")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 8
Type 2 diabetes mellitus development programs in the new regulatory environment with cardiovascular safety requirements
Authors Yang F, Stewart M, Ye J, DeMets D
Received 5 March 2015
Accepted for publication 4 May 2015
Published 20 July 2015 Volume 2015:8 Pages 315—325
DOI https://doi.org/10.2147/DMSO.S84005
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Fred Yang,1 Murray Stewart,2 June Ye,2 David DeMets3
1AstraZeneca, Gaithersburg, MD, USA; 2GlaxoSmithKline, Upper Merion, PA, USA; 3Department of Biostatistics and Medical Informatics, University of Wisconsin–Madison, Madison, WI, USA
Abstract: For type 2 diabetes mellitus treatment and clinical development, proper evaluation of cardiovascular risk has been required by regulatory agencies (eg, the US Food and Drug Administration) since cardiovascular safety is very important in this patient population. The US Food and Drug Administration issued general guidelines for cardiovascular safety evaluation that outlined the requirements considered adequate for cardiovascular safety evaluation. However, there are multiple options to obtain the data and fulfill these requirements. In this paper, we outline the potential pathways and challenges in various aspects of cardiovascular safety evaluation in type 2 diabetes clinical development, including study design, populations, and endpoints. Specifically, we discuss some challenges in statistical analysis which have implications for the design, implementation, and interpretation of these outcome studies.
Keywords: T2DM, cardiovascular outcome, CV risk, noninferiority, superiority, major adverse cardiac event
Background
Diabetes affects an estimated 347 million people worldwide,1 with type 2 diabetes mellitus (T2DM) accounting for more than 90% of diabetes cases.2 Cardiovascular (CV) events, including myocardial infarctions (MIs) and stroke, are major causes of mortality and morbidity in patients with diabetes. In 2003–2006, after adjusting for population age differences, CV disease death rates were approximately 1.7 times higher among adults aged 18 years or older with diagnosed diabetes than among adults without diagnosed diabetes.3 Thus, the risk that a T2DM therapy might increase the rate of these events is a legitimate concern. Given the availability of alternative treatments for treating type 2 diabetes, the US Food and Drug Administration (FDA) issued a formal guidance document in 2008 suggesting that explicit assessment of CV safety should be done as part of the development of all new drugs for type 2 diabetes regardless of their mechanism of action or preclinical and clinical evidence suggesting a possible increased CV risk.4 The key points of these guidelines are outlined below:
- Establishment of an independent CV clinical endpoints committee (CEC) for prospective adjudication of CV events from all Phase II and III trials through a meta-analysis, or from a single large safety trial.
- Events of interest should include CV death, MI, and stroke, and may include hospitalization for acute coronary syndrome (ACS), urgent revascularization procedures, and other major CV clinical endpoints.
- The patient population should include those at higher risk for a CV event (eg, a longer duration of T2DM, advanced age, renal impairment).
- Studies should be designed and conducted such that a meta-analysis could be performed.
- A protocol describing statistical methods for the proposed meta-analysis should be submitted in which a relative risk (RR) of >1.8 should be ruled out at the time of meta-analysis, with subsequent postmarketing trials to provide definitive evidence of a CV RR <1.3.
- The selection of the RR thresholds of 1.8 and 1.3 is based on the following rationale:5
- At the initial filing stage, when glycemic control has been established in short-term studies, there is a higher tolerance for additional uncertainty (capped at a 1.8 RR threshold), as lowering glycated hemoglobin (HbA1c) reduces the associated symptoms of hyperglycemia and reduces long-term microvascular complications.
- The 1.3 RR threshold has been used in other settings for excluding CV risk (eg, for COX-2 inhibitors).
- The 1.3 RR threshold is feasible, but meeting this criterion preapproval would significantly delay new drug availability.
Aspects needing consideration in development plan design
While the FDA guidelines outlined what kind of evidence/data would be considered appropriate for the initial evaluation and final confirmation of CV safety,6 there are many different pathways to obtaining the data/evidence and accordingly multiple ways to analyze the data. When designing the development plan, the following aspects need to be considered:
- Medicine in development (agent)
- Risk factor/biomarker evaluation (biomarkers)
- Patient population selection (cohort)
- Study/program planning (design)
- Safety/clinical outcome (endpoint)
- Completeness of data (follow-up)
- Evaluation of data and statistical analysis (analysis)
In this article, we discuss several challenges in study design, study conduct, and data collection/analysis to obtain sufficient information on CV safety during the development of a T2DM treatment/medication.
Medicine in development (agent) and risk factor/biomarker evaluation (biomarkers)
Before the start of any outcome study, it is essential to have full understanding of the potential biological impact of a new T2DM medication on CV risk factors (biomarkers), such as lipids or blood pressure. A new medication which causes, for example, an increase in lipids or blood pressure should be evaluated more carefully, and further development should include the initiation of a CV outcome study.6 During the study, it is very important to closely monitor CV risk factors/biomarkers, especially if the testing agent is known to increase certain risk factors.
