated antibodies were purchased from Santa Cruz Biotechnology, Inc.
Quantitative real-time (qRT)-PCR
The qRT-PCR was performed as described previously.18 To determine the mRNA levels of STAT3 and USP28, qRT-PCR was performed using SYBR Green qPCR Master Mix (Clontech Laboratories, Inc., Palo Alto, CA, USA) with Roche LightCycler® 480II real-time PCR system (Roche, Basel, Switzerland). The primers used were as follows: STAT3, forward 5′-CAGTGACCAGGCAGAAGA-3′ and reverse 5′-ACTCCATCGCTGACAAAA-3′; USP28, forward 5′-TGGGAAGGATTCTGGTTA-3′ and reverse 5′-GCTGATAGAGCCTGGAGTA-3′; GAPDH, forward 5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse 5′-TGGTGAAGACGCCAGTGGA-3′.
Preparation of shRNA lentivirus
The lentivirus-delivered shRNAs against USP28 (shUSP28) and negative control (shNC) were purchased from Shanghai GeneChem Co., Ltd. (Shanghai, China). The target sequences of shUSP28#1 and shUSP28#2 were 5′-AAGTGGCATGAAGATTATAGT-3′ and 5′-AAGAGAGAGTGTATTCGAAAG-3′, respectively. The viral particles were prepared according to manufacturer’s instructions as described previously.19
Cycloheximide (CHX) chase assay
CHX chase was performed as described previously.20 In brief, Myc-USP28 plasmids or empty vector were transfected into H1299 cells by Lipofectamine®2000 (Invitrogen). Twenty-four hours later, the cells were treated with 50 μg/mL CHX (Sigma-Aldrich Co., St Louis, MO, USA) for the indicated time before being lysed for immunoblotting analysis.
Luciferase assay
STAT3-Luc or Myc-USP28 plasmids along with the internal control renilla were transfected into H1299 cells by Lipofectamine®2000 (Invitrogen) according to the manufacturer’s instruction. Forty-eight hours later, cells were prepared for luciferase assay by using Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA) as described previously.18
Cell growth and viability
NSCLC cells stably infected with shNC, shUSP28#1, or shUSP28#2 were plated at a start density of 2,000 cells per well into 96-well plates. Cells were incubated for indicated time, and the viable cells were evaluated by Cell Counting Kit-8 (CCK-8) staining according to the manufacturer’s instructions (Biotool, Houston, TX, USA).
Xenograft studies
The nude mice (5–6 weeks old, female) were purchased from Shanghai Slac Laboratory Animal Co. Ltd., Shanghai, China. Mice were randomly divided into two groups, and the human NSCLC cells H1299 stably infected with shNC or shUSP28#1 were injected subcutaneously in the right flanks of nude mice at a density of 6 million cells/site/mouse. When tumors were palpable, tumor sizes were measured every other day. At the end of the experiment, tumors were excised for immunoblotting. The animal experiments were conducted according to the University’s Laboratory Animal Center Care Guidelines and were approved by the Review Board of Animal Care and Use of The Affiliated Huai’an No 1 People’s Hospital, Nanjing Medical University.
Statistical analysis
The Student’s t-test was used for comparisons of two groups in the studies. All statistical tests were two-sided, and a P-value <0.05 was considered statistically significant.
Ethics approval and consent to participate
This study was approved by the Review Board and Ethical Committee of The Affiliated Huai’an No. 1 People’s Hospital, Nanjing Medical University, and each patient provided written informed consent to donate tissues for this study after clinical procedures.
Results
USP28 mediates STAT3 signaling in NSCLC cells
The transcription factor STAT3 is often overexpressed or overactivated in many tumors.21,22 In this study, we found that STAT3 and p-STAT3 were overexpressed or overactivated, along with the upregulation of USP28 in four NSCLC cell lines compared with the HBE cell line (Figure 1A). Additionally, STAT3 luciferase activity assays on basal levels of these five cell lines showed that NSCLC cell lines expressed greater STAT3 activity compared with the HBE cell line (Figure 1B). To further confirm whether USP28 regulated STAT3 signaling, the Dual-Luciferase reporter assay was performed. As shown in Figure 1C, overexpression of USP28 significantly upregulated STAT3-derived luciferase activity, but another USP isoform, USP25, had no effect on STAT3 luciferase activity. In addition, the immunoblotting assay also revealed that overexpression of USP28 enhanced the STAT3 signaling in NSCLC cells (Figure 1D). In contrast, knockdown of USP28 suppressed STAT3-derived luciferase activity (Figure 1E), and inhibited the expression of STAT3 in NSCLC cells (Figure 1F).
