")
Back to Journals » International Journal of Nanomedicine » Volume 11
Tumor vascular-targeted co-delivery of anti-angiogenesis and chemotherapeutic agents by mesoporous silica nanoparticle-based drug delivery system for synergetic therapy of tumor
Authors Li X, Wu M, Pan L, Shi J
Received 18 January 2015
Accepted for publication 27 March 2015
Published 29 December 2015 Volume 2016:11 Pages 93—105
DOI https://doi.org/10.2147/IJN.S81156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Xiaoyu Li, Meiying Wu, Limin Pan, Jianlin Shi
State Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai, People’s Republic of China
Abstract: To overcome the drawback of drug non-selectivity in traditional chemotherapy, the construction of multifunctional targeting drug delivery systems is one of the most effective and prevailing approaches. The intratumoral anti-angiogenesis and the tumor cell-killing are two basic approaches in fighting tumors. Herein we report a novel tumor vascular-targeting multidrug delivery system using mesoporous silica nanoparticles as carrier to co-load an antiangiogenic agent (combretastatin A4) and a chemotherapeutic drug (doxorubicin) and conjugate with targeting molecules (iRGD peptide) for combined anti-angiogenesis and chemotherapy. Such a dual-loaded drug delivery system is capable of delivering the two agents at tumor vasculature and then within tumors through a differentiated drug release strategy, which consequently results in greatly improved antitumor efficacy at a very low doxorubicin dose of 1.5 mg/kg. The fast release of the antiangiogenic agent at tumor vasculatures led to the disruption of vascular structure and had a synergetic effect with the chemotherapeutic drug slowly released in the following delivery of chemotherapeutic drug into tumors.
Keywords: mesoporous silica nanoparticles, drug delivery, tumor vasculatures targeting, antiangiogenic agent
Introduction
At present, the effective therapy of cancer is still an unsolved challenge faced by human beings. There are a number of critical drawbacks in traditional chemotherapeutics to overcome, such as low treatment efficiency, harmful side effects, poor pertinence, and so on. So how to substantially enhance the efficacy while diminish the side effects of chemotherapy has been the focus in cancer therapy.1–3
Tumor angiogenesis plays an important role in tumor development, and therefore is also an important sector considered in the treatment of tumors.4–7 Different from normal tissues, the vasculatures of tumors show specific overexpressions of cytokines, and therefore can be used as targets for tumor treatment,8,9 and tumor vasculatures, which are the transport channel of nutrition for the fast proliferating tumor cells, are appropriate therapeutic targets.4 The disruption of tumor vasculatures can lead to the necrosis of tumors. According to the literature,10 drug resistance and other side effects that occur in traditional chemotherapy can be circumvented effectively by focusing on tumor vasculatures in tumor treatments; however, the antitumor efficacy is not satisfactory if only adopting anti-angiogenesis therapy.11 So, the combination of anti-angiogenesis with conventional chemotherapeutics is expected to give substantially improved results in tumor treatments, which can be achieved by delivering both antiangiogenic agent and chemotherapeutic drug under optimal dosages into tumors. Doxorubicin (DOX), as a traditional chemotherapeutic agent, has been proved to be a wide-spectrum anticancer drug and effective for various cancers, such as breast cancer, liver cancer, lung cancer, etc. In addition, combretastatin A4 (CA4) as an effective anti-angiogenesis agent has been proved to be capable of disrupting tumor vasculatures which support tumor growth and spreading.12,13 So some literature points out that coadministration of CA4 and DOX seems to be a good choice.14–16 Unfortunately, the conventional simple coadministration of two different drugs usually results in unsatisfactory therapeutic efficacy, mainly due to the lack of targeting function of the naked drugs and onsite synergetic therapeutic effects.
To effectively integrate anti-angiogenesis and conventional chemical therapy by direct and simultaneous transport of multidrugs, drug carriers become crucial. Compared with organic nanoparticles, inorganic nanocarriers, such as mesoporous silica nanoparticles (MSNs), carbon nanotubes, and gold nanoparticles, are highly stable both chemically and biologically.17–21 Especially, MSNs have a number of unique features, such as uniform and tunable particle size, pore size, and morphology; high surface area and pore volume; facile surface functionalization; etc,1,22 and they have been demonstrated to be one of the most valuable drug carriers.23,24
Herein, we used MSNs as a carrier to construct a tumor vasculature-targeting drug delivery system (DDS) by co-loading DOX and CA4 as the chemotherapeutic drug and anti-angiogenesis agent, respectively, and MSNs were grafted with iRGD sequence (CRGDKGPDC), which was reported to be a ligand showing effective affinity to α2β3 receptor overexpressed on a number of cancer cells as well as endothelial cells in tumor tissues.25 Especially, it has been reported that iRGD-coupled composites are capable of binding to tumor vasculatures and spreading into the extravascular tumor parenchyma; however, conventional RGD peptides can only deliver cargos to the blood vessels.25
Most encouragingly, when the DDS arrives at tumor vasculatures during its bloodstream circulation under guidance by iRGD-containing peptides in a short moment of time, the angiogenesis inhibitor CA4 is able to release rapidly from the constructed DDS at tumor vasculatures under the effective tumor vascular targeting, and during the later, much longer process of DDS uptake by tumor cells, the anticancer drug DOX will be released mostly within the cells of significantly lower pH values, thanks to the special feature of pH-responsive release behavior of DOX from MSNs,23,26 as shown in Figure 1, which is of great benefit in realizing differentiated release rates of anti-angiogenesis and anticancer effects in the pathway of drug delivery from the vascular system to the tumor cells, as recently proposed conceptually by us.1,23
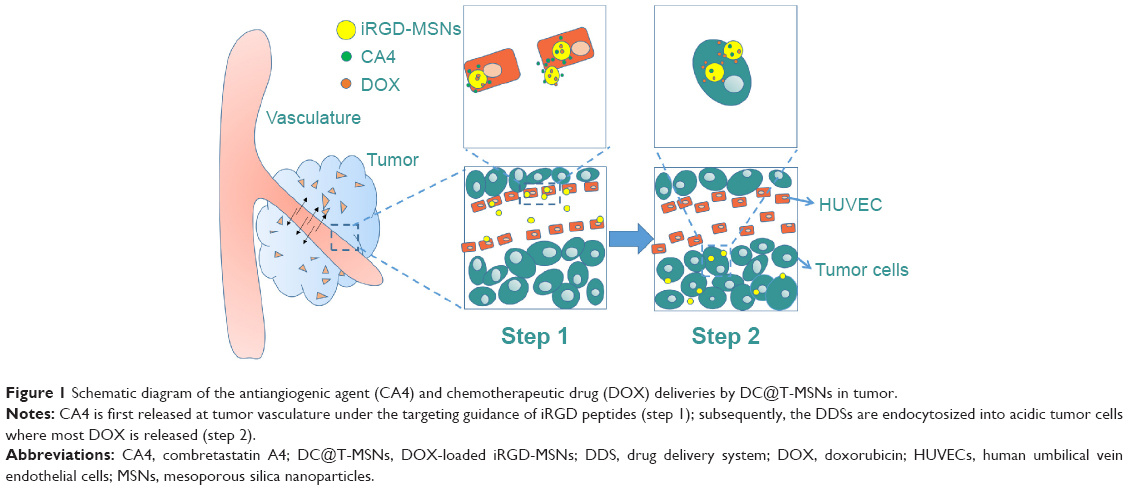 | Figure 1 Schematic diagram of the antiangiogenic agent (CA4) and chemotherapeutic drug (DOX) deliveries by DC@T-MSNs in tumor. |
Experimental methods
Materials
Tetraethyl orthosilicate, triethanolamine, ethanol, methanol, hydrochloric acid ([HCl] 37%), and sodium chloride (NaCl) were purchased from Sinopharm Chemical Reagent Co. Hexadecyltrimethylammonium chloride ([CTAC] 25 wt%), 3-aminopropyltriethoxysilane (APTES), and rhodamine isothiocyanate were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Phosphate-buffered saline (PBS) solution (pH 7.4) was procured from Shanghai Runcheng Biomedical Co., Ltd. The anticancer drug doxorubicin (DOX) was procured from Beijing HuaFeng United Technology Co., Ltd. CA4 was procured by Hangzhou Ruishu Biochemical Co., Ltd. iRGD was synthesized by Shanghai Sangon Biotech Co., Ltd. No further purification was conducted before chemicals were used.