Patient population selection (cohort)
The target T2DM patient populations for clinical studies to evaluate efficacy and tolerability/safety and to evaluate CV safety may or may not be the same. In most of the Phase III studies which provide full evaluation of efficacy (measured by HbA1c) and tolerability/safety, a wide spectrum of patients is recruited into different trials to evaluate the new medication in various treatment combinations (from drug-naïve patients to those on a combination that includes insulin) and against various comparators (placebo or different active controls). Within each study, patients are usually relatively homogeneous in terms of background medication (eg, metformin only), and a wide age range is represented (eg, 18 to 75 years). In general, the number of patients with high CV risk is limited, even when studies are designed to include those patients.
However, to evaluate CV safety, most outcome studies – including PROactive,7 ADVANCE,8,9 ADOPT,10 VADT,11,12 ACCORD,13 RECORD,14 BARI 2D,15,16 EXAMINE,17,18 and SAVOR-TIMI 5319,20 (see Table S1) – enrolled elderly patients with higher CV risk defined by either a previous history of CV events or by the presence of two or more CV risk factors. Some of the studies even enrolled patients with a history of ACS. In general, these are not the “typical” patient populations of Phase III T2DM studies. There are at least two factors that need to be considered for patients to be enrolled for a CV outcome study. On one hand, patients with higher CV risk (eg, an ACS population) would yield more CV events during the study, which is critical for study power and duration. On the other hand, if patients enrolled in the study are at high risk with less modifiable CV disease, their CV events or deaths are driven more by the disease itself and may not be treatment related. The results of the study from such a population, especially for a noninferiority trial, may suffer from a lack of “assay sensitivity” and would not be interpretable or generalizable. However, if the results demonstrate superiority of one treatment over another in a population with less modifiable disease, the data would be considered more generalizable and more meaningful. Table 1 outlines some of the considerations for patient population selection.
 | Table 1 Options and considerations for patient population selection in an outcome study |
Study/program planning (design)
The consideration of design(s) for the CV safety evaluation of a medication in T2DM patients includes two aspects: the overall development program design and the specific outcome study design.
There are many different options for an overall development program, ranging from one big outcome study to multiple traditional Phase III studies and various hybrid approaches dependent on study size, timing, and target population. The initial challenge of ruling out a CV RR >1.8 for regulatory approval may require a single large trial or a meta-analysis of several smaller Phase II/IIIa trials. The ultimate requirement of ruling out a CV RR >1.3 definitely will require a large-scale, long-term CV outcome trial. Demonstration of superiority (when desirable) may require an even larger and/or longer term trial and may not be feasible if the drug already has been approved for use and established as relatively safe. A hybrid approach is the choice for most developers. Figure 1 lists two different hybrid approaches.
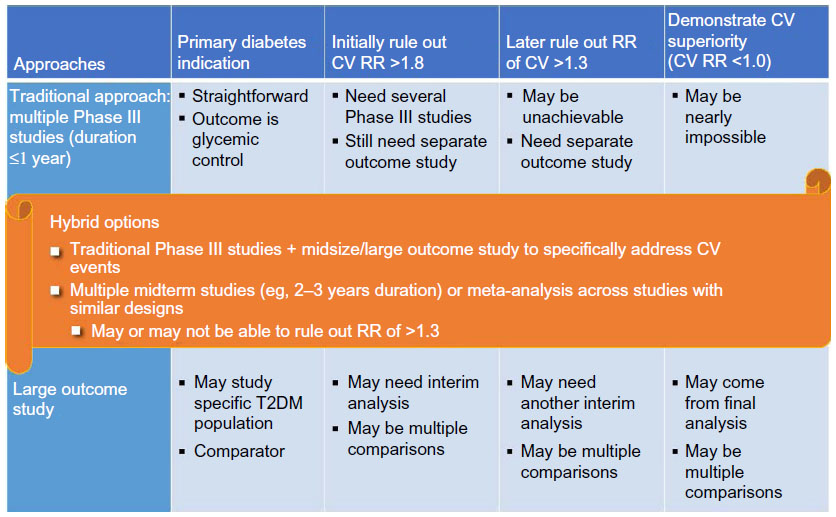 | Figure 1 Issues in research program design. |
It is almost impossible for the traditional approach of multiple Phase III studies to produce a sufficient number of events to provide definitive evidence of CV safety, especially for the RR margin of 1.3. On the other hand, depending on the study design, the single large outcome study approach might not allow researchers to evaluate glycemic control across a broad spectrum of patient populations or against different comparators for risk–benefit evaluations. In addition, the patients included in a single outcome study usually would be patients with high CV risk, differing from general T2DM patients included in the Phase III studies.
There are a number of different hybrid approaches. The options listed in Figure 1 could be considered, with variations as appropriate. Each of them has its own advantages and limitations. Depending on the size of the separate outcome study, the first hybrid option in general would be able to rule out RRs of both >1.8 and >1.3, and even demonstrate superiority. However, with this option, one may encounter the challenge of early data release/publication (see the section “Evaluation of data and statistical analysis [analysis]”). Depending on the study population and sample size, use of the second hybrid option may not accumulate enough events for full evaluation, especially for ruling out an RR of >1.3.