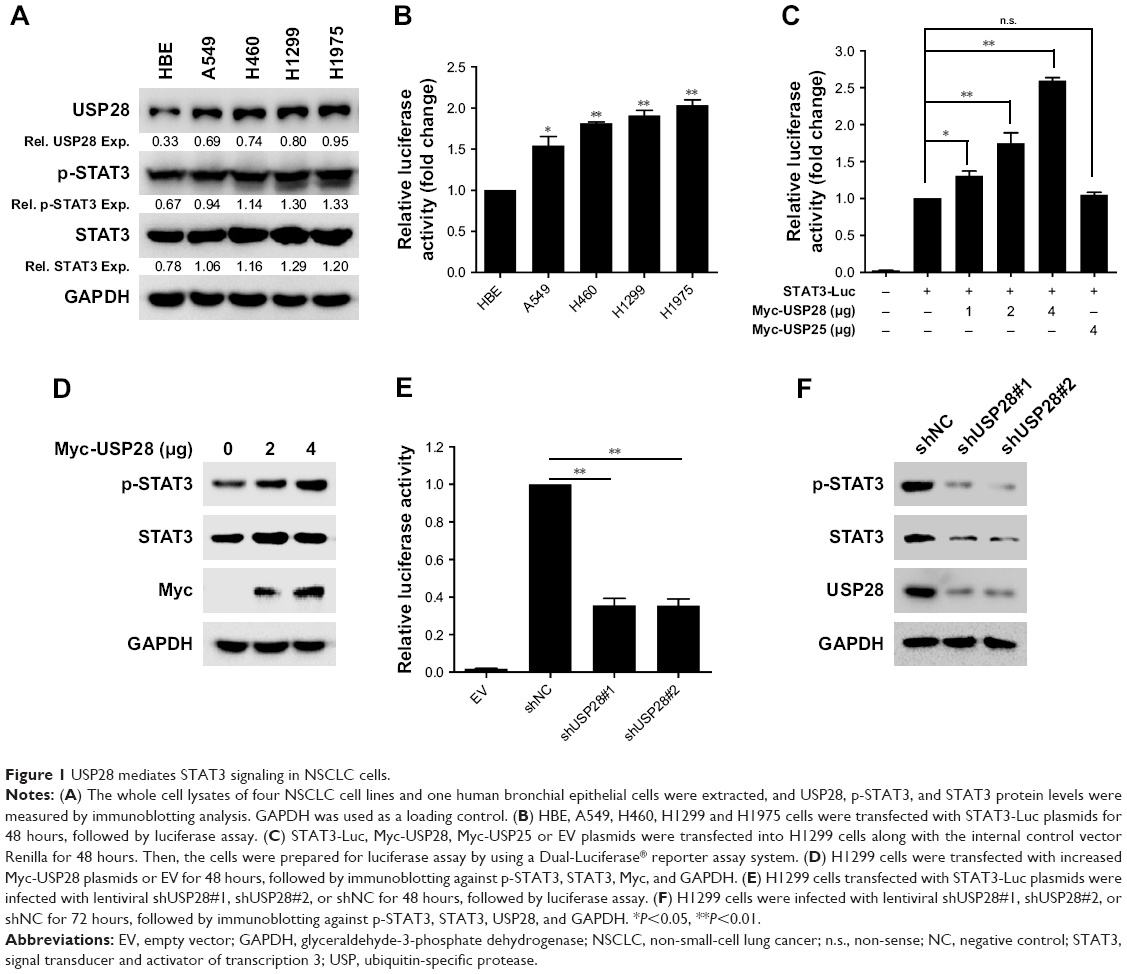 | Figure 1 USP28 mediates STAT3 signaling in NSCLC cells. |
USP28 interacts with STAT3 and decreases its polyubiquitination level
To evaluate whether USP28 interacted with STAT3, the reciprocal Co-IP was performed. As shown in Figure 2A and B, the Co-IP revealed that exogenous USP28 interacted with exogenous STAT3 protein. Whole cell lysates were also prepared for immunoblotting, which showed that USP28 upregulated the expression level of STAT3 (Figure 2A and B). Additionally, the Co-IP in Figure 2C showed that endogenous STAT3 also interacted with endogenous USP28. As USP28 was a deubiquitinase, we then assessed whether USP28 regulated the polyubiquitination of STAT3. As shown in Figure 2C, the IP indicated that STAT3 itself was polyubiquitinated and USP28 deubiquitinated the polyubiquitination of STAT3. The whole cell lysates also revealed that USP28 increased the protein level of STAT3 (Figure 2D).