Preparation of MSNs, targeted-MSNs (T-MSNs), and drug-loaded MSNs
Preparation of MSNs
Briefly, 2 g of CTAC and 0.02 g of triethanolamine were dissolved into 20 g of water with magnetic stirring, respectively. The solution was heated to 80°C for 1 hour, followed by the addition of 1.5 mL tetraethyl orthosilicate dropwise; 20 μL of rhodamine isothiocyanate–APTES was also added dropwise in order to ensure enough –NH2 groups grafted on MSNs for subsequent surface iRGD modification. The magnetic stirring was continued for another 1 hour to achieve a uniform mixture. In order to remove the residual reactants, the nanoparticles collected by centrifugation were washed three times with ethanol and deionized water. The centrifugation was continued for 20 minutes at 18,000 rpm and the particles were redispersed under sonication. Then, the as-received nanoparticles were repeatedly extracted three times with a 1 wt% solution of NaCl in methanol at room temperature for 3 hours to remove the CTAC.
Conjugation of iRGD (T-MSNs)
iRGD was covalently conjugated onto amine-functionalized MSNs through -COOH group by using crosslinking reagents EDC and N-hydroxysuccinimide (NHS). To modify the MSN surface with iRGD peptide, 0.01 mmol iRGD was dissolved in PBS solution (pH 7.4) and 0.2 mmol EDC and 0.5 mmol NHS were added into the solution. The mixture was then stirred at room temperature for 15 minutes. Subsequently, 20 mL PBS solution of MSNs (1 mg/mL) was added to the above solution, and the mixture was stirred for 12 hours at room temperature. Excess EDC, NHS, and iRGD were removed by repeatedly washing the nanoparticles with distilled water several times.
Drug loading in MSNs and T-MSNs
Five milligrams of MSNs or T-MSNs were respectively mixed with 5 mL of DOX solution of water (0.5 mg/mL), CA4 solution of water (2 mg/mL), and DOX plus CA4 mixed solution of water with stirring in darkness for 24 hours. The drug-loaded nanoparticles were collected by centrifugation. The supernatant solution was collected and the residual drug contents were measured by ultraviolet–visible (UV-vis) measurements to evaluate the drug-loading capacity. The residual DOX content was measured by UV-vis spectroscopy at the wavelength of 480 nm, and residual DOX content was measured at a wavelength of 200 nm. The drug-loading capacity is represented as mean ± standard deviation of three independent experiments.
Nanoparticle characterization
The morphology and mesostructure of nanoparticles were observed via a JEM-2010 electron microscope. The accelerating voltage was 200 kV. Dynamic light scattering measurements were conducted on Zetasizer Nanoseries (Nano ZS90). UV-vis spectra were recorded on a Shimadzu UV-3101PC spectroscope.
In vitro drug release
Twenty milligrams of the above-prepared drug-loaded MSNs (D@MSNs, C@MSNs, DC@MSNs) in a 3,500 Ka dialysis bag were immersed in 20 mL pH 7.4 PBS at 37°C and shaken at a speed of 100 rpm. At certain time intervals, 3 mL of the supernatant PBS was taken out to test the drug-released concentration, by virtue of UV-vis absorption technique at different wavelengths, and then was returned to the original PBS. The standard curve was used to calculate the drug release amount. The absorbances of the supernatant PBS were recorded on a Shimadzu UV-3101PC UV-vis absorption spectrophotometer.
Cell culture
HeLa cells and human umbilical vein endothelial cells (HUVECs) were seeded at a concentration of 5,000 cells/cm2 in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (Sijiqing Biological Engineering Materials Co., Ltd., Hangzhou, People’s Republic of China) or HUVEC special medium. All the cells were cultured at 37°C in a 5% CO2 humidified environment until the cell concentration reached 80%. Then, the medium containing MSNs, drug-loaded MSNs, and free drugs in different concentrations were added for various cell experiments. Ninety-six-well culture plates were used for the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and 35 mm glass-bottom dishes were used for confocal laser scanning microscopy imaging.
MTT assay
All cell viability tests were conducted by employing MTT assay. Specifically, HeLa cells were first seeded at a concentration of 5,000 cells/cm2 and then cultured for 48 hours. Then, new culture media containing MSNs and T-MSNs at concentrations of 25, 50, 100, and 200 μg SiO2/mL were added for cell culture, while the culture media of DDSs of different drug-loaded MSNs were introduced at a fixed concentration of DOX. Then, the cells were cultured for an additional 24 and 48 hours. Then, 100 μL of MTT solution with a concentration of 0.5 mg/mL was added and incubated for another 4 hours’ incubation. Then, 100 μL of dimethyl sulfoxide was added and formazan crystals were solubilized. The absorbance was read with a microplate reader (Bio-Tek ELx800) at the wavelength of 490 nm.
Immunohistochemical staining
Twenty-four hours after injection with MSNs and T-MSNs and other materials, the mice were sacrificed. The tumors were fixed in 4% paraformaldehyde. Tumors were stained with CD31 antibodies for microvessel and Ki-67 antibodies for cell proliferation analysis. The nuclei were counterstained with hematoxylin. TUNEL assay was carried out for cellular apoptosis.
In vivo antitumor studies
The healthy adult male Kunming (KM) mice and KM mice bearing HeLa tumors were obtained from Shanghai Laboratory Animal Center, Chinese Academy of Science (Shanghai, People’s Republic of China) (warrant number SCXK [Shanghai] 2007-0005). All animal operations were conducted in accordance with the People’s Republic of China national standards for laboratory animal quality and the Chinese guidelines for the care and use of laboratory animals. Six- to eight-week-old female KM mice were used. All mouse experimental procedures were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People’s Republic of China.
The tumor-bearing mice were allowed to grow until the tumor volume reached 100 mm3 before all the in vivo experiments. Mice were divided into seven groups and every group had five mice, and the groups were injected with PBS solution, DC@MSNs, DC@T-MSNs, C@MSNs, D@MSNs, free DOX plus CA4, and free DOX, respectively. All materials were diluted with saline. The DOX doses were kept identical at 1.5 mg/kg by injection through the tail vein. The injection interval was every 6 days and, in total, four injections were conducted (24 days in all). Tumor sizes were recorded every 2 days with a digital caliper.
Biodistribution study
Mice that received intravenous injections were euthanized 24 hours post-injection, and blood, tumor, and major organs/tissues were collected and wet-weighed. The organs were cut into 1–2 mm2 pieces and incubated in digestion solution containing mixed acids (nitric acid:perchloric acid, V:V=4:1) for 12 hours. Silicon content was determined by inductively coupled plasma optical emission spectrometer (ICP-OES).
Results
Characterization of MSNs
T-MSNs were synthesized by covalently conjugating iRGD onto amine-functionalized MSNs through the -COOH group, as schematically shown in Figure 2A. As can be seen from the transmission electron microscopy images (Figure 2B), we successfully synthesized monodispersed MSNs of about 40–50 nm in diameter without visible aggregation among the particles. The MSNs had a regular spherical morphology, a mesoporous structure, and perfect monodispersivity. Dynamic light scattering measurement results indicated that the MSNs had a well-defined particle size distribution centered at 63 nm in accordance with the above transmission electron microscopy results. DOX and CA4 were successfully loaded into MSNs with different drug-loading capacities, which were confirmed by UV-vis measurements. No visible effects of the grafting of iRGD and drug loading on the morphology, size, and dispersivity of the MSNs could be observed. The Fourier transform infrared spectra of MSNs and T-MSNs are shown in Figure 3A. Compared with the spectrum of MSNs, that of the T-MSNs shows a series of new bands at 1,680 cm–1 (stretching vibration bands of N-H), 1,590 cm–1 (stretching vibration bands of C=O), and 1,400 cm–1 (bending vibration bands of C-N), which should be assigned to the vibration in –NHCO–, and, more specifically, 1,250 cm–1 stretching vibration bands of C-O in COOH. So the Fourier transform infrared spectra provide evidence that iRGD was successfully conjugated on MSNs.