As for the large outcome study design, Table 2 outlines several current ongoing or completed outcome studies that were initiated after the 2008 FDA guidelines.6 Some of the studies were initiated before the end of the main Phase III programs and some of them were initiated after FDA approval. In both of these situations, the patients enrolled in the CV outcome studies were generally different from those in the main Phase III program, with higher CV risk and prior CV events. As an extreme example, the EXAMINE study enrolled diabetic patients with recent ACS.17,18 These patients are usually excluded from Phase III studies in T2DM.
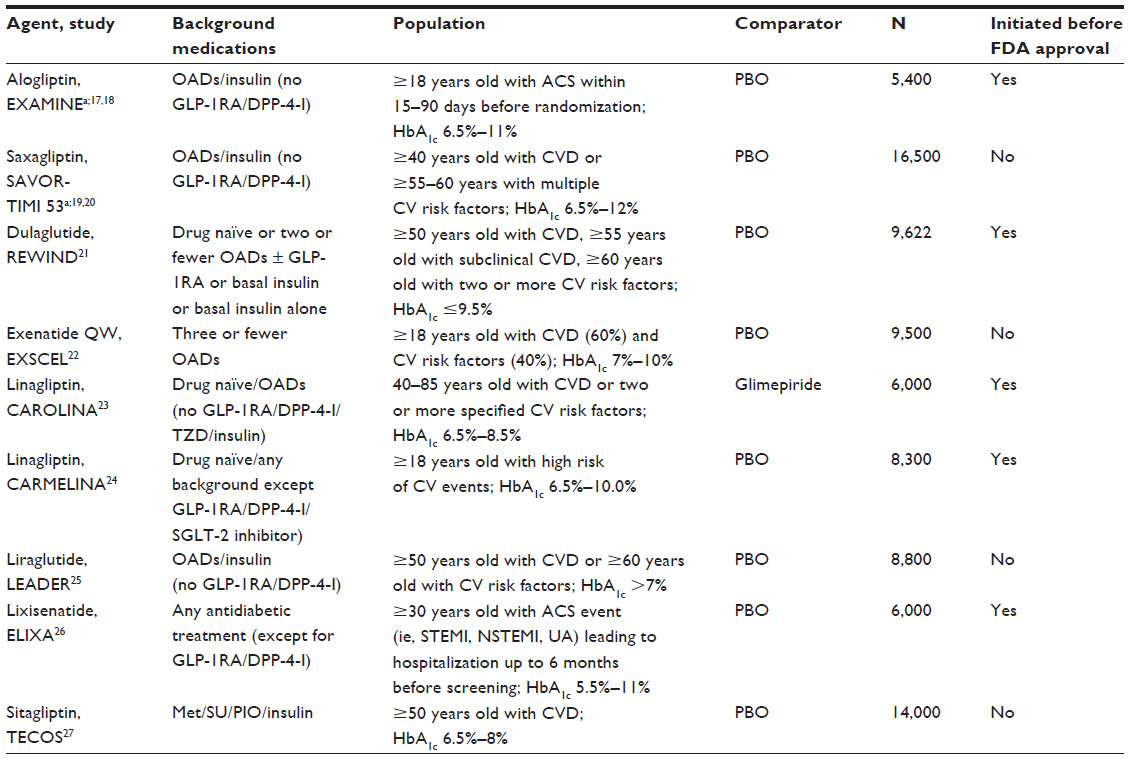 | Table 2 Selected type 2 diabetes mellitus CV outcome studies |
The most important aspects of the design of a specific CV outcome study are the objectives of the study and the choice of comparator. Is safety the only study objective (ie, to rule out a CV RR of >1.3) or is a second objective to demonstrate CV superiority (ie, a clinically meaningful benefit)? Different objectives have different impacts on both the sample size and patient population selection. For example, it might be harder to demonstrate superiority in the ACS patient population, as those patients might not be considered to have CV disease that can be modified by T2DM medication. It also takes much longer to complete a superiority study if the intervention effect size is moderate, which is often the case. As for the selection of comparators, most of the studies17–27 outlined in Table 2 used a placebo added to standard care as the comparator, while the CAROLINA study23 used glimepiride. However, the sponsor of the CAROLINA study also started a separate placebo-controlled study (ie, the CARMELINA study24 of linagliptin versus placebo).
Another important part of study planning is about clinical management of patients which will affect incidence of major CV events. The prospective randomized clinical trials have failed to establish a correlation between patients with tight glycemic control and CV outcomes – eg, ADVANCE and ACCORD.8,9,13 Therefore, more attention should be paid to alterations beyond hyperglycemia, eg, a reduction of endoplasmic reticulum stress, oxidative stress, and/or inflammation through pharmacological or lifestyle interventions seems promising as, given the extensive crosstalk between these three alterations, reducing one can potentially reduce the others.28 With improvement of clinical management of patients, the incidence of major adverse cardiac events (MACE) may be lower than estimated at study design period. The incidence rate should be carefully monitored during the trial so that number of patients or duration of the study could be adjusted accordingly.