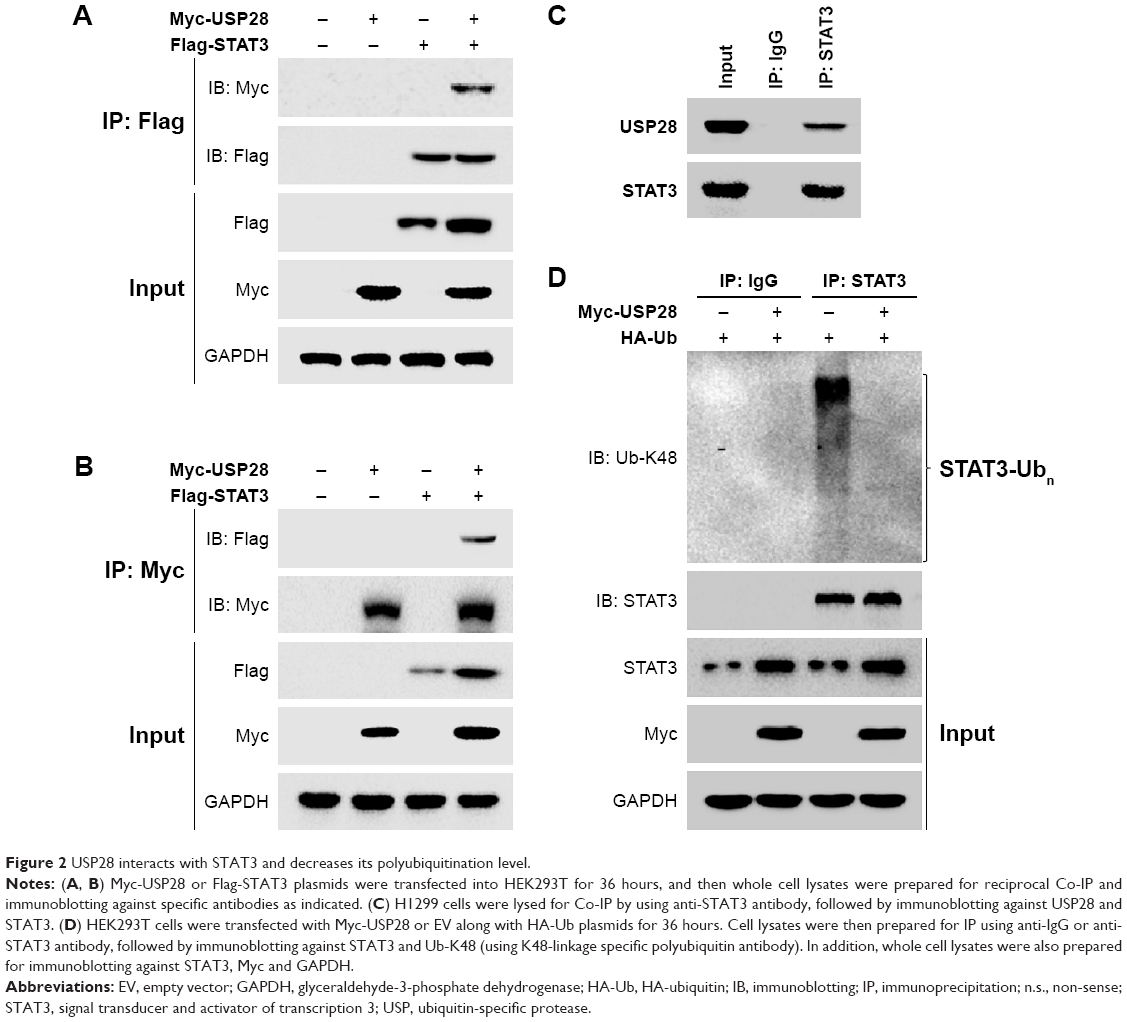 | Figure 2 USP28 interacts with STAT3 and decreases its polyubiquitination level. |
USP28 increases the stability of STAT3
To further confirm whether USP28 stabilized STAT3 protein, an increased dose of Myc-USP28 plasmids was transfected into NSCLC cells. As shown in Figure 3A, overexpression of USP28 upregulated STAT3 protein in a dose-dependent manner, but the mRNA level of STAT3 was not changed obviously (Figure 3B). However, ectopic expression of USP28C171A, a catalytically inactive mutant, could not enhance STAT3 levels, which indicated that USP28 regulated the post-translational modification of STAT3 (Figure 3C). In contrast, knockdown of USP28 downregulated the protein level of STAT3 in NSCLC cells (Figure 3D), but the mRNA level did not show significant change (Figure 3E). Moreover, the CHX chase assay revealed that overexpression of USP28 prolonged the half-life of STAT3 protein in NSCLC cells, which further suggested that USP28 stabilized STAT3 protein (Figure 3F and G).
