")
Back to Journals » International Medical Case Reports Journal » Volume 9
Two cases of Vogt–Koyanagi–Harada’s disease in sub-Saharan Africa
Authors Oluleye TS, Rotimi-Samuel AO, Adenekan A, Ilo OT , Akinsola FB, Onakoya AO, Aribaba OT, Adefule-Ositelu A, Musa KO, Oyefeso Y
Received 11 February 2016
Accepted for publication 9 April 2016
Published 28 November 2016 Volume 2016:9 Pages 373—376
DOI https://doi.org/10.2147/IMCRJ.S106248
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Tunji S Oluleye,1 Adekunle O Rotimi-Samuel,2 Adetunji Adenekan,2 Olubanke T Ilo,2 Folashade B Akinsola,2 Adeola O Onakoya,2 Olufisayo T Aribaba,2 Adebukunola Adefule-Ositelu,2 Kareem O Musa,2 Yele Oyefeso2
1Retina and Vitreous Unit, Department of Ophthalmology, University College Hospital, Ibadan, 2Department of Ophthalmology, Guinness Eye Center, Lagos University Teaching Hospital, Lagos, Nigeria
Abstract: Vogt–Koyanagi–Harada’s (VKH) disease has been reported to be rare in sub-Saharan Africa. Two Nigerians with the disease are presented in this report. The first patient, a 32-year-old pregnant Nigerian woman presented with a 1-month history of bilateral blurring of vision, persistent headache, and alopecia. Presenting visual acuity was 1 m counting fingers in both eyes. Examination revealed vitiligo and poliosis with bilateral panuveitis as well as bilateral exudative retinal detachment. A clinical assessment of complete VKH disease was made. The patient commenced systemic and topical steroids that resulted in remarkable recovery of vision and control of inflammation. The second patient, a 56-year-old Nigerian woman presented with severe headache, tinnitus, and visual loss in both eyes of 2 weeks duration. There was associated redness of both eyes and photophobia. Examination showed visual acuity of Hand motion (HM) and counting fingers at 1 meter (CF). in the right and left eye, respectively, with bilateral panuveitis and bilateral exudative retinal detachment. Subsequent follow-up showed poliosis, vitiligo, and sunsetting fundus appearance. The patient improved with systemic and topical corticosteroids. Developing a high index of suspicion is necessary in diagnosing VKH disease, even in sub-Saharan Africa. Prompt institution of appropriate treatment prevents blindness.
Keywords: Vogt–Koyanagi–Harada’s disease, exudative retinal detachment, vitiligo, sub-Saharan Africa
Introduction
Vogt–Koyanagi–Harada’s (VKH) disease is a multisystemic disease reported to be rare in sub-Saharan Africa. Vogt, Koyanagi, and Harada first described the initial set of patients at different stages of the disease.1–3 The disease was said to be common in Asian patients.4 Here we describe two Nigerian patients with the disease.
Case presentation
Patient 1
Patient 1 is a 32-year-old Nigerian woman who presented to us with a 1-month history of bilateral blurring of vision, red eyes, persistent headache, and tinnitus. There was no associated meningism or cranial nerve palsies. Also, there was no significant systemic disease. The patient was 16 weeks pregnant at presentation. General examination showed significant alopecia, and skin depigmentation on the forehead (Figure 1A and B). Her blood pressure was 130/80 mmHg. Ophthalmic examination showed visual acuity of counting fingers at 1 m in both eyes. The right eye showed poliosis. Conjunctival congestion was seen in both eyes. Slit-lamp examination showed multiple medium-sized keratic precipitates on the corneal endothelium, with significant cells (++) and flare (++) in the anterior chamber. Both pupils showed posterior synechiae. Examination of the posterior segment showed vitreous cells with bilateral exudative retinal detachment with disc edema (Figure 1C and D). Intraocular pressure was 16 mmHg in both eyes. An assessment of complete VKH disease was made to rule out other causes of uveitis. Full blood count and differential count were normal; erythrocyte sedimentation rate was 25 mm; retroviral (human immunodeficiency virus 1 and 2) and venereal disease research laboratory screening were negative; and Mantoux test was not significantly reactive (2 mm). Chest radiograph was normal. Consultation with the obstetricians was sought before commencing systemic steroids. Periocular triamcinolone 20 mg and topical steroids (dexamethasone four hourly) were also commenced. Cycloplegic agent (0.5% tropicamide three times daily [tid]) was added. At 2 weeks follow-up, the patient showed remarkable improvement. Vision improved to 6/24 in both eyes with reduction of the exudative detachment; however, disc edema persisted. At 1 month, her vision improved to 6/18 and 6/12 in the right and left eye, respectively. The exudative detachment almost completely resolved, and disc edema remarkably reduced (Figure 1E and F). Intraocular pressure was 12 and 14 mmHg in the right and left eye, respectively. However, the cutaneous manifestation of poliosis and vitiligo increased. The neurological complaints had resolved.