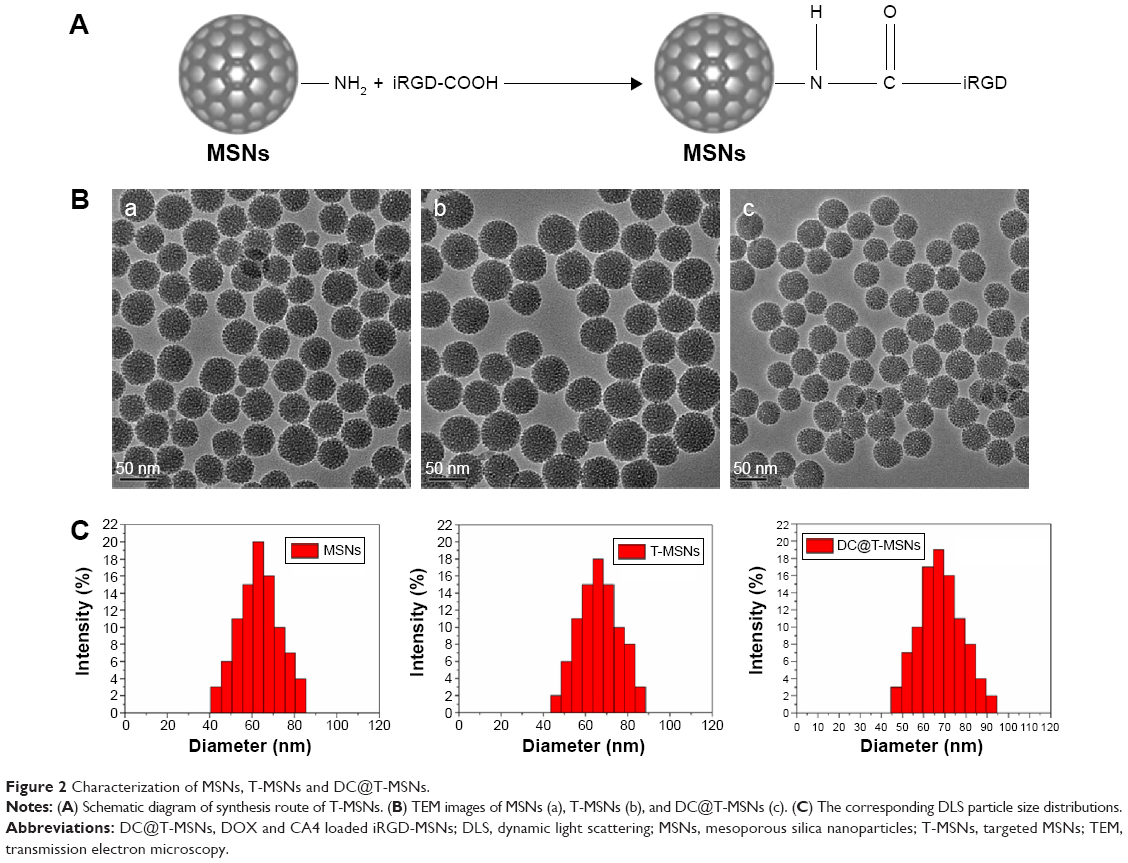 | Figure 2 Characterization of MSNs, T-MSNs and DC@T-MSNs. |
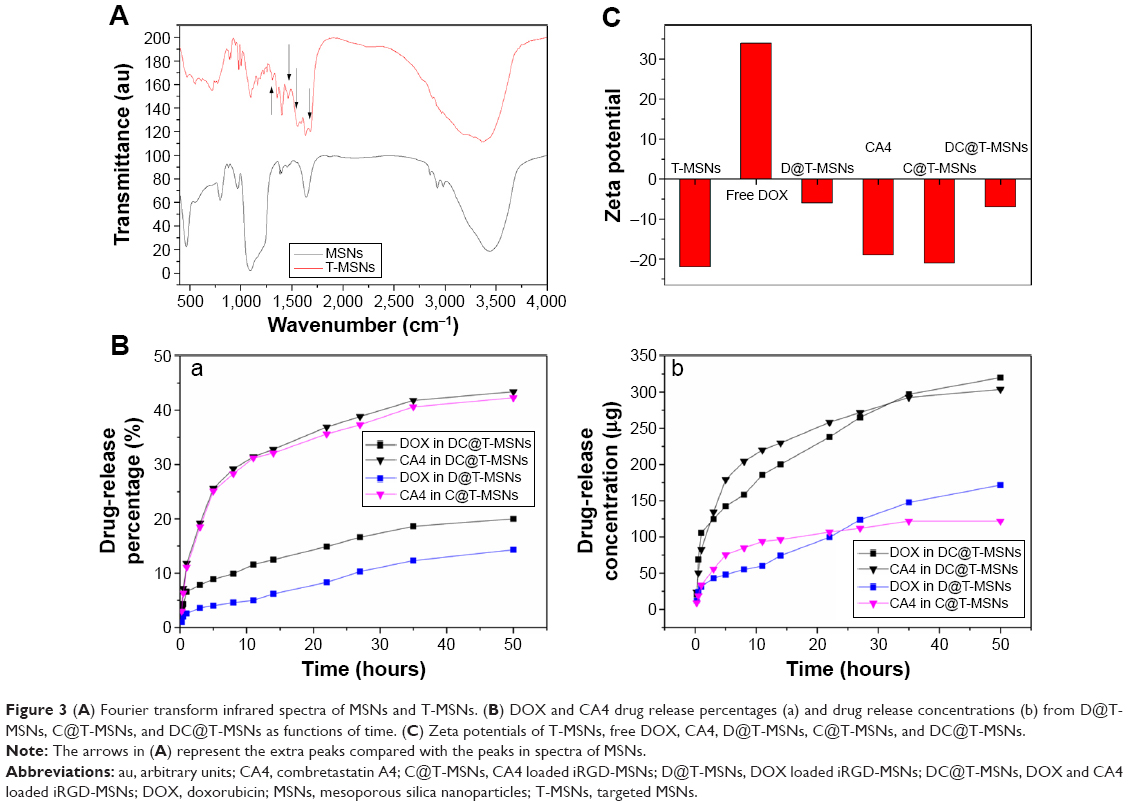 | Figure 3 (A) Fourier transform infrared spectra of MSNs and T-MSNs. (B) DOX and CA4 drug release percentages (a) and drug release concentrations (b) from D@T-MSNs, C@T-MSNs, and DC@T-MSNs as functions of time. (C) Zeta potentials of T-MSNs, free DOX, CA4, D@T-MSNs, C@T-MSNs, and DC@T-MSNs. |
The amounts of DOX in single DOX-loaded MSNs (D@T-MSNs, with iRGD grafting, the same hereinafter) and double DOX plus CA4-loaded MSNs (DC@T-MSNs) were determined to be 12%±0.9% and 16%±0.7%, while those of CA4 in single CA4-loaded C@T-MSNs) and double DC@T-MSNs were 3%±0.2% and 7%±0.5%, respectively. The promoted loading amounts of both DOX and CA4 in the double-loaded DDS can be attributed to the electrostatic interaction and hydrogen bonding among positively charged DOX molecules, negatively charged CA4 molecules, and inner-mesopore surfaces. Positively charged DOX can be easily adsorbed onto the negatively charged surface, mainly the inner pore surface of MSNs, and the hydrogen bonding between the OH groups on the MSN surface and the -OH groups in DOX molecules will further promote the DOX loading.27 So MSNs can load more DOX than CA4, while more CA4 can be loaded in DC@MSNs than in C@MSNs due to CA4 electrostatic interaction with the positively charged DOX.