Safety/clinical outcome (endpoint)
Traditionally, an adjudicated “MACE” is considered as a fully validated clinical outcome for CV outcome studies, where MACE is defined as CV death, MI, or stroke. In the 2008 FDA guidelines for evaluating CV risk in the diabetes population, additional CV-related events (referred to as MACE+ in some literature) also could be considered for adjudication by the CEC.6 Note that the general guidance provided by the FDA later indicated that even though the adjudication could include events beyond MACE (such as congestive heart failure), the recommended MACE+ endpoint should be defined as MACE plus hospitalization due to unstable angina. All events need to be adjudicated by an independent and blinded CEC (see Table S2).
While the most important aspect of clinical endpoint selection is clinical meaningfulness, statistically it is important to understand the appropriateness of the selection: Is the endpoint so narrow that it may not cover the CV safety aspect of the targeted patient population? Or is it so wide that it would not allow the detection of safety signal? That is, a narrow outcome might miss key results that the intervention might negatively affect. A very wide outcome might include results that would never be affected by the intervention and thus could obscure the components that are affected, resulting in an overall safety signal not being detected.
Completeness of data (follow-up)
In all outcome studies, completeness of data and follow-up are critical for final data analysis and interpretation. A study with too many patients being lost to follow-up limits and may distort inferences about the population, especially if the loss may be related to adverse events or a life event that would impact the primary analysis. Most analysis methods assume that the missing data are independent of the disease process or underlying risk. However, there are examples where the loss to follow-up, or failing to follow patients when they stop taking their medication, appears related to the underlying risk and thus is a process referred to as informative censoring, eg, the APPROVe and APPROVe+1 studies.29,30
Even if the loss to follow-up is not considered related to the endpoint, the amount of lost information does not allow proper comparison of the new intervention with the comparator. Such loss could introduce bias “if the unavailability of data is associated with the likelihood of outcome events” or reduce power “because the effective sample size is reduced”.31 Decreased power may have an impact on the interpretation of noninferiority study outcomes.
Recognizing the importance of data completeness, studies should consider proper rescue procedures which would allow patients to remain for follow-up. Even if patients withdraw from active treatment, they still can be followed up in the study for safety information. As T2DM progresses, patients need either to switch to or add on new antidiabetic medications (ie, rescue medications) in order to follow general diabetes treatment guidelines. Proper specification of rescue procedures provides good glycemic control throughout the study, allowing more complete follow-up and therefore also allowing a proper evaluation of CV effects without the confounding factor of a glycemic difference, especially in placebo-controlled studies.
Evaluation of data and statistical analysis (analysis)
Like other clinical studies, data and statistical analysis are involved in every aspect of a CV safety evaluation. Notably, there also are some special statistical challenges throughout the planning and analysis of CV safety data in T2DM drug development. Among them is the practical consideration of study conduct and integrity in the implementation of a stepwise sequential approach for two noninferiority hypotheses, using 1.8 and 1.3 as the RR safety margins. The role of the independent data monitoring committee (IDMC) in the whole process of regulatory submission and data dissemination and also type I error control for superiority and noninferiority tests across different study designs are discussed in this section.
One of the most important recommendations from the FDA pertains to the two-step sequential approach for ruling out CV risk. Specifically, the FDA recommended that the sponsor should rule out a CV RR of >1.8 at the time of regulatory approval for drug marketing and rule out a CV RR of >1.3 in postmarketing trials to provide longer term definitive evidence of CV safety.6 While it is clear that these are two separate hypotheses, the sources of the data that could be used to test these hypotheses can vary: the data could come from 1) a combined set of studies (eg, Phase III studies in combination with an outcome study) or 2) two different studies (eg, Phase III studies ruling out a CV RR >1.8 and a separate CV outcome study ruling out a CV RR >1.3) (see Figure 2). A significant challenge is posed when a single CV study is used to rule out both an RR of >1.8 and an RR of >1.3. If drug approval is sought after ruling out a CV RR of 1.8 or higher, these data will likely be released/made public by the regulatory approval process. This could very likely have a negative effect on the continuation of the single trial to rule out a longer term CV RR of 1.3 or higher. That is, patients will be aware that general efficacy, based on HbA1c control, and early CV safety (ie, the initial finding of an RR <1.8) have been established and that the medication is now available to them, but they are being encouraged to remain on the blinded assigned study drug (active or placebo) until an RR of 1.3 or higher has been ruled out. This may appear to add pressure to the participant and thus pose an ethical dilemma for both physician and patient.
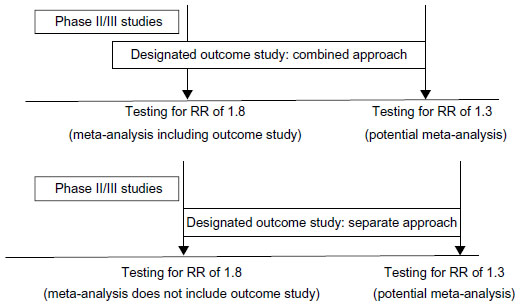 | Figure 2 Graphic illustration of different trial plans and data analysis strategies. |
In the two-step approach, one or more trials to rule out a CV RR of 1.8 or higher would be used to establish regulatory approval. Potential patients in the second CV trial would be aware of the first trial results during the recruitment and consent process and would have the opportunity to enroll or not enroll in the second trial without pressure. It is likely that patients enrolled into the trial to rule out a CV RR of 1.3 or higher would not be the same as those enrolled in the trial(s) to rule out an RR of 1.8 or higher.