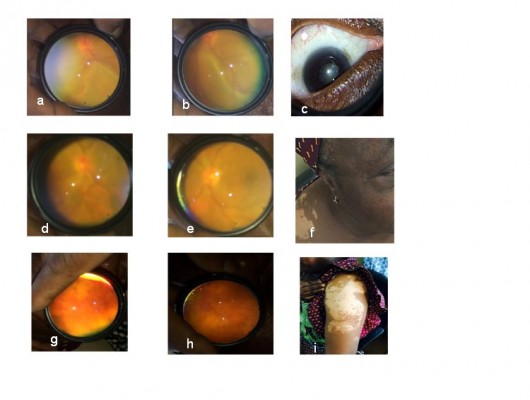
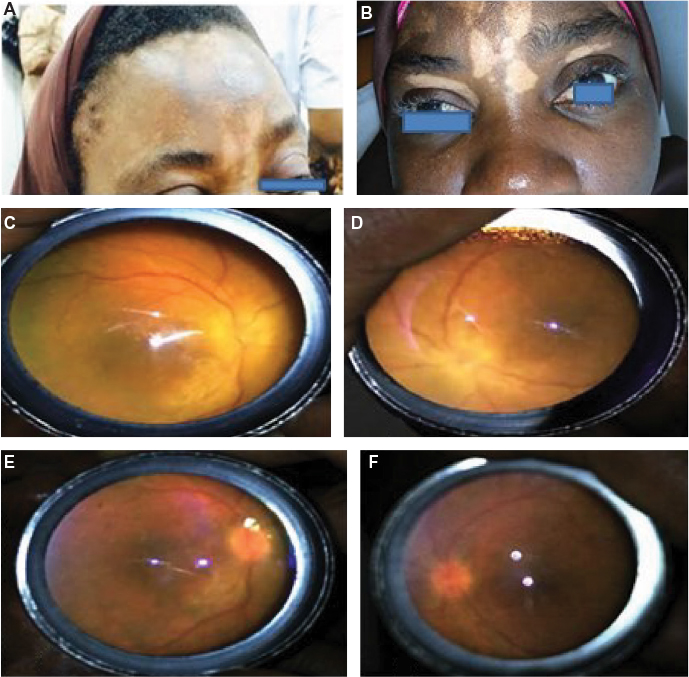 | Figure 1 Patient 1. Notes: Alopecia and vitiligo at presentation (A and B); disc edema and exudative detachment before (C and D) and after (E and F) treatment. |
Patient 2
Patient 2 is a 56-year-old Nigerian woman who presented with severe headache, tinnitus, and visual loss in both eyes of 1 week duration. There was associated redness of both eyes and photophobia, and no associated neck stiffness or previous systemic disease.