Differentiated releases of DOX and CA4
Figure 3B shows the drug release profiles (release amount and concentration) in a constant release medium within an interval of 50 hours. The sustained release behaviors of the two different drugs can be well observed. The release percentages of CA4 from C@T-MSNs and DC@T-MSNs were both around 30% in 10 hours, while those of DOX from D@T-MSNs and DC@T-MSNs were as low as 10% and 5% in the same period (Figure 3A), respectively. Such a difference in release rate between CA4 and DOX remained up to the end of experiments of 50 hours. The much slower release of DOX than that of CA4 can also be attributed to the electrostatic interaction and hydrogen bonding between positively charged DOX molecules and mesopore surface of MSNs. To verify this, zeta potentials were measured and the data are shown in Figure 3C. It can be seen that pure MSN and CA4 were negatively charged and DOX was positively charged. Because of the electrostatic interaction between the oppositely charged MSN and drugs, the CA4 loading capacity of DC@T-MSN was higher than that in C@T-MSN, and the different drug release rates were also related to the different zeta potentials. Besides, in respect of the accumulative release amount shown in Figure 3C, both agents released much higher amounts from double-loaded DC@T-MSNs than single-loaded D@T-MSNs or C@T-MSNs due to the much higher drug-loading amounts in the double-loaded DDS than those in the single-loaded DDSs. The above results demonstrate that, considering the drug releases from double-loaded DC@T-MSNs, a large part of CA4 will be released first and most DOX is still within MSNs in 2 or 3 days of incubation, which means most DOX is retained within MSNs while most CA4 is released from DC@T-MSNs during the early period of drug delivery. Nevertheless, DOX will be released much faster once the DDS has been taken up in a low pH environment such as tumor cells owing to the pH-responsive release behavior of DOX from MSNs.28,29
It should be noted that the fast and prior release of CA4 to DOX from DC@T-MSNs is fortunately in favorable coincidence with the expectation of the sequential drug delivery in vivo: the first fast-released CA4 from DC@T-MSNs at the vascular wall will play a first role in anti-angiogenesis once the DDS reaches the endothelial system under the guidance of tumor vascular targeting, then the later released DOX within tumor cells of lower pH values will be responsible for killing cancer cells in the later stage when the DDS has been taken up by the tumor cells.
Cytotoxicity
Next, the cytotoxicities of carrier MSNs and T-MSNs at different concentrations and for different incubation time durations against HeLa cells and HUVECs were evaluated by MTT assay. As can be seen from Figure 4, MSNs and T-MSNs exhibited no significant cytotoxic effects up to 48 hours. The cytotoxicities of MSNs loaded with different drugs were also evaluated by the same method. All of the three drug-loaded MSNs showed significant cytotoxicities, which became more significant at increased drug concentrations and incubation durations. Though only a partial amount of drug was released, the cytotoxicity of DC@MSNs remained higher than that of D@MSNs and C@MSNs under the same conditions. By comparison, C@MSNs showed the lowest cytotoxicity. It can also be seen that the cytotoxicity difference between DOX@T-MSNs and DOX@MSNs was larger at low concentrations than that at elevated concentrations, which can be attributed to the targeted drug delivery by DOX@T-MSNs.
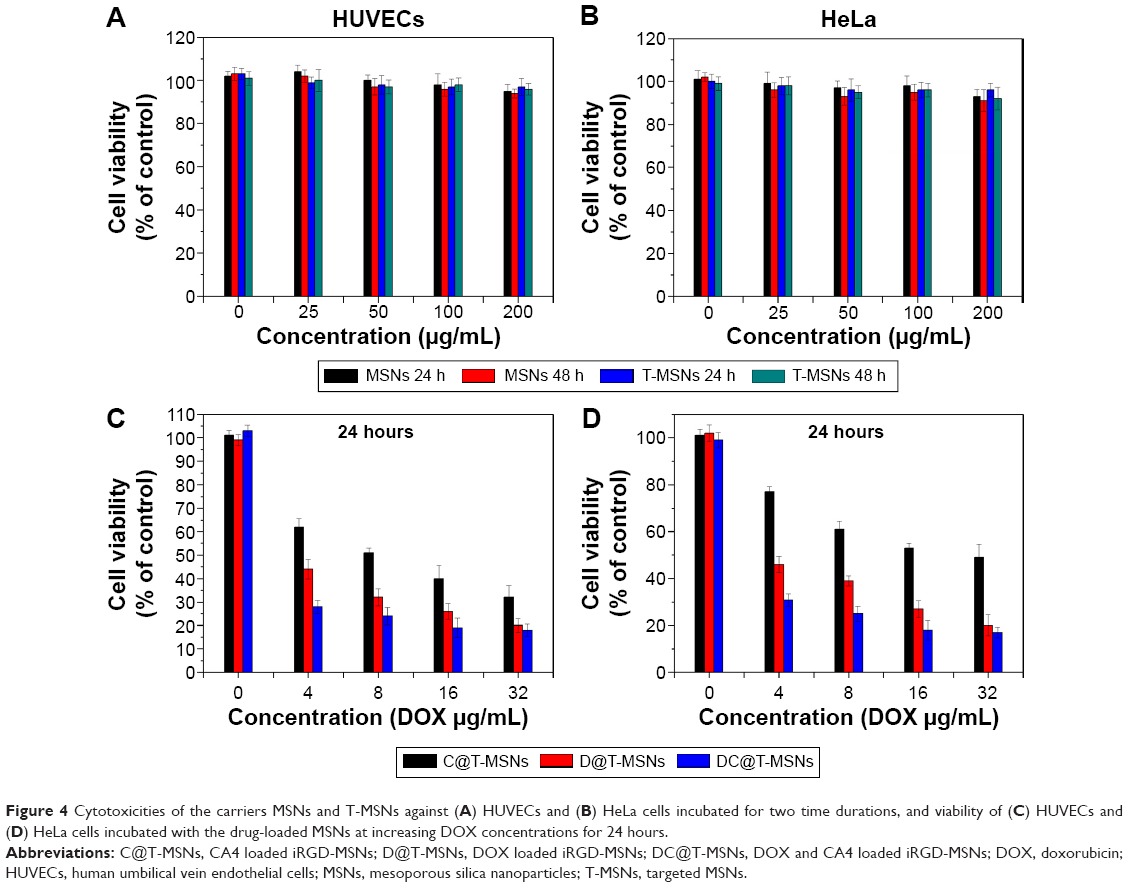 | Figure 4 Cytotoxicities of the carriers MSNs and T-MSNs against (A) HUVECs and (B) HeLa cells incubated for two time durations, and viability of (C) HUVECs and (D) HeLa cells incubated with the drug-loaded MSNs at increasing DOX concentrations for 24 hours. |
Cellular uptake of DDSs
The cellular uptake behavior of MSNs and T-MSNs was observed by using HUVECs and HeLa cells. The time-dependent uptakes of these two types of MSNs at 100 μg/mL were monitored by confocal laser scanning microscopic imaging (Figure 5). In HeLa cells, we can see that longer incubation favors the particles’ uptake, no matter whether MSNs or T-MSNs. Thanks to the overexpression of α2β3, HeLa cells can uptake more T-MSNs than MSNs. When free iRGD peptides (50 μM) were added into the medium, the amount of T-MSNs taken up by HeLa cells decreased significantly, confirming the targeting effect of the iRGD.