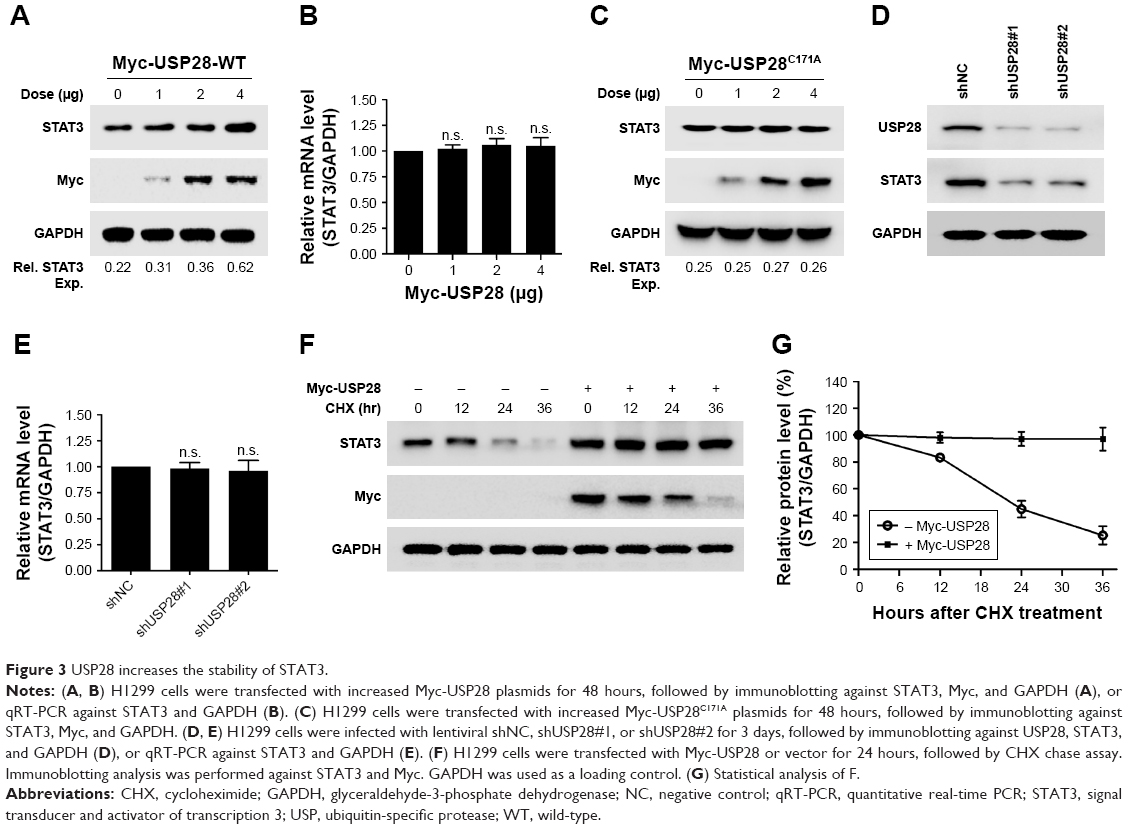 | Figure 3 USP28 increases the stability of STAT3. |
USP28 is upregulated and predicts a poor prognosis in NSCLC
As stated before, USP28 mediated STAT3 signaling by stabilizing STAT3 protein, which suggested that USP28 was functional in NSCLC cells. First, the public database The Cancer Genome Atlas revealed that USP28 expression was significantly higher in lung squamous cell carcinoma compared with that of controls (P<0.05) (Figure 4A). Then, we collected the normal paracancerous tissues or cancerous tissues from NSCLC patients, and the expression level of USP28 was detected by qRT-PCR and immunoblotting. As shown in Figure 4B and C, the level of USP28 was markedly elevated in these tumor tissues, and most tumor cell lines examined (Figure 1A). It is noteworthy that the Kaplan–Meier plotter also showed that the lung cancer patients with high level of USP28 had a significant poor overall survival compared with these with low USP28 expression (Figure 4D).