General examination showed an apprehensive woman with blood pressure of 110/70 mmHg. Visual acuity was Hand motion (HM) and counting fingers at 1 meter (CF). close to face in the right and left eye, respectively. Both conjunctivas were hyperemic with no discharge. Slit-lamp examination showed medium-sized keratic precipitates in both eyes. Both anterior chambers had moderate cells (++) and flare (++). Pupils reacted slowly with posterior synechiae. Dilated fundoscopy showed bilateral exudative retinal detachment (Figure 2A–C). Intraocular pressure was 11 and 13 mmHg in the right and left eye, respectively.
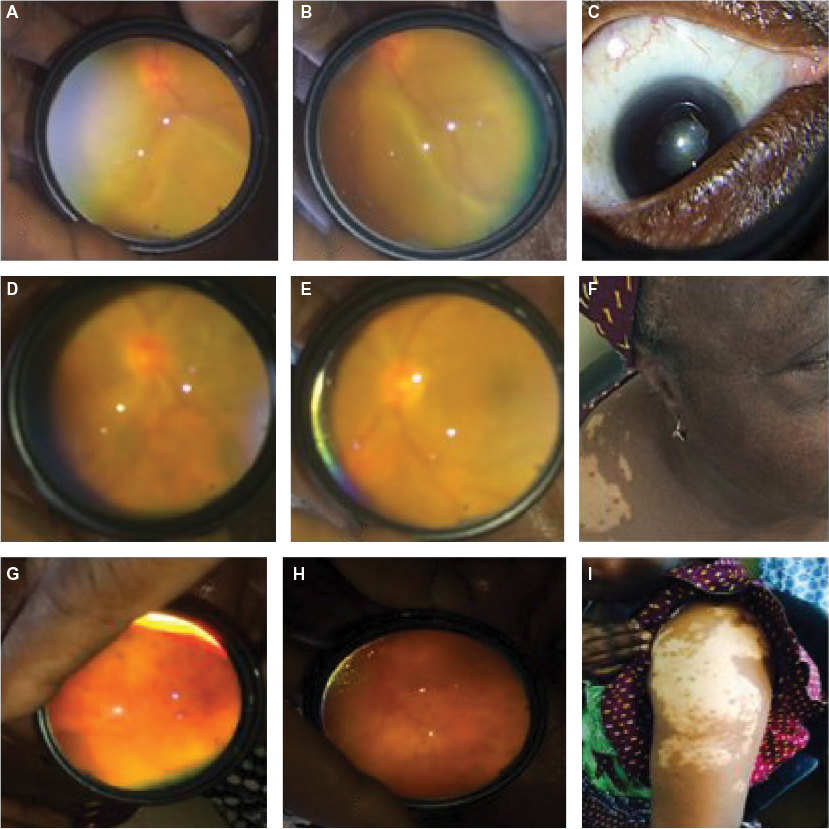 | Figure 2 Patient 2. Notes: Bilateral exudative retinal detachment (A and B) and posterior synechiae (C) at presentation. Two weeks after treatment with steroids, exudative retinal detachment resolved (D and E). Subsequent follow-up visits revealed vitiligo (F), sun setting appearance of fundi (G and H), and increasing vitiligo (I). A and B= right and left eyes; D and E= right and left eyes; G and H= right and left eyes. |
An assessment of VKH was considered, as possibly an incomplete/probable disease. Full blood count, erythrocyte sedimentation rate, and chest radiograph were normal. Mantoux test and venereal disease research laboratory test were negative.
The patient was commenced on systemic intravenous methylprednisolone 1 g daily for 3 days followed by oral prednisolone 1 mg/kg/day, and four hourly topical dexamethasone and tropicamide 0.5% tid as cycloplegic. Two weeks later, the inflammation and exudative retinal detachment reduced significantly (Figure 2D and E). Visual acuity improved to 6/36 and 6/24 in the right and left eye, respectively.
Subsequent follow-up over 3-month visits revealed development of increasing poliosis, vitiligo, and sunset fundus (Figure 2F–I), thereby revealing complete VKH syndrome. Final visual acuity at last follow-up was 6/24 and 6/18 in the right and left eye, respectively. Intraocular pressure was 15 and 12 mmHg in the right and left eye, respectively.