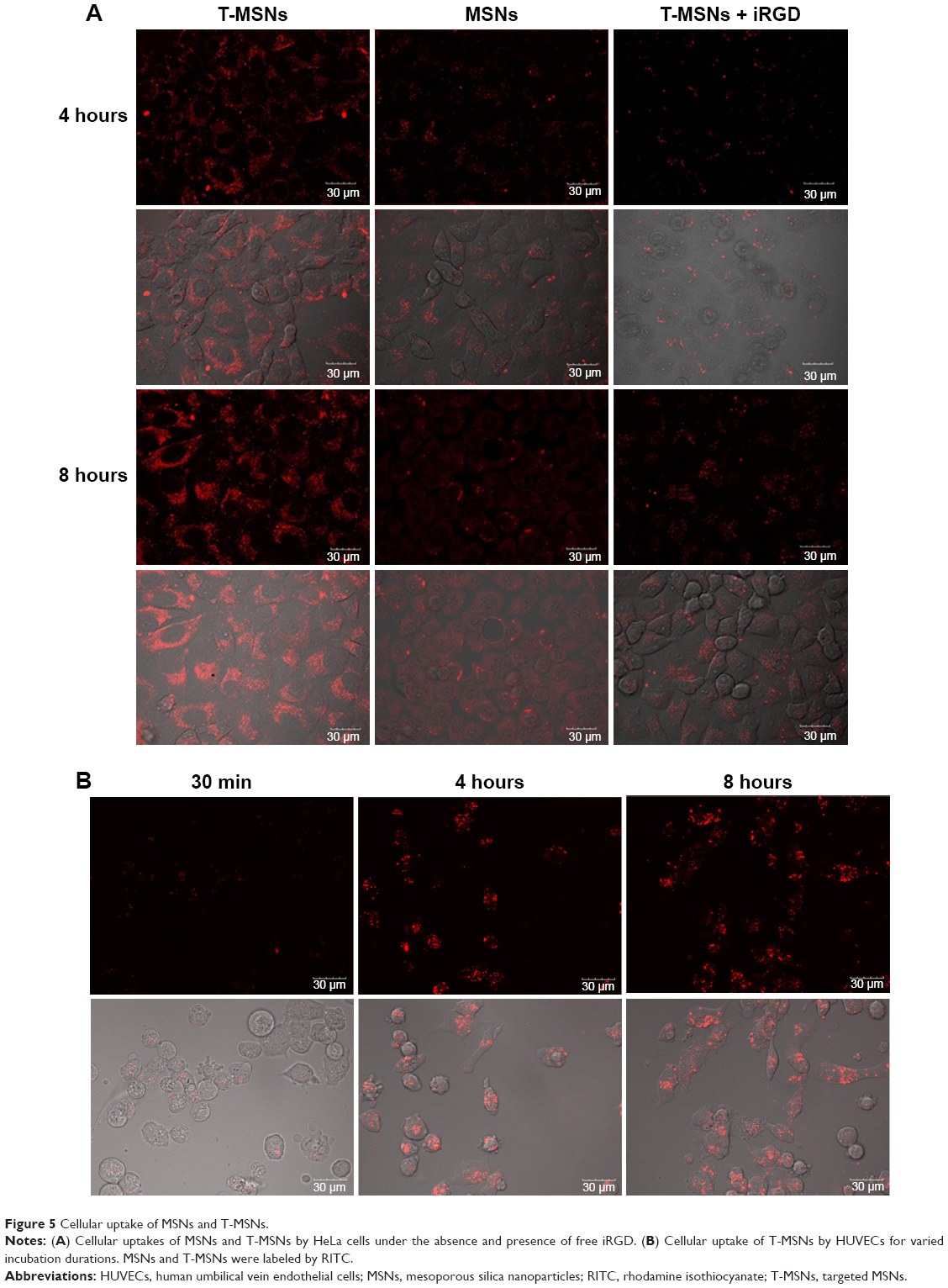 | Figure 5 Cellular uptake of MSNs and T-MSNs. |
In 30 minutes, HUVECs had uptaken a considerable amount of MSNs, which became much more significant in 24 hours of incubation (Figure 5). So we can see that the dual drug-loaded DDS carrying both anti-vasculature and antitumor agents can be uptaken quickly and in large amount by cancer cells and endothelial cells.
In vitro evaluation of treatment efficacy for endothelial cells by DC@MSNs
The capability of C@MSNs and DC@MSNs in disrupting the HUVEC β-tubulin structure was then investigated. Cultured respectively with C@MSNs, DC@MSNs, and the MSNs in 24 hours, the β-tubulin structure of HUVECs was dyed with green fluorescence. The results are shown in Figure 6. MSN-treated HUVECs showed no significant β-tubulin damage, while the HUVECs treated with C@MSNs and DC@MSNs presented significant β-tubulin disruption, and, especially, HUVECs treated with DC@MSNs were destroyed completely.
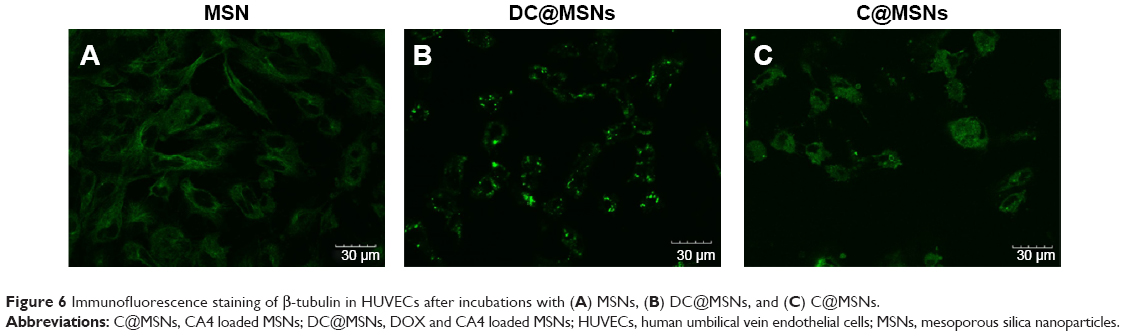 | Figure 6 Immunofluorescence staining of β-tubulin in HUVECs after incubations with (A) MSNs, (B) DC@MSNs, and (C) C@MSNs. |
In vivo evaluation of antitumor effect
To evaluate the antitumor effect of DC@T-MSNs and other drug-loaded MSNs, the tumor growth behaviors in HeLa tumor-bearing mice were recorded. Figure 7 shows the results of the tumor volume and body weight changes in different mouse groups treated with single DOX or CA4, or dual DOX–CA4-loaded DDSs. From the results, tumor growth was inhibited by DC@T-MSNs most effectively; actually, the tumors showed no significant growth in around 20 days of vein injection in this case, as compared to the retarded tumor growth by the other DDSs or free drugs at the same DOX dose of 1.5 mg/kg. The fact that body weight increased slowly in both control and the DC@T-MSN groups indicates that the mice were in a relatively normal state. Non-targeting drug-loaded MSNs and free drugs showed much weaker inhibiting effects on tumor growth than the targeting groups, in correspondence to the body weight losses of mice in these groups.