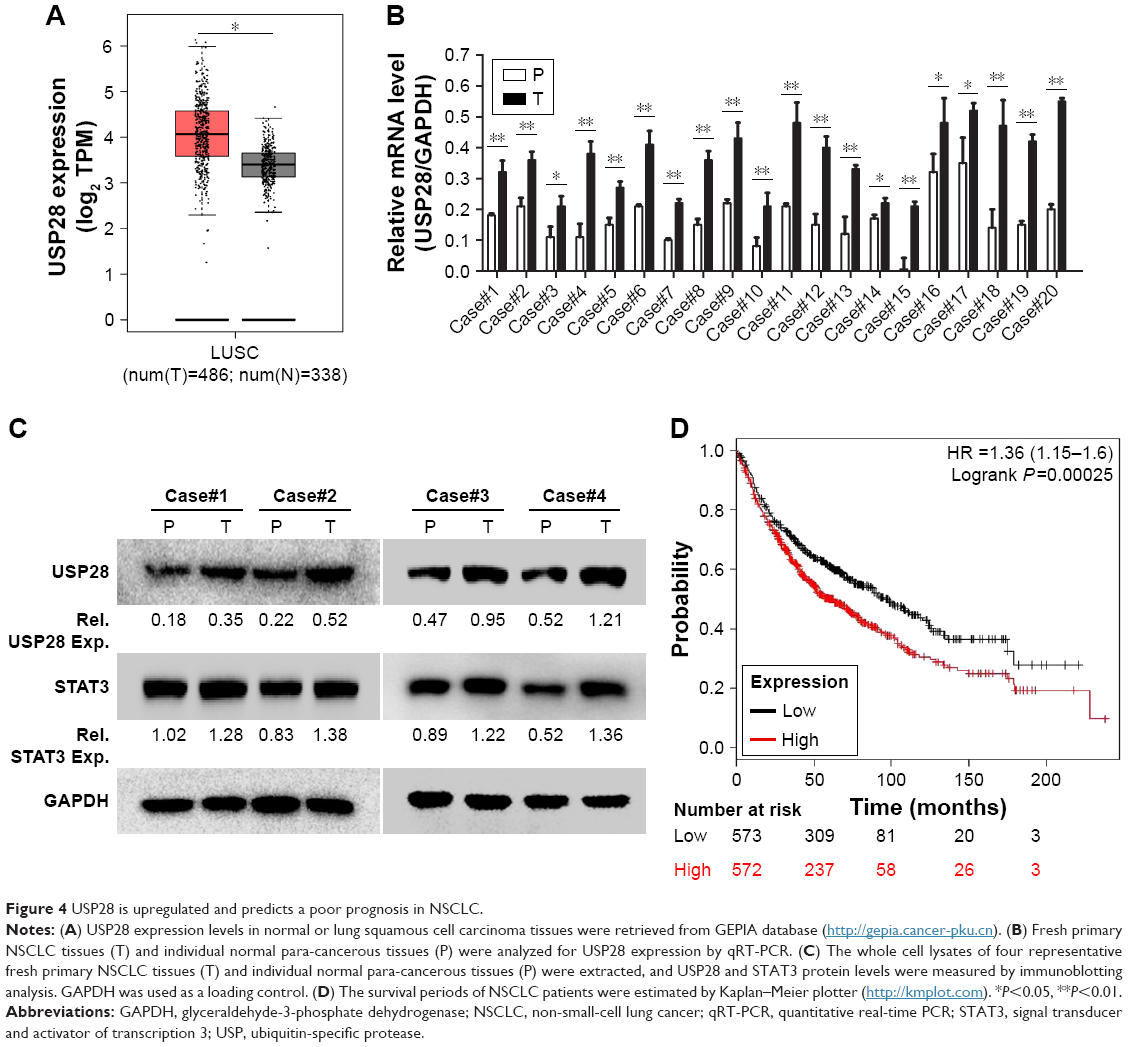 | Figure 4 USP28 is upregulated and predicts a poor prognosis in NSCLC. |
Knockdown of USP28 inhibits NSCLC cell growth
The increased expression of USP28 in NSCLC primary tissues and cell lines prompted us to further investigate the roles of USP28 in NSCLC cells. NSCLC cells A549 and H1299 were stably infected with lentiviral shNC, shUSP28#1, or shUSP28#2 for indicated time, followed by CCK-8 assay at different days. As shown in Figure 5A and B, knockdown of USP28 significantly inhibited NSCLC cell growth in both A549 and H1299 cells. Next, to further analyze the function of USP28 in vivo, a xenograft model was established. H1299 cells stably infected with lentiviral shNC or shUSP28#1 were subcutaneously injected into the right flanks of each nude mouse, and tumor sizes were monitored every other day for continuously 3 weeks when tumors were palpable. The studies in nude mice showed that knockdown of USP28 delayed tumor growth of NSCLC in vivo (Figure 5C and D). In addition, the immunoblotting also showed that knockdown of USP28 downregulated STAT3 expression in vivo (Figure 5E).