This dilemma is still not resolved and continues to be a challenge for sponsors and investigators conducting T2DM trials. Perhaps the only immediate solution to this problem is to not submit early data for regulatory approval, although this delays the availability of new interventions for patients.
Another important aspect is to further clarify the role of the IDMC (also referred to as the data safety monitoring board) in various development programs/options. The traditional responsibility of the IDMC is to protect the safety of study participants and to assess the overall risk-to-benefit ratio. However, when the overall program is designed to use a combination of studies (eg, preapproval Phase III studies in combination with partial information from a single large outcome study started prior to approval) to evaluate CV safety, the IDMC may be asked to provide interim data analyses (prespecified) to facilitate the decision on various regulatory submissions, ie, provide data analysis results to show that a CV RR of >1.8 is ruled out while the large CV study is still ongoing to rule out an RR >1.3. In this scenario, the responsibility is transferred to the IDMC, which will be a new challenge for the IDMC, sponsor, and regulatory agency alike.
Last but not least, an important consideration in outcome studies is around multiplicity control for primary endpoints and other secondary endpoints. The outcome study to rule out an RR of 1.3 usually requires 600~700 MACE, which will be translated into thousands of patients being followed up for several years. It will be a good opportunity to collect more information to explore some potential benefits which can only be evaluated by such large trials due to the low incidence, eg, composite microvascular endpoints or MACE superiority. However, strict multiplicity control for those secondary endpoints would make it impossible to claim marginal benefit, which may discourage the investigators to collect less information and to do selective analysis. Two studies, SAVOR-TIMI 5319,20 and EXAMINE,17,18 both using MACE as the primary endpoint and only including the MACE+ type of endpoint as a secondary endpoint, took sequential testing approaches similar to some published approaches (see Table 3).32 That is, after success of the noninferiority test for CV safety by ruling out an RR >1.3, CV superiority and specified secondary endpoints were to be tested based on a predefined sequence. Note that in the SAVOR-TIMI 53 study, glycemic control-related endpoints are included under “other efficacy endpoints” and evaluated at a nominal type I error rate.19,20 No glycemic-related endpoint was included in the efficacy endpoints listed in the EXAMINE study.17,18 In the CAROLINA study, only the noninferiority and superiority of the primary endpoint (MACE plus hospitalization due to unstable angina) are included in the testing sequence (also see Table 3).23 Glycemic endpoints are included in the “secondary objectives” and, per study design, “all other secondary analyses will be exploratory with no correction for multiple hypotheses.” Published results from this study are not available yet.
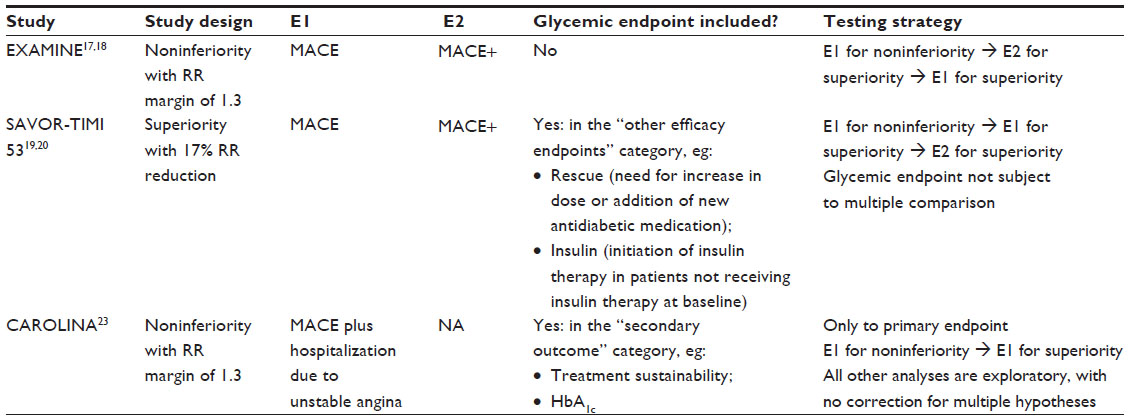 | Table 3 Study design and testing strategy for selected CV outcome studies |
Based on the trial information outlined above, it seems reasonable to separate the test of glycemic control endpoints from CV endpoints to allow for evaluation of the glycemic-related endpoints without multiple comparison adjustments with respect to the CV endpoints.