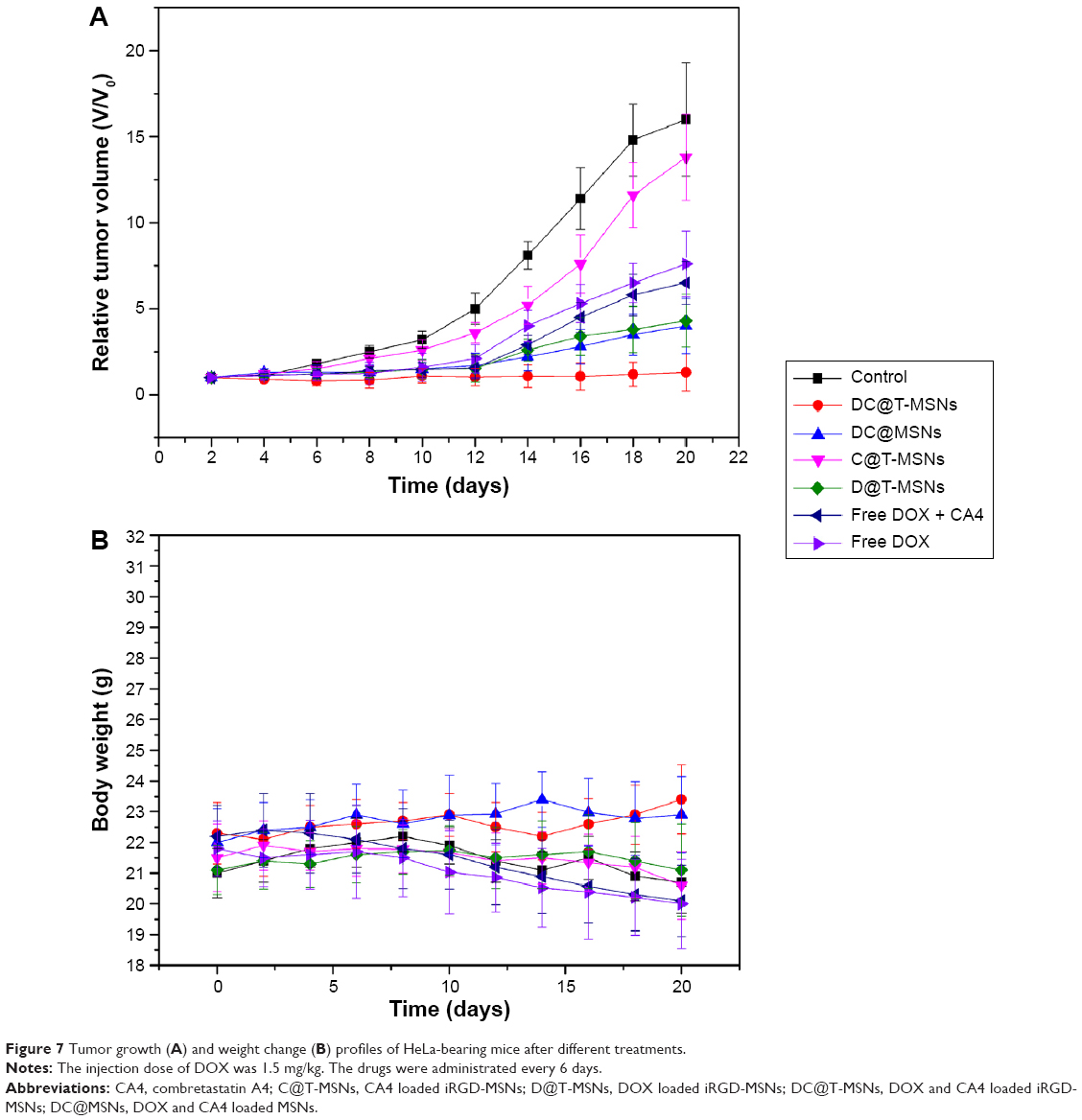 | Figure 7 Tumor growth (A) and weight change (B) profiles of HeLa-bearing mice after different treatments. |
In vivo tumor biodistributions of MSNs and T-MSNs
Tumor blood vessels were detected by immunofluorescence stain (CD31). When T-MSNs were injected alone, the red fluorescence of T-MSNs was found along with the green fluorescence of the tumor blood vessels (Figure 8), whereas in tumors treated by MSNs, there was no red fluorescence observable at the tumor blood vessels.
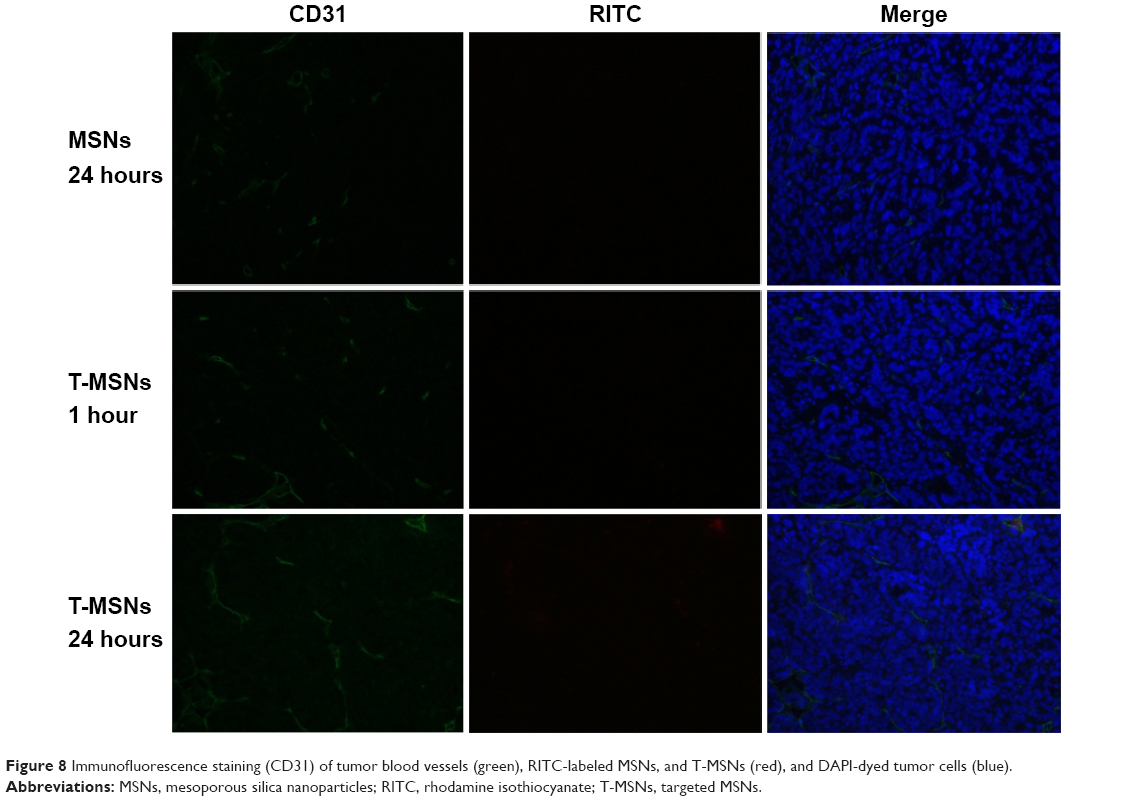 | Figure 8 Immunofluorescence staining (CD31) of tumor blood vessels (green), RITC-labeled MSNs, and T-MSNs (red), and DAPI-dyed tumor cells (blue). |
Two groups of four HeLa tumor-bearing mice were each intravenously injected with MSNs and T-MSNs for biodistribution studies. To obtain quantitative dosages of MSNs and T-MSNs uptaken in different organs in the following 24 hours after intravenous injection, the silicon concentrations in the tumor, heart, liver, spleen, lung, and kidney were analyzed by ICP-OES. The results are shown in Figure 9. Tumors treated by T-MSNs showed substantially enhanced amounts of Si uptake in 24 hours compared with tumors treated by MSNs, showing the prominent targeting effect of T-MSNs. Mice treated with both targeting and non-targeting MSNs showed relatively high Si uptakes both in the liver and lung, confirming the remarkable capture of nanoparticles by the reticuloendothelial system (RES).