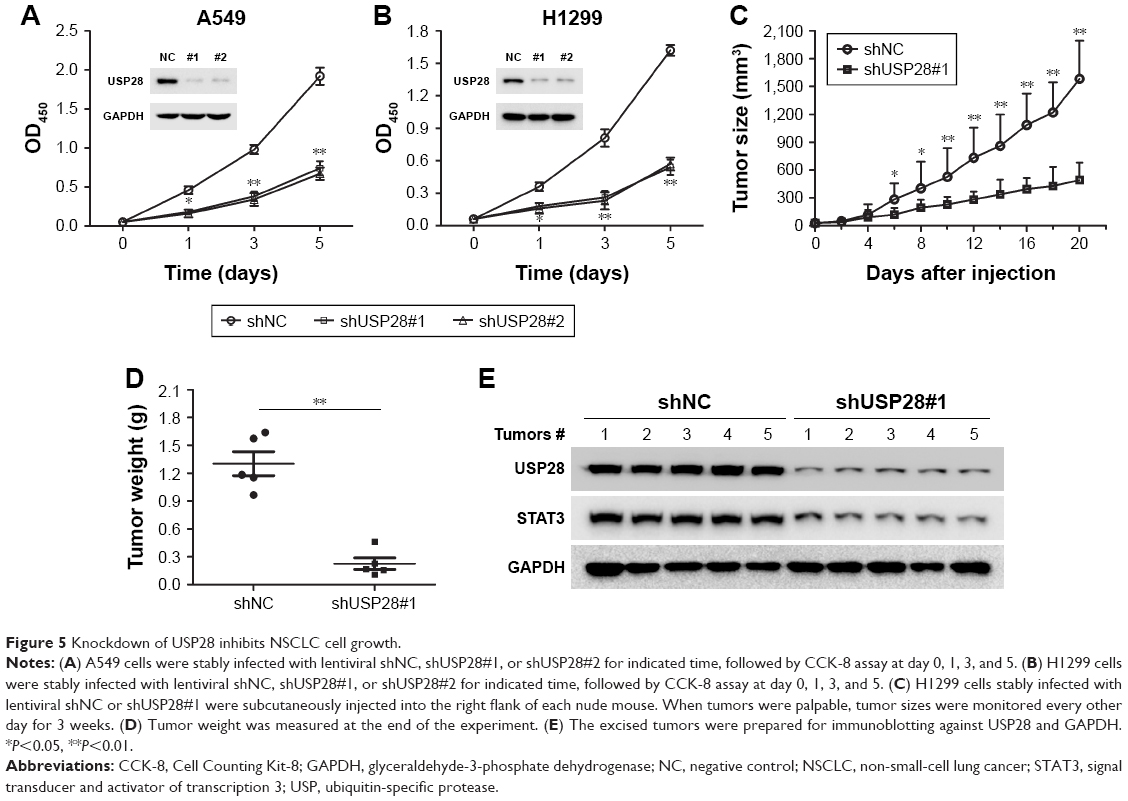 | Figure 5 Knockdown of USP28 inhibits NSCLC cell growth. |
Discussion
NSCLC remains a leading cause of cancer-related mortality worldwide due to its poor prognosis in clinic.23–25 Although strategies for NSCLC therapy have been focused on new targeted therapies against EGFR, immune checkpoints or angiogenesis, the overall survival for NSCLC patients is still low in clinic.26 Therefore, there is an urgent demand to identify novel and effective targets or drugs to improve systemic therapy for NSCLC patients. One of the possible strategies is to target the UPP for NSCLC therapy, such as DUBs.27 DUBs are responsible for cleavaging the ubiquitin chain of the substrate proteins, which balances ubiquitination and deubiquitination for determining protein fate.28 USP28 is one of the most functionally important DUBs and several potential substrates of USP28 have been identified, which play significant roles in cancer-related pathways, such as tumour progression, DNA-damage response, cell cycle, etc.5
Previous study has reported that USP28 was overexpressed in NSCLC and overexpression of USP28 promoted NSCLC cell growth, but its mechanism was still unknown in NSCLC cells.29 In this study, we confirmed that USP28 was highly expressed in NSCLC and predicted a poor prognosis for NSCLC patients (Figures 1 and 4). We also found that USP28 interacted with STAT3, and stabilized STAT3 by decreasing its polyubiquitination, which indicated that STAT3 was a potential substrate of USP28 (Figures 2 and 3). Previous studies have reported several other important substrates of USP28. USP28 was required to stabilize Chk2 and 53BP1 in response to DNA damage, thereby regulating the Chk2-p53-PUMA pathway.8 USP28 was also required for Myc stability, and bound to Myc through an interaction with Fbw7α, an F-box protein that was a part of an Skp1-Cullins-F-box (SCF)-type ubiquitin ligase.6 Therefore, reduced growth of the USP28 knockdown may be due to reduced STAT3 or reduced levels of other proteins involved in growth promoting pathways, such as Chk2 and Myc. Additionally, based on the findings that USP28 is required for oncoproteins’ stability, USP28 inhibition may represent a novel strategy for cancer treatment, and several dual inhibitors of the USP25/28 DUB subfamily were first identified.30
USP28 was also involved in drug resistance in cancer therapy. It was reported that USP28 functioned through a feedback loop to destabilize RAF family members.31 In a proportion of melanoma patients, USP28 was deleted, and loss of USP28 enhanced MAPK activity through the stabilization of RAF family members, which suggested that USP28 was a key factor in BRAF inhibitor resistance.31 In this study, we found that USP28 mediated STAT3 signaling and promoted NSCLC cell growth (Figures 1 and 5). In addition, STAT3 signaling also contributes to drug resistance in tumor therapy.32,33 These suggest that USP28/STAT3 axis may be another mechanism for drug resistance in the process of tumor therapy, which will be elucidated in our future work. Additionally, a recent publication showed that USP22 modulated the activity of STAT3 indirectly by stabilizing EGFR, which indicated that USP22 and USP28 may have redundant effects in NSCLC.34