For other endpoints (eg, microvascular events, renal parameters), the picture is not that clear. As pointed out earlier, the CV outcome study in T2DM patients is not only a study to evaluate CV safety, but also a great opportunity to evaluate microvascular benefit and other health outcome benefits, in addition to evaluating long-term glycemic control. However, important clinical endpoints, such as microvascular benefit, were not included in the list of secondary endpoints in the examples above. This may have been due to the fact that the studies were not long enough and were not powered to evaluate microvascular events. Nevertheless, if researchers also are interested in the effect of medication on microvascular benefits (as well as CV benefit/superiority) and would like to make formal inferences for these endpoints, a testing strategy to have proper multiplicity control will be needed in addition to adequate sample size. The testing procedures outlined in the above examples – 1) CV noninferiority → CV superiority → secondary endpoints (eg, microvascular) or 2) CV noninferiority → secondary endpoints (eg, microvascular) → CV superiority – are all mathematically valid, but do not make logical sense. One might ask why testing CV superiority should be contingent on the success of microvascular endpoints or vice versa. In a broader sense, the question really is about whether a superiority test that followed the success of a noninferiority test should be subject to multiplicity control with other secondary endpoints when the superiority test is not the primary objective from a clinical point of view. Under a traditional approach, once noninferiority is achieved, CV superiority and other secondary endpoints would be tested, either sequentially using gatekeeping procedures or by other means of type I error adjustment, such as the methods of Hochberg and Hommel, as reported by Hung and Wang.32 The former procedure has weaknesses as discussed, and the latter procedure has its own drawbacks, since some endpoints may be tested at a reduced alpha level, depending on the significance of other endpoints.
Discussion
In this paper, we have explored the different options to assess CV outcome. We considered several factors that impact the CV outcome studies in diabetes. These include risk factors such as lipids and blood pressure, as well as treatment. These need to be assessed when exploring the CV data from different development programs, which can vary by population and different background of previous CV events. In addition, statistical testing procedures also have an impact on the design of study.
There are certain limitations of the paper. First of all, the paper discussed several aspects of CV outcome study design and interpretation; there are many factors which would have an impact on CV outcome study which are beyond the scope of what has been outlined or are not known to the researchers. Second, the paper considered several aspects of the trial design in terms of proper statistical planning; the clinical outcome and data from clinical study would also mostly depend on the biological plausibility/rationale and may not be fully explained statistically. Third, the first and second considerations outlined earlier, focused on the evaluation of CV efficacy and safety in a clinical trial setting. This has bias, as, in the trial setting, subjects are included within certain criteria and are often excluded when there are several comorbidities. Furthermore, the behavior of participants and their physicians in the trial results in greater adherence to guidelines for control of lipids and blood pressure resulting in better control and less CV events than would be seen in the real-world setting. The evaluation of CV efficacy and safety in the “real-world” clinical practice setting might be considered more important and should be considered further. Last but not least, the paper outlined some challenges of statistical evaluation of secondary endpoints and more work/guidance is needed on resolving the multiple comparison issues.
Conclusion
The FDA guidelines outline the scope of evidence/data that would be considered appropriate to determine CV safety during the development of a T2DM medication. There are many different pathways to obtaining the data and proper evidence, and accordingly multiple ways to analyze the data. For example, there are at least 23 (or 16) different development pathways upfront in the planning stage, considering only the following (previously discussed) four factors:
- patient population selection (ACS population versus general high-risk patients);
- overall development program planning (independent outcome study versus simultaneous Phase III/outcome study);
- outcome study planning (noninferiority alone versus superiority also); and
- safety/clinical endpoint selection (MACE versus MACE+).
Each of the pathways has its pros and cons. Depending on the medicine in development (agent) and potential biological effects (biomarkers), and the stage of development (before or after approval), one should choose one pathway over the other after careful consideration.
These options give certain challenges for statistics, including the changing role of the IDMC. When outcome studies are conducted simultaneously with other Phase III studies, the IDMC would be involved, eg, asked to provide data analysis and input to the sponsor for the submission decision or to satisfy regulatory agency requests. This is different from the traditional IDMC role, and the implication of the release of data halfway through the study needs further evaluation. Finally, the statistical analysis and type I error control/multiple comparisons in various design options also need to be carefully evaluated. Multiple comparison adjustment has special importance as it would be a key factor to influence the study outcome and design, especially if the study includes a secondary endpoint other than glycemic control. All this information is critical to patients, clinicians, and health care providers. To address those objectives and make formal statistical inferences from the data, one would need a testing strategy to have proper multiplicity control.
Acknowledgments
Dr Jill Donaldson (GlaxoSmithKline) provided editorial support for the manuscript. This manuscript was prepared according to the International Society for Medical Publication Professionals’ “Research Methods & Reporting. Good publication practice for communicating company sponsored medical research: the GPP2 guidelines”. Funding for this work was provided by GlaxoSmithKline. Editorial assistance (copy editing, formatting, and preparation of submission package) was provided by PharmaWrite LLC of Princeton, NJ, USA and was funded by GlaxoSmithKline.
Disclosure
Fred Yang is a former employee of GlaxoSmithKline. The authors report no other conflicts of interest in this work.