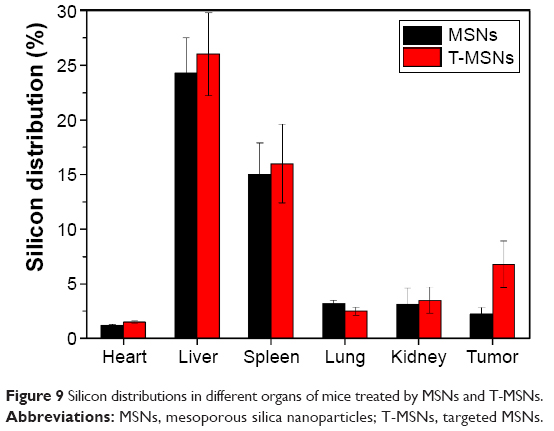 | Figure 9 Silicon distributions in different organs of mice treated by MSNs and T-MSNs. |
Histological evaluation after different treatments
TUNEL results are shown in Figure 10, showing the apoptosis of the cancer cells that received different treatments. Tumor cells treated with DC@T-MSNs showed the most significant TUNEL staining, while the other groups showed much weaker staining, indicating that DC@T-MSNs had the highest drug efficacy.
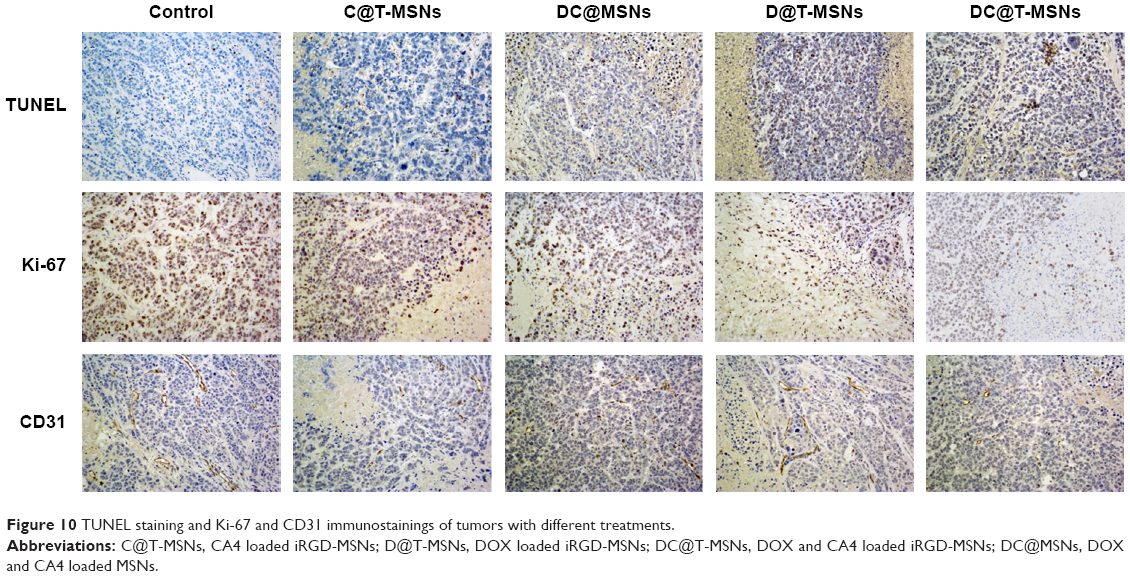 | Figure 10 TUNEL staining and Ki-67 and CD31 immunostainings of tumors with different treatments. |
Tumor cellular proliferative activity was measured by Ki-67 immunostaining. The group treated with DC@T-MSNs showed the least proliferative activity, but other drug-loaded MSN-treated groups showed much higher proliferative activity according to the stronger staining. So DC@T-MSNs presented the most proliferation inhibition effect on cancer cells.
Vasculatures in tumors treated with different DDSs were dyed by CD 31 immunostaining. Figure 10 shows that tumors treated with PBS maintained the apparent normal structure of vasculatures. Comparatively, vasculatures in tumors treated with D@MSNs were discontinued, and those treated with either C@MSNs or DC@MSNs were more significantly damaged, showing the substantial effect of CA4.
Discussion
In clinics, antiangiogenic agents are used usually as adjuvant therapy because of the low and incomplete killing effect of tumors and because traditional chemotherapy will lead to serious side effects and drug resistance. Reports have evidenced that the combination of two therapeutic protocols, such as anti-angiogenesis and chemotherapy or chemotherapy and thermal therapy, would have synergistic effects against malignant cancers. However, the conventional simple coadministration of two different drugs is incapable of guaranteeing these drugs reaching the same tumor site at respectively desired time points. Hence, the elaborate combination of different drugs’ deliveries of anti-vasculature and anticancer agents through a controllable and targetable DDS could be of great potential for cancer therapy.
In this study, MSNs were chosen as a drug support to deliver two different drugs, and iRGD was used as the targeting peptide. The wide-spectrum chemotherapeutic drug DOX and vascular disrupting agent CA4 were selected as chemotherapeutic agent and antiangiogenic agent, respectively. The iRGD peptide conjugated on MSNs can target α2β3 integrin receptors overexpressed in cancer cells and tumor vascular cells. As shown in Figure 2, the uniform and consistent morphology and small size of MSNs favors their prolonged blood circulation, which enables better targeting to the vessel wall of tumor sites. After peptide conjugation and drug loading, the morphology, dispersity, and hydraulic diameter have no significant changes, ensuring the long circulation of the drugs in the body.
Effective drug targeting is one of the keys in cancer chemotherapy. In the present study, the targeting effect was verified by in vivo tumor biodistributions of MSNs and T-MSNs. Tumors treated by T-MSNs showed a substantially enhanced amount of Si uptake in 24 hours compared with tumors treated by MSNs, showing the prominent targeting effect of T-MSNs by iRGD grafting.
The release behaviors of the two drugs (DOX and CA4) are in line with expectations. After injection into the vascular system, the double drug-loaded MSNs will circulate in the bloodstream and gradually accumulate in the targeted tumor under the guidance of iRGD peptide to the α2β3 receptors on the angiogenes in tumors, and then firstly release most of CA4 -loaded in MSNs and a small fraction of DOX due to the electrostatic interaction and hydrogen bonding between positively charged DOX molecules and negatively charged mesopores. Sequentially, when the particles permeate deep into the interior of the tumor, then are uptaken by the tumor cells, a large amount of DOX will be released quickly owing to the promoted release of DOX in the tumor cells of lowered pH value, though DOX releases very slowly in a neutral environment. Thus, the differentiated releases of the antiangiogenic agent and then the chemotherapeutic anticancer drug, respectively, at tumor vasculature and within tumor cells can be realized, which, we believe, will be highly favorable for enhancing the anticancer efficacy.
Thus, the greatly enhanced therapeutic efficacy of the double-loaded DC@MSNs compared to the single-loaded D@MSNs or C@MSNs can be attributed to the synergetic effect between sequentially released antiangiogenic agent CA4 and chemotherapeutic anticancer drug DOX: CA4 is first released from DC@MSNs to the tumor vascular system under the effective iRGD targeting. As a vascular disrupting agent, the released CA4 would result in significant tumor vascular disruption, tumor hemorrhage, and shutdown of tumor blood flow. Then, the damaged tumor vasculatures would facilitate the permeation of the DDS, which is carrying most of the unreleased DOX, into the tumor microenvironment and the accumulation of DOX molecules within the tumor cells with high concentration through intracellular endocytosis of the DDS. Finally, the DOX release is promoted in the acidic environment in tumor cells, and the released DOX efficiently induces the apoptosis of cancer cells therein. Such a synergetic effect during the drug delivery by DC@MSNs is responsible for the greatly enhanced cytotoxic effect of DOX to cancer cells and the in vivo antitumor efficacy.
The greatly enhanced in vivo antitumor efficacy, including either antiangiogenic or anticancer effects, has been evidenced by the immunohistochemical staining, CD 31 staining, and Ki-67 staining results, as shown in Figure 10. It can be seen that DC@T-MSNs are able to induce widespread tumor vasculature disruption (CD31 staining), extensive cytotoxicity (TUNEL), and strong proliferation inhibition of tumor cells (Ki-67 staining). Compared with the groups treated with non-targeted MSNs or single drug-loaded MSNs, DC@T-MSNs induced the strongest disruption and apoptosis of the tumor cells.