Discussion
To the best of the authors’ knowledge, the above two cases may be the first set of cases of VKH disease to be reported from Nigeria. The disease is said to be uncommon in Africans. Females are more affected. We have presented two Nigerian women with the disease in this report. Informed consent was sought and obtained from both patients who consented to being included in this report and have their images included.
The International Committee on Nomenclature Classification and the American Uveitis Association laid down the criteria for the diagnosis of VKH syndrome, and Ethical Approval was obtained from the Lagos University Teaching Hospital Review Board.5 Patients present initially with neurological complaints before the ocular and integument manifestations. The neurological manifestations include meningismus – malaise, fever, headaches, and neck stiffness. Tinnitus is an auditory sign. The integument signs are poliosis, vitiligo, and alopecia.
To diagnose VKH, the condition must be bilateral with no evidence of other ocular disease, trauma, or surgery. Three categories have been identified: complete, incomplete, and probable VKH disease. The two patients presented with complete VKH disease with full manifestations. The etiology of the disease is linked to cell-mediated autoimmunity. Antibodies are directed at melanocytes on the skin, uvea, inner ear, and meninges, hence the multisystemic manifestation.6
The first patient was in her second trimester of pregnancy at presentation. VKH in pregnancy has been reported. In a study, the use of systemic steroids in midtrimester did not cause any abnormalities in the baby.7 In another study, high-dose systemic steroids produced low birth weight in the baby.8 The other systemic effects noted in the baby were reported to be unconnected to the steroids. Consultation was sought from the obstetrician in our case, who gave the go ahead for systemic steroids. The patient is on follow-up with the ophthalmologist and obstetrician.
The second patient presented with the ophthalmic manifestations before the integument signs. Sunsetting appearance of the fundus represents retinal pigment epithelial depigmentation. In both patients, vitiligo and poliosis increased with improvement in vision while on treatment. The neurological symptoms mostly resolve earlier than the ophthalmic manifestations.
Differential diagnosis of bilateral disc edema, exudative retinal detachments, and panuveitis should be considered. Posterior scleritis, uveal effusion syndrome, and toxemia of pregnancy can mimic VKH disease. Ocular tuberculosis with granulomatous uveitis is a close differential in Africa.
Conclusion
VKH disease is not common in Nigeria and sub-Saharan Africa. High index of suspicion is recommended for early detection and prompt treatment to avoid irreversible visual loss.
Disclosure
The authors report no conflicts of interest in this work.
References
Vogt A. Early graying of cilia and remarks about the so-called sudden entry of this change. Klin Monatsbl Augenh. 1906;44:228–242. | ||
Harada E. On the acute diffuse choroiditis. Acta Soc Ophthalmol Jpn. 1926;30:356–378. | ||
Koyanagi Y. Dysakusis, Alopecia und Poliosis bei schwerer Uveitis nicht traumatischen Ursprungs. Klin Monatsbl Augenh. 1929;82:194–211. | ||
Yang P, Ren Y, Li B, Fang W, Meng Q, Kijlstra A. Clinical characteristics of Vogt-Koyanagi-Harada syndrome in Chinese patients. Ophthalmology. 2007;114(3):606–614. | ||
Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131(5):647–652. | ||
Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase family proteins are antigens specific to Vogt-Koyanagi-Harada disease. J Immunol. 2000;165(12):7323–7329. | ||
Miyata N, Sugita M, Nakamura S, et al. Treatment of Vogt-Koyanagi- Harada’s disease during pregnancy. Jpn J Ophthalmol. 2001;45(2):177–180. | ||
Doi M, Matsubara H, Uji Y. Vogt-Koyanagi-Harada syndrome in a pregnant patient treated with high-dose systemic corticosteroids. Acta Ophthalmol Scand. 2000;78(1):93–96. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.