References
Danaei G, Finucane MM, Lu Y, et al; Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (Blood Glucose). National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet. 2011;378(9785):31–40. | |
Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycemia: Report of a WHO/IDF Consultation. World Health Organization; International Diabetes Federation; 2006. Available from: http://whqlibdoc.who.int/publications/2006/9241594934_eng.pdf?ua=1. Accessed January 31, 2015. | |
National Diabetes Statistics Report, 2014: Estimates of Diabetes and Its Burden in the United States. Division of Diabetes Translation, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention; 2014. Available from: http://www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf. Accessed December 31, 2014. | |
Brass EP. The Food and Drug Administration and the future of drug development for the treatment of diabetes. Diabetes Spectr. 2014;27(2):75–77. | |
Fleming TR. Identifying and addressing safety signals in clinical trials: some issues and challenges. In: Fleming TR, Weir BS, editors. Proceedings of the Fourth Seattle Symposium in Biostatistics: Clinical Trials. New York, NY: Springer; 2013:137–156. | |
Guidance for Industry: Diabetes Mellitus – Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2008. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071627.pdf. Accessed January 31, 2012. | |
Dormandy JA, Charbonnel B, Eckland DJ, et al; PROactive investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. | |
Patel A, Chalmers J, Poulter N. ADVANCE: action in diabetes and vascular disease. J Hum Hypertens. 2005;19 Suppl 1:S27–S32. | |
Patel A; ADVANCE Collaborative Group, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet. 2007;370(9590):829–840. | |
Viberti G, Lachin J, Holman R, et al; ADOPT Study Group. A Diabetes Outcome Progression Trial (ADOPT): baseline characteristics of type 2 diabetic patients in North America and Europe. Diabet Med. 2006;23(12):1289–1294. | |
Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360(2):129–139. | |
Meyers CD, McCarren M, Wong ND, Abraira C, Duckworth WC, Kashyap ML; VADT Investigators. Baseline achievement of lipid goals and usage of lipid medications in patients with diabetes mellitus (from the Veterans Affairs Diabetes Trial). Am J Cardiol. 2006; 98(1):63–65. | |
ACCORD Study Group, Buse JB, Bigger JT, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol. 2007;99(12A):21i–33i. | |
Home PD, Pocock SJ, Beck-Nielsen H, et al; RECORD Study Team. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373(9681):2125–2135. | |
Bypass Angioplasty Revascularization Investigation 2 Diabetes Study Group. Baseline characteristics of patients with diabetes and coronary artery disease enrolled in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Am Heart J. 2008;156(3):528–536. | |
BARI 2D Study Group, Frye RL, August P, et al. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360(24):2503–2515. | |
White WB, Bakris GL, Bergenstal RM, et al. EXamination of CArdiovascular OutcoMes with AlogliptIN versus Standard of CarE in Patients with type 2 diabetes mellitus and acute coronary syndrome (EXAMINE): a cardiovascular safety study of the dipeptidyl peptidase 4 inhibitor alogliptin in patients with type 2 diabetes with acute coronary syndrome. Am Heart J. 2011;162(4):620–626. | |
White WB, Cannon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–1335. | |
Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369(14):1317–1326. | |
Scirica BM, Bhatt DL, Braunwald E, et al. The design and rationale of the saxagliptin assessment of vascular outcomes recorded in patients with diabetes mellitus-thrombolysis in myocardial infarction (SAVOR-TIMI) 53 study. Am Heart J. 2011;162(5):818–825. | |
Eli Lilly and Company. Researching cardiovascular events with a weekly incretin in diabetes (REWIND). Available from: http://www.clinicaltrials.gov/ct2/show/NCT01394952. NLM identifier: NCT01394952. Accessed June 30, 2014. | |
AstraZeneca. Exenatide study of cardiovascular event lowering trial (EXSCEL): a trial to evaluate cardiovascular outcomes after treatment with exenatide once weekly in patients with type 2 diabetes mellitus. Available from: https://clinicaltrials.gov/ct2/show/NCT01144338. NLM identifier: NCT01144338. Accessed October 24, 2011. | |
Boehringer Ingelheim. CAROLINA: cardiovascular outcome study of linagliptin versus glimepiride in patients with type 2 diabetes. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01243424. NLM identifier: NCT01243424. Accessed June 30, 2014. | |