In cancer therapy, in order to improve the efficacy of treatment, high doses of chemotherapeutic agents are used. But high doses of chemotherapeutic agents will bring about large numbers of problems, such as drug resistance and damage to other organs, which are the most important reasons leading to high mortality. So, low-dose but high-efficiency chemotherapeutic agents are greatly needed. Encouragingly, by intravenously administrating our targeting and dual-loaded MSN delivery system, DC@T-MSNs, the therapeutic effect is so remarkable that the tumor growth can actually be almost totally inhibited in nearly 3 weeks at a rather low DOX dose of 1.5 mg/kg, in great contrast to other single-loaded and/or non-targeting counterparts. This demonstrates the great potential of combining dual-drug delivery and tumor vascular targeting in future cancer therapy.
Conclusion
In this study, we designed and constructed an MSN-based DDS with iRGD peptide surface modification for tumor vascular targeting and sequential antiangiogenic agent (CA4) and chemotherapeutic drug (DOX) co-delivery. The double-loaded and targeting DDSs were featured with significant releases at different rates of the antiangiogenic agent and the chemotherapeutic drug at tumor vasculature and within the tumor cells, respectively. As a result, both antiangiogenic and anticancer effects, such as vascular disruption, enhanced permeability of the vessel wall, extensive cytotoxicity, cellular proliferation inhibition, and apoptosis induction, were achieved in vivo, which finally resulted in the complete suppression of tumor growth in nearly 3 weeks at an extremely low DOX dose of 1.5 mg/kg. A synergetic effect between the antiangiogenic effect by CA4 and the anticancer effect by DOX was manifested and discussed in the in vivo tumor experiments. Thus, we envision that the present DDS featured with dual drug co-delivery and tumor vascular-targeting effect will be of great potential in the future clinical treatment of cancer.
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (grant numbers 51132009, 51102259).
Disclosure
The authors report no conflicts of interest in this work.
References
He Q, Gao Y, Zhang L, et al. A pH-responsive mesoporous silica nanoparticles-based multi-drug delivery system for overcoming multi-drug resistance. Biomaterials. 2011;32(30):7711–7720. | ||
Simberg D, Duza T, Park JH, et al. Biomimetic amplification of nanoparticle homing to tumors. Proc Natl Acad Sci U S A. 2007;104(3):932–936. | ||
Pearce TR, Shroff K, Kokkoli E. Peptide targeted lipid nanoparticles for anticancer drug delivery. Adv Mater. 2012;24(28):3803–3822. | ||
Chang DK, Chiu CY, Kuo SY, et al. Antiangiogenic targeting liposomes increase therapeutic efficacy for solid tumors. J Biol Chem. 2009;284(19):12905–12916. | ||
Gosk S, Moos T, Gottstein C, Bendas G. VCAM-1 directed immunoliposomes selectively target tumor vasculature in vivo. Biochim Biophys Acta. 2008;1778(4):854–863. | ||
Mann AP, Tanaka T, Somasunderam A, Liu X, Gorenstein DG, Ferrari M. E-selectin-targeted porous silicon particle for nanoparticle delivery to the bone marrow. Adv Mater. 2011;23(36):H278–H282. | ||
Gu G, Gao X, Hu Q, et al. The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials. 2013;34(21):5138–5148. | ||
Perabo F, Sharma S, Gierer R, et al. Circulating intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and E-selectin in urological malignancies. Indian J Cancer. 2001;38(1):1–7. | ||
Shi S, Yang K, Hong H, et al. Tumor vasculature targeting and imaging in living mice with reduced graphene oxide. Biomaterials. 2013;34(12):3002–3009. | ||
Liu D, Liu F, Liu Z, Wang L, Zhang N. Tumor specific delivery and therapy by double-targeted nanostructured lipid carriers with anti-VEGFR-2 antibody. Mol Pharm. 2011;8(6):2291–2301. | ||
Huang S, Shao K, Liu Y, et al. Tumor-targeting and microenvironment-responsive smart nanoparticles for combination therapy of antiangiogenesis and apoptosis. ACS Nano. 2013;7(3):2860–2871. | ||
Zhang YF, Wang JC, Bian DY, Zhang X, Zhang Q. Targeted delivery of RGD-modified liposomes encapsulating both combretastatin A-4 and doxorubicin for tumor therapy: in vitro and in vivo studies. Euro J Pharm Biopharm. 2010;74(3):467–473. | ||
Xuan R, Lin L, Jiang D. [Vascular targeting agent-combretastatin A4]. Chinese Journal of New Drugs. 2007;16(17):1336–1341. Chinese. | ||
Yang T, Wang Y, Li Z, et al. Targeted delivery of a combination therapy consisting of combretastatin A4 and low-dose doxorubicin against tumor neovasculature. Nanomedicine. 2012;8(1):81–92. | ||
Nelkin BD, Ball DW. Combretastatin A-4 and doxorubicin combination treatment is effective in a preclinical model of human medullary thyroid carcinoma. Oncol Rep. 2001;8(1):157–160. | ||
Mitrus I, Sochanik A, Cichoń T, Szala S. Combination of combretastatin A4 phosphate and doxorubicin-containing liposomes affects growth of B16-F10 tumors. Acta Biochim Pol. 2009;56(1):161–165. | ||
Zhang F, Huang X, Zhu L, et al. Noninvasive monitoring of orthotopic glioblastoma therapy response using RGD-conjugated iron oxide nanoparticles. Biomaterials. 2012;33(21):5414–5422. | ||
Shenoi MM, Iltis I, Choi J, et al. Nanoparticle delivered vascular disrupting agents (VDAs): use of TNF-alpha conjugated gold nanoparticles for multimodal cancer therapy. Mol Pharm. 2013;10(5):1683–1694. | ||
Chen Y, Chen H, Zeng D, et al. Core/shell structured hollow mesoporous nanocapsules: a potential platform for simultaneous cell imaging and anticancer drug delivery. ACS Nano. 2010;4(10):6001–6013. | ||
Hauck TS, Ghazani AA, Chan WC. Assessing the effect of surface chemistry on gold nanorod uptake, toxicity, and gene expression in mammalian cells. Small. 2008;4(1):153–159. | ||
Lebold T, Jung C, Michaelis J, Bräuchle C. Nanostructured silica materials as drug-delivery systems for doxorubicin: single molecule and cellular studies. Nano Lett. 2009;9(8):2877–2883. | ||
He Q, Gao Y, Zhang L, et al. One-pot self-assembly of mesoporous silica nanoparticle-based pH-responsive anti-cancer nano drug delivery system. J Mater Chem. 2011;21(39):15190–15192. | ||
He Q, Shi J. MSN anti-cancer nanomedicines: chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv Mater. 2014;26(3):391–411. | ||
Chen Y, Chen H, Shi J. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv Mater. 2013;25(23):3144–3176. | ||
Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. | ||
Ma M, Chen H, Chen Y, et al. Au capped magnetic core/mesoporous silica shell nanoparticles for combined photothermo-/chemo-therapy and multimodal imaging. Biomaterials. 2012;33(3):989–998. | ||
He Q, Zhang J, Chen F, Guo L, Zhu Z, Shi J. An anti-ROS/hepatic fibrosis drug delivery system based on salvianolic acid B loaded mesoporous silica nanoparticles. Biomaterials. 2010;31(30):7785–7796. | ||
Gao Y, Yang C, Liu X, Ma R, Kong D, Shi L. A multifunctional nanocarrier based on nanogated mesoporous silica for enhanced tumor-specific uptake and intracellular delivery. Macromol Biosci. 2012;12(2):251–259. | ||
He Q, Shi J. Mesoporous silica nanoparticle based nano drug delivery systems: synthesis, controlled drug release and delivery, pharmacokinetics and biocompatibility. J Mater Chem. 2011;21(16):5845–5855. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.