Conclusion
USP28 is highly expressed and mediates STAT3 signaling by stabilizing STAT3 in NSCLC cells, which suggests that USP28 can be a potential target for NSCLC therapy in the future.
Acknowledgments
The authors are thankful for support from Jiangsu Provincial Key Medical Discipline (Laboratory) (No. ZDXKB2016007), the Clinical Medicine Center of Suzhou (No. SZZX201502), Suzhou Key Laboratory for Respiratory Medicine (No. SZS201617) and the Key Technology Applications Research Projects of Suzhou (No. SS201630).
Disclosure
The authors report no conflicts of interest in this work.
References
Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78(1):363–397. | ||
Guo YC, Zhang SW, Yuan Q. Deubiquitinating enzymes and bone remodeling. Stem Cells Int. 2018;2018(9):1–9. | ||
Valero R, Bayés M, Francisca Sánchez-Font M, et al. Characterization of alternatively spliced products and tissue-specific isoforms of USP28 and USP25. Genome Biol. 2001;2(10):Research0043. | ||
Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–563. | ||
Wang X, Liu Z, Zhang L, et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018;9(2):186. | ||
Popov N, Wanzel M, Madiredjo M, et al. The ubiquitin-specific protease USP28 is required for Myc stability. Nat Cell Biol. 2007;9(7):765–774. | ||
Popov N, Herold S, Llamazares M, Schülein C, Eilers M. Fbw7 and USP28 regulate Myc protein stability in response to DNA damage. Cell Cycle. 2007;6(19):2327–2331. | ||
Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126(3):529–542. | ||
Fong CS, Mazo G, Das T, et al. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife. 2016;5:e16270. | ||
Kitamura H, Ohno Y, Toyoshima Y, et al. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017;108(10):1947–1952. | ||
Xu X, Han K, Zhu J, et al. An inhibitor of cholesterol absorption displays anti-myeloma activity by targeting the JAK2-STAT3 signaling pathway. Oncotarget. 2016;7(46):75539–75550. | ||
Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–248. | ||
Yao S, Xu F, Chen Y, et al. Fbw7 regulates apoptosis in activated B-cell like diffuse large B-cell lymphoma by targeting STAT3 for ubiquitylation and degradation. J Exp Clin Cancer Res. 2017;36(1):10. | ||
Im CN, Yun HH, Song B, et al. BIS-mediated STAT3 stabilization regulates glioblastoma stem cell-like phenotypes. Oncotarget. 2016;7(23):35056–35070. | ||
Yang L, Wang R, Ma Z, et al. Porcine reproductive and respiratory syndrome virus antagonizes JAK/STAT3 signaling via NSP5, which induces STAT3 degradation. J Virol. 2017;91(3):e02087–16. | ||
Xu X, Han K, Tang X, et al. The ring finger protein RNF6 induces leukemia cell proliferation as a direct target of pre-B-cell leukemia homeobox 1. J Biol Chem. 2016;291(18):9617–9628. | ||
Han K, Xu X, Chen G, et al. Identification of a promising PI3K inhibitor for the treatment of multiple myeloma through the structural optimization. J Hematol Oncol. 2014;7:9. | ||