Boehringer Ingelheim. Cardiovascular and renal microvascular outcome study with linagliptin in patients with type 2 diabetes mellitus (CARMELINA). Available from: http://www.clinicaltrials.gov/ct2/show/NCT01897532. NLM identifier: NCT01897532. Accessed June 30, 2014. | |
Novo Nordisk A/S. Liraglutide effect and action in diabetes: evaluation of cardiovascular outcome results – a long term evaluation (LEADER®). Available from: http://www.clinicaltrials.gov/ct2/show/NCT01179048?term=NCT01179048&rank=1. NLM identifier: NCT01179048. Accessed October 25, 2011. | |
Sanofi. Evaluation of cardiovascular outcomes in patients with type 2 diabetes after acute coronary syndrome during treatment with AVE0010 (lixisenatide) (ELIXA). Available from: http://www.clinicaltrials.gov/ct2/show/NCT01147250. NLM identifier: NCT01147250. Accessed June 30, 2014. | |
Merck Sharp & Dohme Corp. Sitagliptin cardiovascular outcome study (MK-0431-082) (TECOS). Available from: http://www.clinicaltrials.gov/ct2/show/NCT00790205. NLM identifier: NCT00790205. Accessed June 30, 2014. | |
Badimon L, Hernández Vera R, Vilahur G. Determinants of cardiovascular risk in diabetes beyond hyperglycemia. J Cardiovasc Dis. 2013;1(2):53–62. | |
Baron JA, Sandler RS, Bresalier RS, et al. Cardiovascular events associated with rofecoxib: final analysis of the APPROVe trial. Lancet. 2008;372(9651):1756–1764. | |
Bresalier RS, Sandler RS, Quan H, et al; Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–1102. | |
Akl EA, Briel M, You JJ, et al. Potential impact on estimated treatment effects of information lost to follow-up in randomised controlled trials (LOST-IT): systematic review. BMJ. 2012;344:e2809. | |
Hung HM, Wang SJ. Some controversial multiple testing problems in regulatory applications. J Biopharm Stat. 2009;19(1):1–11. |
Supplementary materials
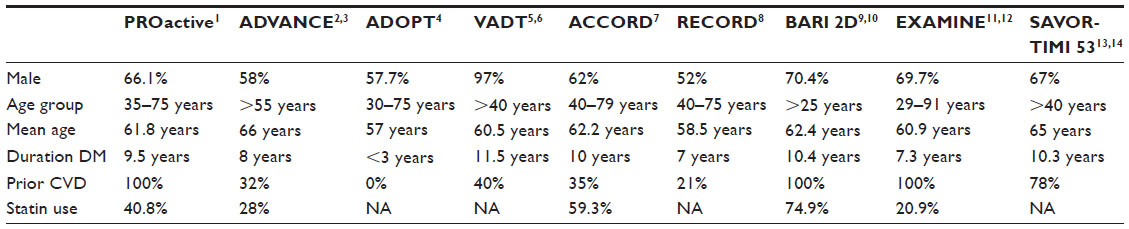 | Table S1 Patient population/risk factor comparison for outcome studies |
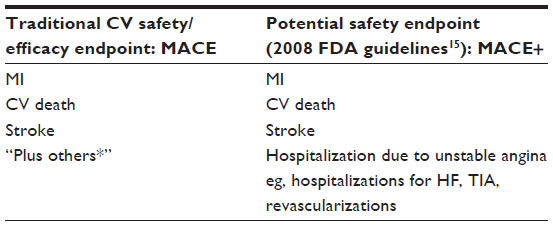 | Table S2 Endpoint selection for CV safety evaluation |
References
Dormandy JA, Charbonnel B, Eckland DJ, et al; PROactive investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. | |
Patel A, Chalmers J, Poulter N. ADVANCE: action in diabetes and vascular disease. J Hum Hypertens. 2005;19 Suppl 1:S27–S32. | |
Patel A; ADVANCE Collaborative Group, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet. 2007;370(9590):829–840. | |
Viberti G, Lachin J, Holman R, et al; ADOPT Study Group. A Diabetes Outcome Progression Trial (ADOPT): baseline characteristics of type 2 diabetic patients in North America and Europe. Diabet Med. 2006;23(12):1289–1294. | |
Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360(2):129–139. | |
Meyers CD, McCarren M, Wong ND, Abraira C, Duckworth WC, Kashyap ML; VADT Investigators. Baseline achievement of lipid goals and usage of lipid medications in patients with diabetes mellitus (from the Veterans Affairs Diabetes Trial). Am J Cardiol. 2006;98(1):63–65. | |
ACCORD Study Group, Buse JB, Bigger JT, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol. 2007;99(12A):21i–33i. | |
Home PD, Pocock SJ, Beck-Nielsen H, et al; RECORD Study Team. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373(9681):2125–2135. | |
Bypass Angioplasty Revascularization Investigation 2 Diabetes Study Group. Baseline characteristics of patients with diabetes and coronary artery disease enrolled in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Am Heart J. 2008;156(3):528–536. | |
BARI 2D Study Group, Frye RL, August P, et al. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360(24):2503–2515. | |
White WB, Bakris GL, Bergenstal RM, et al. EXamination of CArdiovascular OutcoMes with AlogliptIN versus Standard of CarE in Patients with type 2 diabetes mellitus and acute coronary syndrome (EXAMINE): a cardiovascular safety study of the dipeptidyl peptidase 4 inhibitor alogliptin in patients with type 2 diabetes with acute coronary syndrome. Am Heart J. 2011;162(4):620–626. | |
White WB, Cannon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–1335. | |
Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369(14):1317–1326. | |
Scirica BM, Bhatt DL, Braunwald E, et al. The design and rationale of the saxagliptin assessment of vascular outcomes recorded in patients with diabetes mellitus-thrombolysis in myocardial infarction (SAVOR-TIMI) 53 study. Am Heart J. 2011;162(5):818–825. | |
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Diabetes Mellitus – Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes. Silver Spring: US Food and Drug Administration; 2008. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.