Xu X, Wang J, Han K, et al. Antimalarial drug mefloquine inhibits nuclear factor kappa B signaling and induces apoptosis in colorectal cancer cells. Cancer Sci. 2018;109(4):1220–1229. | ||
Zhang J, Wu H, Yi B, et al. Ring finger protein 38 induces gastric cancer cell growth by decreasing the stability of the protein tyrosine phosphatase SHP-1. FEBS Lett. 2018;592(18):3092–3100. | ||
Chen G, Xu X, Tong J, et al. Ubiquitination of the transcription factor c-Maf is mediated by multiple lysine residues. Int J Biochem Cell Biol. 2014;57:157–166. | ||
Tong M, Wang J, Jiang N, Pan H, Li D. Correlation between p-STAT3 overexpression and prognosis in lung cancer: a systematic review and meta-analysis. PLoS One. 2017;12(8):e0182282. | ||
Peyser ND, Freilino M, Wang L, et al. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene. 2016;35(9):1163–1169. | ||
Remon J, Vilariño N, Reguart N. Immune checkpoint inhibitors in non-small cell lung cancer (NSCLC): approaches on special subgroups and unresolved burning questions. Cancer Treat Rev. 2018;64:21–29. | ||
Janning M, Loges S. Anti-angiogenics: their value in lung cancer therapy. Oncol Res Treat. 2018;41(4):172–180. | ||
Han B, Yang L, Wang X, Yao L. Efficacy of pemetrexed-based regimens in advanced non-small cell lung cancer patients with activating epidermal growth factor receptor mutations after tyrosine kinase inhibitor failure: a systematic review. Onco Targets Ther. 2018;11:2121–2129. | ||
Meyers DE, Bryan PM, Banerji S, Morris DG. Targeting the PD-1/PD-L1 axis for the treatment of non-small-cell lung cancer. Curr Oncol. 2018;25(4):e324–e334. | ||
Chen X, Yang Q, Chen J, et al. Inhibition of proteasomal deubiquitinase by silver complex induces apoptosis in non-small cell lung cancer cells. Cell Physiol Biochem. 2018;49(2):780–797. | ||
Yeasmin Khusbu F, Chen FZ, Chen HC. Targeting ubiquitin specific protease 7 in cancer: a deubiquitinase with great prospects. Cell Biochem Funct. 2018;36(5):244–254. | ||
Zhang L, Xu B, Qiang Y, et al. Overexpression of deubiquitinating enzyme USP28 promoted non-small cell lung cancer growth. J Cell Mol Med. 2015;19(4):799–805. | ||
Wrigley JD, Gavory G, Simpson I, et al. Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem Biol. 2017;12(12):3113–3125. | ||
Saei A, Palafox M, Benoukraf T, et al. Loss of USP28-mediated BRAF degradation drives resistance to Raf cancer therapies. J Exp Med. 2018;215(7):1913–1928. | ||
Nagathihalli NS, Castellanos JA, Lamichhane P, et al. Inverse correlation of STAT3 and MEK signaling mediates resistance to Ras pathway inhibition in pancreatic cancer. Cancer Res. 2018;78(21):6235–6246. | ||
Wang L, Wang Q, Gao M, et al. STAT3 activation confers trastuzumab-emtansine (T-DM1) resistance in HER2-positive breast cancer. Cancer Sci. 2018;109(10):3305–3315. | ||
Zhang H, Han B, Lu H, et al. USP22 promotes resistance to EGFR-TKIs by preventing ubiquitination-mediated EGFR degradation in EGFR-mutant lung adenocarcinoma. Cancer Lett. 2018;433:186–198. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.